

Abstract
Diphthamide biosynthesis involves a carbon-carbon bond forming reaction catalyzed by a radical S-adenosylmethionine (SAM) enzyme that cleaves a C-S bond in SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. Using rapid freezing, we have captured an organometallic intermediate with an Fe-C bond between ACP and the enzyme’s [4Fe-4S] cluster. In the presence of the substrate protein, elongation factor 2, this intermediate converts to an organic radical, formed by addition of the ACP radical to a histidine side chain. Crystal structures of archaeal diphthamide biosynthetic radical SAM enzymes reveal that the carbon of the SAM C-S bond being cleaved is positioned near the unique cluster Fe, able to react with the cluster. Our results explain how selective C-S bond cleavage is achieved in this radical SAM enzyme.
One Sentence Summary:
An unusual radical enzyme forms an iron-carbon bond as the first step in modification of a protein side chain.
Diphthamide is a post-translationally modified histidine residue on archaeal and eukaryotic translation elongation factor 2 (EF2), a protein essential for ribosomal protein synthesis (1–3). Diphthamide biosynthesis involves at least four steps and seven proteins (4). The first step of the biosynthesis is the transfer of a 3-amino-3-carboxypropyl (ACP) group from S-adenosyl-L-methionine (SAM) to a histidine residue of EF2 (Fig. 1). The enzyme that performs this reaction is a Dph2 (diphthamide biosynthesis protein 2) homodimer in archaea, such as Pyrococcus horikoshii (PhDph2) (5) or a Dph1-Dph2 heterodimer in eukaryotes (6). Dph2 binds an essential [4Fe-4S] cluster and is thought to use the reduced (1+) state of the cluster to cleave the Cγ,Met-S bond of SAM to generate a 3-amino-3-carboxypropyl (ACP) radical. The formation of the ACP radical is supported by the PhDph2-catalyzed generation of 2-aminobutyric acid and homocysteine sulfinic acid in the absence of the substrate protein, PhEF2 (5). Additional support for a radical mechanism was provided by the reaction with a carboxyallyl SAM analogue, SAMCA (7). However, no radical intermediate has been directly observed in the reaction with SAM itself, and the detailed reaction mechanism remains unknown.
Fig. 1.
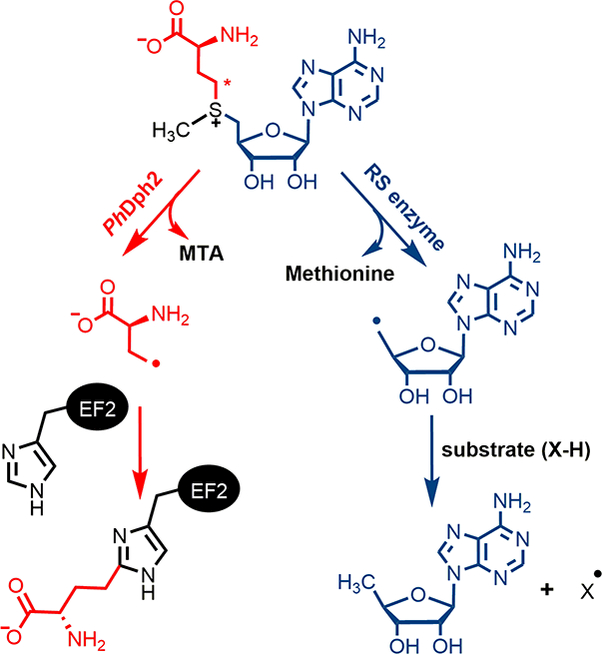
PhDph2 and 5′-dA• forming Radical SAM enzyme catalyzed reactions. The red asterisk labels Cγ,Met of SAM.
Despite using the same components, a [4Fe-4S] cluster and SAM, PhDph2 is structurally unrelated to the much larger family of 5′-deoxyadenosine radical (5′-dA•) forming radical SAM (RS) enzymes. In those enzymes, the C5′,Ade-S bond of SAM is cleaved, generating a 5′-dA• that then initiates downstream reactions (Fig. 1) (8). The question of how PhDph2 homodimer or eukaryotic Dph1-Dph2 bind SAM and achieve a different SAM cleavage pattern remains unanswered. Herein we report biochemical and spectroscopic studies of two kinetically competent intermediates that, together with X-ray crystal structures of archaeal Dph2 homodimers in complex with SAM, allow us to propose a reaction mechanism for this unique class of RS enzymes.
We used rapid freeze-quench (RFQ) to arrest reaction mixtures, which were then analyzed by electron paramagnetic resonance (EPR) and electron-nuclear double resonance (ENDOR) spectroscopies for radical intermediates. Since the optimum temperature of PhDph2 is above 60 ℃, we opted to use the yeast Dph1-Dph2 system, which is active at room temperature and thus more convenient for RFQ experiments. A solution containing Dph1-Dph2 and SAM was mixed with dithionite and freeze-quenched at 500 ms to 5 min. Each sample displayed a new EPR signal with g|| = 2.036, g⊥= 2.005 at 12 K (Fig. S1). The intensity of this signal reached a maximum at 2 s quench time and decreased at longer delays (Fig. 2A). This species is not an ACP radical, which would not show such a high g-shift and would exhibit resolvable 1H hyperfine splittings from the methylene radical protons; nor is it an S-based radical, which could exhibit such g-values but again would give resolved hyperfine 1H splittings. Based on these observations and experience from previous work using SAMCA (7), we interpreted this new species as having an iron-sulfur cluster-based organometallic structure. Importantly, the EPR spectrum and g-values of this species resemble those of the organometallic intermediate that was detected in the RS enzyme, pyruvate formate lyase-activating enzyme (PFL-AE) and assigned to an Fe-5′-C bond between the deoxyadenosyl group and the unique cluster iron (9). As confirmation, when this intermediate was generated with 57Fe-enriched enzyme, its EPR spectrum exhibited 57Fe-hyperfine line broadening (Fig S1). Thus, the reaction catalyzed by Dph1-Dph2 produces an organometallic intermediate. We note that its g-values follow the pattern of a [4Fe-4S]3+ cluster, g|| > g⊥ > 2(10), where g|| is sensitive to coordination at the unique Fe, suggesting a possible formal description of the intermediate as containing a carbanion bound to an oxidized cluster.
Fig. 2.
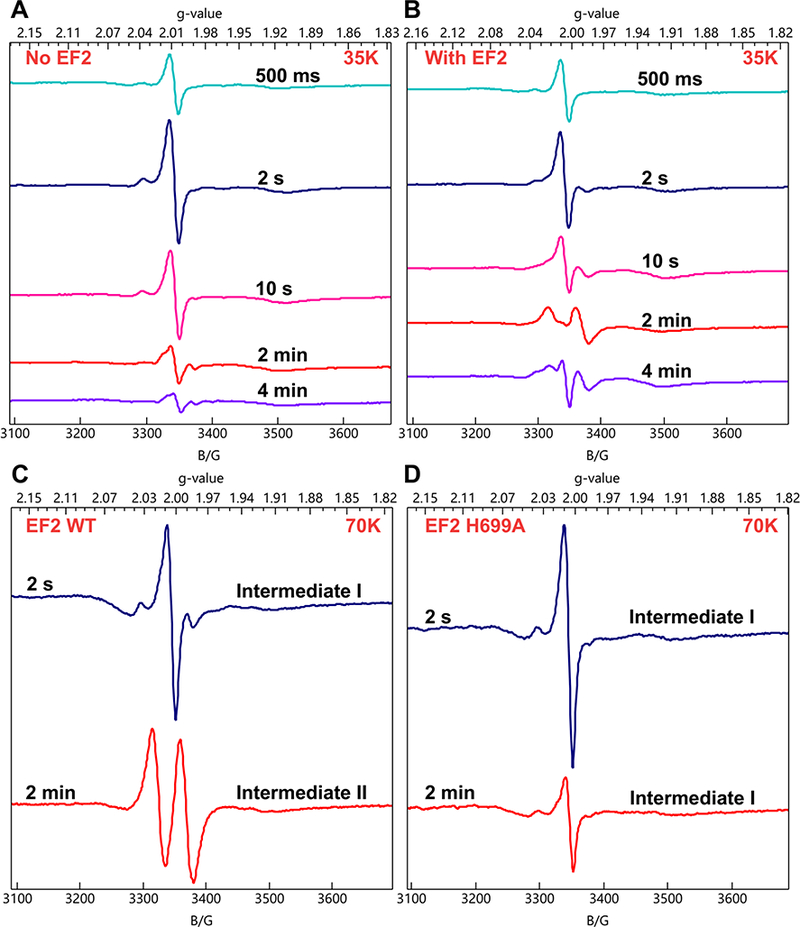
X-band CW EPR spectra of RFQ samples showing the formation of the organometallic intermediate I and organic radical intermediate II. (A) Reaction of reduced Dph1-Dph2 with SAM quenched at various time points (T = 35 K). (B) Reaction of reduced Dph1-Dph2 with SAM and EF2 quenched at various time points (T = 35 K). (C) Reaction of reduced Dph1-Dph2 with SAM and wild type EF2 quenched at 2 s and 2 min (T = 70 K). (D) Reaction of reduced Dph1-Dph2 with SAM and EF2 H699A mutant quenched at 2 s and 2 min (T = 70 K). We note that in early-time spectra a variable contribution at g-2 from an unidentified organic radical enhances the intensity of the g⫠ feature of Intermediate I relative to that at g|| and distorts its shape; Figure S1 presents spectra with little of the radical and thus a more faithful lineshape of Intermediate I.
To establish the structure of this intermediate, we collected RFQ ENDOR samples prepared with (methionine-13C5)-SAM, in which the methionine carbons of SAM were uniformly labeled with 13C. Field-modulated continuous wave (CW) ENDOR spectra obtained at 2 K of this sample exhibits a 13C doublet, with a 13C hyperfine coupling constant of A ≈ 7.8 MHz (Fig. 3A). This coupling constant is comparable to Aiso = 9.4 MHz observed for the Fe–[5′−13C]-deoxyadenosyl bond in the PFL-AE intermediate, and together with the 57Fe hyperfine broadening, provides strong support that this intermediate contains an Fe-C bond. However, in this case, the Fe-bonded carbon must originate from the 13C-labeled methionine, presumably the Cγ,Met.
Fig. 3.
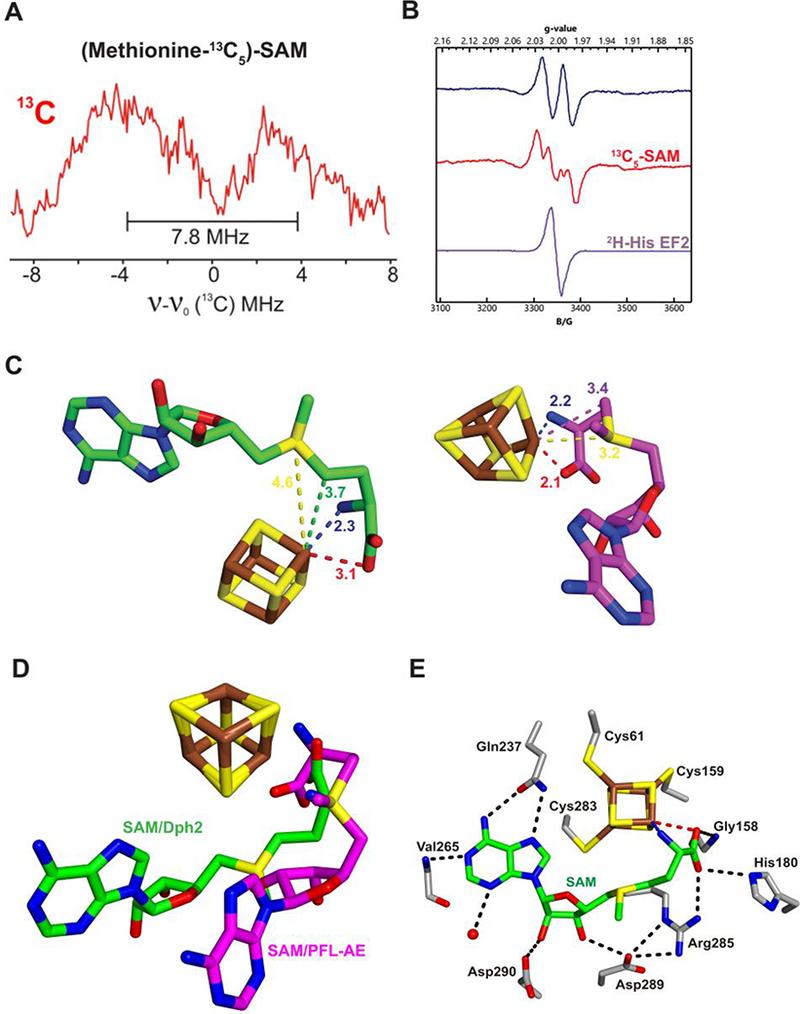
A) 35 GHz CW 13C ENDOR spectra for intermediate I with (methionine-13C5)-SAM. B) Isotope EPR study of the organic radical intermediate II on EF2 quenched at 2 min (T=70 K). C) Cluster and bound SAM in the structure of CmnDph2 (left), in comparison with cluster and bound SAM in the structure of PFL-AE (right, PDB 3CB8) (13). Distances are given in angstroms. The black and brown asterisks label Cγ,Met of SAM and the differentiated iron of the [4Fe-4S] cluster, respectively. D) Overlay of SAM and cluster in CmnDph2 and PFL-AE structures. E) CmnDph2 active site showing the binding of SAM. Hydrophobic interactions with purine ring of SAM are not shown for clarity.
In testing whether this organometallic species (denoted intermediate I) was an active reaction intermediate, we collected a RFQ EPR time-course in the presence of the substrate protein, EF2. Intermediate I accumulated in the first 10 s (Fig. 2B). At later time points, the intermediate I signal diminished concomitantly with the appearance of a doublet radical signal centered at g = 2 (Fig. 2B). This new signal, denoted intermediate II, reached a maximum at 2 min quench time and decreased at 5 min (Fig. 2B) in a process that ultimately leads to product formation.
The intermediate II EPR signal was distorted by saturation effect at 35 K (Fig. 2B), but was readily observed at 70 K (Fig. 2C), where spin relaxation is faster. Tentative assignment of the doublet splitting to coupling to a single proton with a large hyperfine splitting (A ≈ 120 MHz) led us to hypothesize that this intermediate is an organic radical generated when the organometallic intermediate I reacted with the enzymatic target, histidine 699 (H699) of yeast EF2. To test this, we repeated the RFQ experiment with the EF2 His699 to alanine mutant (H699A), in which the target histidine residue was mutated to Ala. With this EF2 mutant, only the organometallic intermediate I was detected at both 2 s and 2 min (Fig. 2D) and no intermediate II signal was detected. This experiment provides strong support that intermediate I is chemically competent.
To test the proposed structure, intermediate II was RFQ-trapped with (methionine-13C5)-SAM. The resulting X-band EPR spectrum showed additional splitting of Aiso(13C) ≈ 59 MHz and broadening compared to that of the natural-abundance SAM (Fig. 3B), demonstrating that the radical incorporates the ACP group of SAM. The EPR-resolved doublet, A(1H) ≈ 120 MHz, observed for intermediate II collapsed to a singlet when intermediate II was prepared with 2H5-His labeled EF2 protein (Fig. 3B), confirming that the splitting observed with the natural-abundance EF2 was associated with 1H of the histidine residue on EF2. This was further confirmed by 1H ENDOR of intermediate II with EF2, which exhibited a non-exchangeable 1H signal with the same coupling (Fig. S2); along with an exchangeable coupling, A(1H) ≈ 15 MHz not resolved in the EPR spectrum was also seen. These results confirm that intermediate II is a radical produced by the reaction of an ACP radical from Intermediate I with the imidazole ring of His699 of the EF2 substrate.
Given the structure of the final diphthamide product, a candidate for the structure of intermediate II is an ACP modified histidine radical, as shown in Fig. 4. The shape of the 1H doublet in the EPR spectrum (Fig. 3B) and the strongly-coupled 1H ENDOR signal (Fig. S2) suggest that the proton coupling is essentially isotropic, as expected for a proton β to the spin in the N-2p-π orbital (Fig. 4)(11); the resolved 13C splitting in the EPR spectrum of intermediate II (Fig. 3B) prepared with (methionine-13C5)-SAM must come from the carbon of the ACP fragment bound to the His, again β to the spin site. The individual peaks of the 1H doublet are broad enough to contain unresolved 14N splitting introduced by the spin density on the 14Nδ of His (Fig. 4). As shown in Fig. S3, the EPR spectrum of the natural-abundance radical is well simulated with a strong, nearly-isotropic 1H coupling (A1= A2= 122 MHz, A3=103 MHz) and a highly anisotropic 14N hyperfine coupling (A1=A2=4 MHz, A3=54 MHz). 15N features seen in the ENDOR of the 15N-His EF2 sample (Fig. S4) likely are a superposition of signals associated with the A1/A2 components of this histidine nitrogen (assigned as Nδ in Fig. 4), and those from the less strongly coupled His 15Nε. Finally, weakly coupled 13C ENDOR signals seen when intermediate II was prepared with 13C-His EF2 (Fig. S4, S5) are assigned to His carbons bonded to the spin-bearing Nδ (Fig 4.), and account for the slight broadening in the components of the 1H doublet in this isotopologue; an exchangeable 1H seen in ENDOR (Fig S2) is thought to reside on Nε of His.
Fig. 4.
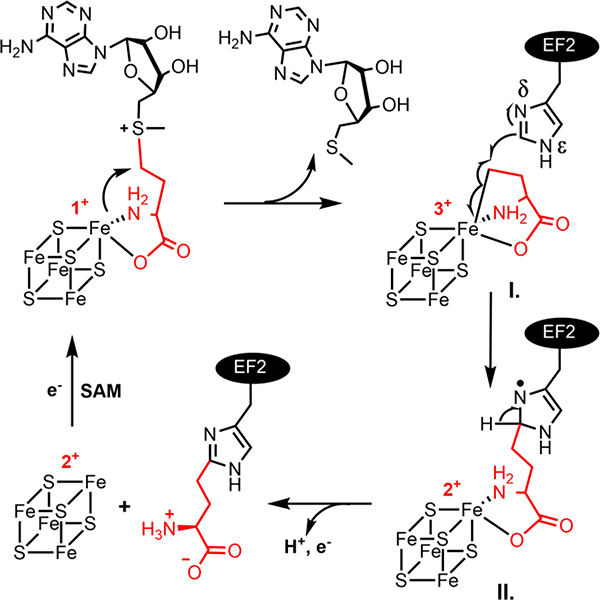
The proposed reaction mechanism of diphthamide biosynthetic RS enzymes.
The time-scale for intermediate II formation measured by EPR tracked that of intermediate I decay, while the reaction product, ACP modified EF2 as quantified by mass spectrometry, accumulates on the same time scale as the intermediate II decay (Fig. S7). These results indicate that both Intermediate I and Intermediate II are formed in a kinetically competent fashion. Thus, we have trapped two sequential intermediates, an organometallic intermediate I, which gives rise to an organic radical intermediate II, which goes on to complete the first step of diphthamide biosynthesis.
To gain further understanding into the reaction mechanism, we obtained crystal structures of the RS enzymes involved in diphthamide biosynthesis in complex with SAM. A 2.3 Å resolution structure of [4Fe-4S]2+-loaded PhDph2 in complex with SAM showed some evidence of SAM cleavage. A 2.1 Å resolution structure of Candidatus Methanoperedens nitroreducens Dph2 (CmnDph2) showed mostly uncleaved SAM. Both structures showed average N-Fe and O-Fe distances of 2.3 and 3.1 Å, respectively (Fig. 3C and Fig. S8), suggesting that similar to 5′-dA• forming RS enzymes, the amino and carboxylate groups of SAM coordinate the unique Fe of the [4Fe-4S] cluster in Dph2. This is consistent with our recent study showing that both the amino and carboxylate of SAM are important for the PhDph2 and Dph1-Dph2 catalyzed reactions (12).
However, SAM in both PhDph2 and CmnDph2 binds with a distinct geometry compared to that in 5′-dA• forming RS enzymes such as PFL-AE(13) (Fig. 3D). We further obtained additional crystal structures using both PhDph2 and CmnDph2 that support the geometry shown in Fig. 3C and 3D (see supplementary information Fig. S9-S11). The SAM binding site of CmnDph2 is shown in Fig. 3E. Gln237 forms hydrogen bonds with N6 and N7 of the adenine ring and the amide nitrogen of Val265 hydrogen bonds to N1. The adenine ring is sandwiched between Phe58 and Ile286. Asp289 and 290 form hydrogen bonds with the hydroxyl groups of the ribosyl moiety, while Arg285, His180, and Gly158 form hydrogen bonds with the carboxylate of SAM.
Most strikingly, the Cγ,Met distance to the unique Fe, as averaged over the PhDph2 and CmnDph2 structures, is 3.7 Å, which is closer than SMet to the Fe (the SMet-Fe distance is 4.6 Å). In contrast, in the PFL-AE structure, the SMet is closer to the unique Fe than C5′,Ade, with an SMet-Fe distance of 3.2 Å. The observation that in PhDph2 and CmnDph2, Cγ,Met was closer than SMet to the unique Fe suggests that during the Cγ,Met-SMet bond cleavage, the electron is transferred from the Fe-S cluster via Cγ,Met, while for PFL-AE (and generally believed for all 5′-dA• forming RS enzymes), the electron is transferred from the Fe-S cluster via SMet (8).
Based on the two intermediates we have detected and the structures of PhDph2 and CmnDph2 in complex with SAM, we propose a reaction mechanism for the first step of diphthamide biosynthesis (Fig. 4). The unique iron of a [4Fe-4S]+ cluster in an archaeal Dph2 homodimer or eukaryotic Dph1-Dph2 heterodimer attacks the γ carbon of the methionine in SAM, generating a 3-amino-3-carboxypropyl-[4Fe-4S]3+ organometallic intermediate I and 5′-methylthioadenosine (MTA). The organometallic intermediate I is essentially a stabilized ACP radical. In the presence of the substrate EF2, the Fe-C bond can break homolytically to react with the imidazole ring of the histidine residue of EF2 and generate the organic radical, intermediate II. Intermediate II then loses a proton and an electron to form an ACP-modified histidine.
In the study of PFL-AE, it was noted that the Fe-[5′-C]-deoxyadenosyl organometallic intermediate could be generated through a radical reaction or nucleophilic reaction (9). In our case, the formation of intermediate I could occur via homolytic reductive-cleavage of SAM to form an ACP radical, which then reacts with the unique Fe (stepwise one-electron transfer) to form the organometallic intermediate I. Alternatively, a concerted one-step, two-electron transfer (nucleophilic attack on Cγ,Met) can form intermediate I directly. The fact that Cγ,Met was only 3.7 Å away from the unique Fe, whereas the sulfonium moiety was 4.6 Å away (Fig. 3C), favors the latter mechanism, although we cannot eliminate the homolytic cleavage mechanism.
The structure also provides a simple solution to the question, how does archaeal Dph2 homodimer or eukaryotic Dph1-Dph2 heterodimer cleave a different C-S bond in SAM compared to all 5′-dA• forming RS enzymes? If the cluster interacts with and transfers electrons to Cγ,Met instead of SMet, the only C-S bond that can be broken in SAM is the Cγ,Met-S bond. Previously, Kampmeier proposed that a stereo-electronic control mechanism could explain the different cleavage patterns (14). This model rationalizes the C-S bond cleavage in RS enzymes using a radical displacement reaction, with the formation of a Fe-S bond accompanying the cleavage of the C-S bond (Fig. S12). This radical displacement reaction also requires that the C-S-Fe atoms involved in the release of a radical on carbon are arranged roughly co-linearly. Thus, if SAM in archaeal Dph2 homodimer or eukaryotic Dph1-Dph2 is bound in a conformation with SMet close to the Fe-S cluster and co-linear Cγ,Met-S-Fe arrangement, it would lead to the cleavage of Cγ,Met-S. However, our structure indicates that in archaeal Dph2 homodimer, the Cγ,Met is close to the unique Fe. The Cγ,Met mediates electron transfer from the Fe-S cluster, dictating which bond is cleaved. This is an important difference between diphthamide biosynthetic RS enzymes and 5′-dA• forming RS enzymes. Why does Nature choose this way to control the bond cleavage in diphthamide biosynthetic RS enzymes? Perhaps conformational constraints on SAM prevent the Cγ,Met-S-Fe co-linear arrangement proposed by Kampmeier, while the roughly co-linear S-Cγ,Met-Fe arrangement as revealed by the structures is feasible (Fig. S12).
In summary, this study has explained how diphthamide biosynthetic RS enzymes create the EF2 product through cleavage of the Cγ,Met-S bond of SAM in a process that involves first an organometallic intermediate then a radical intermediate that collapses to product. Together with the recent study showing an organometallic intermediate in a 5′-dA• forming RS enzyme PFL-AE, these studies further suggest that an organometallic intermediate may serve as a stabilized form of the highly reactive primary organic radical, and provide a strong parallel between RS enzymes and adenosylcobalamin-dependent enzymes.
Supplementary Material
Acknowledgments:
We thank Dr. Minkui Luo at Memorial Sloan–Kettering Cancer Center for providing the methionine adenosyltransferase I plasmid. We thank Dr. Peter Doan for help in confirming the organometallic nature of intermediate I and Matthew Oliver Ross for assistance in collecting ENDOR spectra. We thank Dr. Sheng Zhang (Proteomics and Metabolomics Facility at Cornell) for the help with the mass spectrometry experiment. M. D. thanks Drs. Tyler L. Grove and R. David Britt for helpful discussion.
Funding: This work was supported by grants from the National Institute of Health National Institute of General Medical Sciences (GM088276 to H.L., P41GM103521 to J.H.F., P41GM103403 to S.E.E, GM 111097 to B.M.H., and GM124908 to K.M.L.). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines. The Pilatus 6M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Footnotes
Author contributions: M.D. contributed to experimental design, performed the biochemical experiments, and prepared the proteins for spectroscopic studies. V.K. and A.S. performed the ENDOR studies. M.K., A.T.T., and Y.Z. carried out the crystallization and structure determination of all the pr1teins. J.D.C. and K.M.L. provided critical support for the RFQ experiments and EPR simulation and preformed the fitting of kinetic data. M.D., B.D. and J.H.F performed the X-band EPR measurements. S.E.E. directed the X-ray structural studies; B.M.H. directed the ENDOR studies and contributed to experimental design; H.L. directed the biochemical studies, contributed to experimental design, and coordinated collaborations. M.D., H.L., B.M.H., and S.E.E. wrote the manuscript with the inputs from V.K., M.K.F., A.T.T., Y.Z., and J.D.C. All authors reviewed, edited, and approved the manuscript.
Competing interests: The authors declare no competing financial interests.
Data and materials availability: Crystallographic models have been deposited in the Protein Data Bank under accession codes 6BXK for PhDph2/MTA, 6BXL for PhDph2/SAM, 6BXM for CmnDph2/SAM1, 6BXN for CmnDph2/SAM2, and 6BXO for CmnDph2/SAH. Raw data and materials associated with the main text and the supplementary materials are available for sharing upon request. Supplementary Materials:
References and Notes:
- 1.Robinson EA, Henriksen O, Maxwell ES, Elongation factor 2. Amino acid sequence at the site of adenosine diphosphate ribosylation. J. Biol. Chem 249, 5088–5093 (1974). [PubMed] [Google Scholar]
- 2.Van Ness BG, Howard JB, Bodley JW, ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J. Biol. Chem 255, 10717–10720 (1980). [PubMed] [Google Scholar]
- 3.Van Ness BG, Howard JB, Bodley JW, ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem 255, 10710–10716 (1980). [PubMed] [Google Scholar]
- 4.Schaffrath R, Abdel-Fattah W, Klassen R, Stark MJ, The diphthamide modification pathway from Saccharomyces cerevisiae--revisited. Mol. Microbiol 94, 1213–1226 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y et al. , Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 465, 891–896 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong M et al. , Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. J. Am. Chem. Soc 136, 1754–1757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong M et al. , Organometallic Complex Formed by an Unconventional Radical S-Adenosylmethionine Enzyme. J. Am. Chem. Soc 138, 9755–9758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broderick JB, Duffus BR, Duschene KS, Shepard EM, Radical S-adenosylmethionine enzymes. Chem. Rev 114, 4229–4317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horitani M et al. , Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-C]-deoxyadenosyl bond. Science 352, 822–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cutsail GE 3rd, Telser J, Hoffman BM, Advanced paramagnetic resonance spectroscopies of iron-sulfur proteins: Electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM). Biochim. Biophys.Acta 1853, 1370–1394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.A. M. Carrington AD, Introduction to Magnetic Resonance with Applications to Chemistry and Chemical Physics. (Harper & Row, New York, 1967). [Google Scholar]
- 12.Dong M et al. , Substrate-Dependent Cleavage Site Selection by Unconventional Radical S-Adenosylmethionine Enzymes in Diphthamide Biosynthesis. J. Am. Chem. Soc 139, 5680–5683 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vey JL et al. , Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc.Natl. Acad. Sci. U. S. A 105, 16137–16141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kampmeier JA, Regioselectivity in the homolytic cleavage of S-adenosylmethionine. Biochemistry 49, 10770–10772 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Frazzon J, Dean DR, Formation of iron-sulfur clusters in bacteria: an emerging field in bioinorganic chemistry. Curr.Opin.Chem.Biol 7, 166–173 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Beinert H, Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem 131, 373–378 (1983). [DOI] [PubMed] [Google Scholar]
- 17.Su X et al. , YBR246W is required for the third step of diphthamide biosynthesis. J. Am. Chem. Soc 134, 773–776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du J, Jiang H, Lin H, Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 48, 2878–2890 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Wang R, Zheng W, Luo M, A sensitive mass spectrum assay to characterize engineered methionine adenosyltransferases with S-alkyl methionine analogues as substrates. Anal. Biochem 450, 11–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caranto JD, Vilbert AC, Lancaster KM, Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. Proc. Natl. Acad. Sci. U. S. A 113, 14704–14709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werst MM, Davoust CE, Hoffman BM, Ligand spin densities in blue copper proteins by q-band proton and nitrogen-14 ENDOR spectroscopy. J. Am. Chem. Soc 113, 1533–1538 (1991). [Google Scholar]
- 22.C. E. D. Davoust PE; Hoffman BM, J. Magn. Reson 119, 38 (1996). [Google Scholar]
- 23.Otwinoski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode. Method. Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Painter J, Merritt EA, Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D62, 439–450 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Chen VB et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenwick MK et al. , Non-canonical active site architecture of the radical SAM thiamin pyrimidine synthase. Nat. Commun 6, 6480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta AP et al. , Anaerobic 5-Hydroxybenzimidazole Formation from Aminoimidazole Ribotide: An Unanticipated Intersection of Thiamin and Vitamin B12 Biosynthesis. J. Am. Chem. Soc 137, 10444–10447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenwick MK, Li Y, Cresswell P, Modis Y, Ealick SE, Structural studies of viperin, an antiviral radical SAM enzyme. Proc. Natl. Acad. Sci. U. S. A(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanzelmann P et al. , Characterization of MOCS1A, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis. J. Biol. Chem 279, 34721–34732 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Mehta AP et al. , Catalysis of a new ribose carbon-insertion reaction by the molybdenum cofactor biosynthetic enzyme MoaA. Biochemistry 52, 1134–1136 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy AJ et al. , Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grove TL et al. , A substrate radical intermediate in catalysis by the antibiotic resistance protein Cfr. Nat. Chem. Biol 9, 422–427 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silakov A et al. , Characterization of a Cross-Linked Protein–Nucleic Acid Substrate Radical in the Reaction Catalyzed by RlmN. J. Am. Chem. Soc 136, 8221–8228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.