

Abstract
Acute kidney injury (AKI) is defined by a rapid decline in renal function. Regardless of the initial cause of injury, the influx of immune cells is a common theme during AKI. While an inflammatory response is critical for the initial control of injury, a prolonged response can negatively affect tissue repair. In this review, we focus on the role of macrophages, from early inflammation to resolution during AKI. These cells serve as the innate defense system by phagocytosing cellular debris and pathogenic molecules; and bridging communication with the adaptive immune system by acting as antigen-presenting cells and secreting cytokines. While many immune cells function to initiate inflammation, macrophages play a complex role throughout AKI. This complexity is driven by their functional plasticity: the ability to polarize from a “pro-inflammatory” phenotype to a “pro-reparative” phenotype. Importantly, experimental and translational studies indicate that macrophage polarization holds promise as a novel therapeutic strategy to promote repair during AKI. A thorough understanding of the biological roles these phagocytes play during both injury and repair is necessary to understand the limitations while furthering the therapeutic application.
Keywords: AKI, zebrafish, macrophage, neutrophils, polarization
Introduction
Acute kidney injury (AKI) encompasses a spectrum of disease mechanisms that ultimately result in a rapid decline in renal function. Clinically, the etiologies are classified as pre-renal (e.g., hypovolemia and sepsis), intrinsic (e.g., drugs and toxins), and post-renal causes (e.g., obstruction or malignancy). Despite the array of initial insults, activation of the immune defense systems is a common thread, with immune cells playing a prominent role from initiating injury to promoting tissue repair [1].
The first phase of immune cell recruitment begins when injured renal tubular epithelial cells (RTECs) increase expression of damage-associated molecular pattern (DAMP) molecules, Toll-like receptors (TLRs), and other alarmins [1]. These signals recruit the first responders of the innate immune system – neutrophils, natural killer cells, activated macrophages, and resident macrophages to the site of injury within 24 hours [2]. Most innate immune cells extravasate from the vascular system to the site of injury to engulf cellular debris, as well as further propagating immune response to recruit adaptive immune cells. Unlike most innate immune cells, resident macrophages reside in the kidney even during steady state for immunosurveillance [3]. During injury, resident macrophages will act to engulf cellular debris as additional bone-marrow derived macrophages and monocytes are recruited [4]. The rapid recruitment of immune cells is critical for the regenerative response in the kidney and is also conserved across multiple organ systems [5].
The timeline and balance of invading leukocytes appears to be critical and it has been shown that persistence of the immune response can lead to further damage. In particular, different subtypes of macrophages (called polarization), which will be discussed in this review, have gathered much attention for their roles in progression to chronic diseases in various organs, including the kidney [6, 7]. Without proper regulation of macrophage driven inflammation, tissue repair cannot be successful. This review will outline the major recruitment pathways involved in macrophage polarization, the roles macrophages play in tissue repair, clinical trials focused on pathways important for macrophage polarization, an alternative experimental model using zebrafish, and future directions for further expanding our knowledge of the immune response during AKI.
Macrophage origin and function
Macrophages were once viewed as a single population of phagocytic cells derived from the bone marrow [8]. Later studies determined there were two distinct lineages from which macrophages originate: embryonic and bone marrow [3]. These two populations have varying roles not only during mammalian development but also in response to injury in adults, where various lineages of macrophages play complex roles in both response to the initial insult and resolution of the damaged tissue.
Embryonic macrophages express the PU.1 transcription factor and are derived from the hematopoietic stem cell (HSC) lineage within the yolk sac. Later in development HSCs migrate from the yolk sac to the bone marrow, which becomes the site of hematopoiesis in mammals. Embryonic macrophages function in innate immune protection and regulate fetal architecture by promoting vascularization, clearing apoptotic cells, supplying cellular matrix components (laminin, triggrin, type IV collagen, and proteoglycans), and providing cues for RBC maturation [9]. Another crucial function for embryonic macrophages is to migrate to sites of developing organs and mature into various types of resident macrophages, such as the Kupffer cells in the liver. In each organ, resident macrophages have specific roles to maintain the steady state, including clearing cells undergoing apoptosis and engulfing cells to remodel tissue architecture during development. Over the course of an organism’s life resident macrophages self-replenish, carry out immunosurveillance, and other organ-specific functions [10]. For example, Kupffer cells play a role in liver detoxification, and in the brain microglia carry out synaptic pruning during development and adulthood [3]. Resident macrophages, along with the bone marrow-derived macrophages, assume different phenotypes during injury events to initiate and prolong inflammation, increase phagocytosis, promote recruitment of other immune cells, and ultimately resolve the injury. The ensuing sections will review known contributions of macrophages in each step of injury and resolution.
Initial macrophage response during injury
The current understanding of macrophage function during AKI has mainly been through studies in several types of murine AKI models including ischemia reperfusion (IR), nephrotoxin, rhabdomyolysis, and unilateral urinary obstruction. IR-AKI, one of the most widely used injury models, is performed by blocking blood flow to the kidney, which results in endothelial cells displaying signs of vasoconstriction expressing Endothelin-1, Angiotensin II, thromboxane A2, and adenosine [11, 12], which stimulate leukocyte migration to the kidneys by increasing expression of ICAM-1 [13]. In turn, the lack of oxygen results in the neighboring RTECs to express DAMPs and Hypoxia-inducible-factors (HIFs) [14]. The increased vascular rarefaction results in leukocyte migration and subsequent inflammation within 24 hours [15]. In nephrotoxic AKI, RTECs are directly targeted. A widely used chemotherapeutic reagent, cisplatin, directly damages RTECs by entering cells via transporters such as Ctr1 and OCT2, and causes cell death through DNA damage [16]. An in vitro study found cisplatin treatment of murine peritoneal macrophages significantly increased pro-inflammatory cytokines and NO expression via MAPK pathway [17]. While this may explain the severity of injury associated with cisplatin-AKI, whether this is true in vivo has not yet been proven. Current literature has not compare differences in macrophage responses among various models of AKI.
With the onset of injury, damaged RTECs release DAMPs and pathogen-associated molecular patterns (PAMPs), which act as an initial injury signal to sentinel immune cells within the tissue, such as resident macrophages and dendritic cells. Macrophages recognize the initial damage signals through pattern recognition receptors (PRRs), a family of receptors that recognize DAMPs and PAMPs. Recognition of DAMPs/PAMPs via PRRs results in downstream stimulation of macrophage phagocytosis, phagolysosomes maturation, antigen presentation, and production of the pro-inflammatory cytokine, tumor necrosis factor-alpha (TNFα) [18]. After the initial injury response, resident macrophages further prolong inflammation by recruiting other leukocytes to the site of injury. Among those recruited are: neutrophils, bone marrow derived monocytes and macrophages, and lymphocytes. Macrophage expressed chemokines and cytokines target different stages of leukocyte migration to increase recruitment. For example, TNFα, IL-1β, and histamines target endothelial cells to increase expression of trafficking molecules (selectins, integrin ligands), whereas chemokines CXC1, CXCL2, and CCL2, directly recruit neutrophils to extravasate from the circulatory system into the interstitium [19]. The initial inflammatory macrophage events are subsequently followed by modulation and then inhibition of the inflammatory response. These phenotypic changes occur via tissue-specific, complex pathways and will be discussed in the ensuing sections.
Macrophage polarization
During injury, macrophages acquire a spectrum of phenotypes—from highly inflammatory at the beginning of recruitment to highly reparative towards the resolution of the injury. Pro-inflammatory (classically-activated/M1) macrophages are the first responders to injury. These macrophages phagocytose cellular debris and secrete the cytotoxic reagents such as nitric oxide synthase (NOS) and reactive oxygen species (ROS), which induce mitochondrial damage and apoptosis [20]. The inflammatory milieu is reversed by the infiltration of cells that promote a reparative microenvironment by secreting anti-inflammatory cytokines. These cells include pro-reparative (alternatively activated/M2) macrophages, CD4+ and CD8+ T-cells, and regulatory T-cells that predominate between days 3 and 5 post-injury in rodent models of AKI [21]. While macrophage polarization occurs as a spectrum, for the purposes of simplicity in this review we will refer to the subtypes of activated macrophages as: M1 and M2 macrophages, but acknowledge that this phenomenon occurs as a broad range of subtypes.
M1 macrophages
Recruited macrophages become activated by LPS, IFNγ, and granulocyte monocyte-colony stimulating factor (GM-CSF) released from the damaged microenvironment [22]. In the kidney, M1 specific cytokines increase in expression within the first 24 hours post IR-AKI and significantly decrease at 3 days post injury (dpi) [21]. Upon activation, they secrete inflammatory cytokines such as IL-1β, TNFα, IL-12, IL-18, and IL-23 [2] (Fig. 1). As a result, the site of injury continues to gain other inflammatory cells, including T helper cells [22]. The M1 macrophages also secrete molecules for destruction of pathogenic particles such as nitric oxide generated by inducible nitric oxide synthase (iNOS). Once pathogens or damaged cells are cleared, a rapid change in macrophage polarization is necessary to stop further damage to surrounding cells. Prolonged activation of inflammatory macrophages has negative effects on injury recovery, since the released cytotoxic agents do not discriminate self from pathogenic particles [23, 24]. Various experimental models have concluded that prolonged inflammatory macrophage activation imposes negative consequences in injury resolution due to extensive inflammation. A study in the cardiovascular field reported that atherosclerotic lesions with a higher number of inflammatory macrophages correlated with a higher likelihood for sudden major cardiovascular ischemia [25]. In the kidney, perdurance of M1 macrophages contributes to a worsened outcome after renal ischemia. Clodronate-induced depletion of all macrophages and subsequent transplantation of IFNγ stimulated M1 macrophages prior to renal ischemia resulted in more severe tubular damage [21]. Depletion of macrophages before injury reduced blood urea nitrogen levels and post-injury histological markers of tubular injury, whereas depletion during reparative stages resulted in a significant increase in injury markers [26]. Taken together, these studies suggest that while the initial inflammatory response from macrophages is necessary for removal of damaged and pathogenic particles, prolonged inflammatory activity results in further tissue damage, ultimately inhibiting the reparative phase of injury resolution.
Figure 1. Various pathways identified to be critical for macrophage polarization from M1 (pro-inflammatory) to M2 (pro-regenerative).
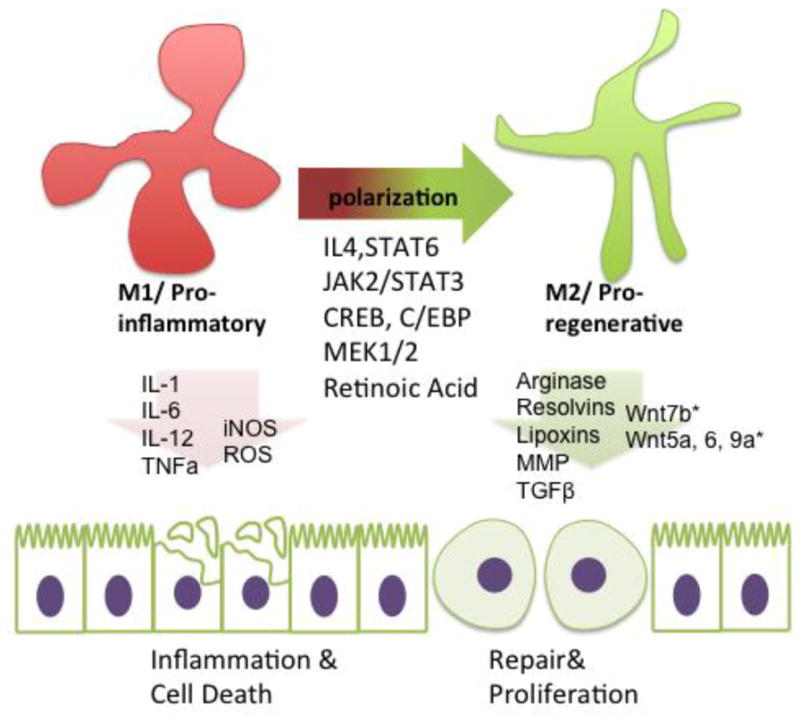
Experimental studies of several pathways have elucidated critical pathways for driving macrophage polarization from M1 to M2. Among them are IL-4/STAT6, JAK2/STAT3, CREB/C/EBP, and Mitogen-activated protein kinase 1/2 (MEK1/2). Each macrophage phenotype has signature expression of certain cytokines and secreted products. M1 macrophages secrete inflammatory cytokines and products and chemoattractants, such as: IL-1, IL-6, IL-12, TNFa, iNOS, and ROS. M2 macrophages secrete anti-inflammatory cytokines and pro-reparative secretions such as: Arginase, Resolvins, Lipoxins, Matrix metalloproteinases, TGFB, and Wnt ligands. *There seems to be tissue-specificity to types of Wnt ligands secreted, specifically Wnt7b in kidney macrophages and Wnt5a, Wnt 6, and 9a in intestinal macrophages.
M2 macrophages
M2 macrophages do not become activated until later in the initial injury phase. In IR-AKI, an increase in M2 markers is observed at 3 dpi, peaking in expression at 7 dpi [21]. M2 macrophages originate from newly recruited monocytes dispatched from the circulatory system, as well as initially recruited M1 macrophages [27]. The phenotypic conversion from monocytes and M1 macrophages to M2 macrophages requires specific cytokine stimulation and subsequent transcriptional changes. M2 macrophages are activated by macrophage colony stimulating factor (M-CSF), IL-4, IL-10, IL-13, and TGF-β [22]. Upon activation, M2 macrophages express mannose receptor (MR), which recognizes and downregulates high levels of inflammatory glycoproteins previously produced by the inflammatory response [28]. M2 macrophages also produce Arginase, an enzyme necessary to produce ornithine and polyamine, building blocks for extracellular matrix architecture. Furthermore, M2 macrophages secrete resolvins, lipoxins, TGFβ, and matrix metalloproteinases that target and cleave chemokines and chemoattractants, resulting in inhibition of inflammatory immune cell activity [19](Fig. 1). Over the past decade, M2 macrophages have been categorized into four subtypes based on their in vitro upstream activators and downstream gene expression patterns [29]. M2a are activated by IL-4, IL-13, M2b are activated by IL-1, LPS, M2c are activated by IL-10 and TGFβ, and glucocorticoids, and M2d are activated by IL-6 and adenosine.
Researchers have examined M2 macrophage ability to curtail inflammation as well as for their inherent reparative capacity. Saha et al. demonstrated that macrophages are crucial for normal repair after an acute injury to the intestines [30]. The post-injury intestinal stem cells required macrophage-derived extracellular vesicles for increased proliferation and repopulation of damaged cells and they found that the Wnt ligands, Wnt5a, Wnt6, and Wnt9a, are critically important factors mediating this effect. In lung injury, mitogen activated protein kinase1/2 inhibition resulted in an increased M2 population, leading to better recovery weight and increased macrophage efferocytosis of inflammatory cells [31]. An AKI study demonstrated that a macrophage-derived Wnt is required for normal kidney repair. Lin et al. reported that kidney specific macrophages secrete Wnt7b, a canonical Wnt ligand [32]. In depleting kidney-specific Wnt7b, the study showed this Wnt is necessary for improved tubular repair, and reduced fibrosis by bypassing G2/M cell cycle arrest. Finally, genetic ablation of a pathway required for M2 polarization, IRAK-M, resulted in increased M1 macrophages during AKI. Early injury response did not vary between wild type and IRAK-M−/−, but in the long-term, the deletion resulted in increased fibrosis and an inability to regenerate kidney mass [33].
Experimentally manipulating macrophage polarization
Studies have begun to focus on macrophage polarization as a means to enhance regeneration in post-AKI models (Table 1). In various experimental models of AKI, increased M2 macrophage polarization has been found to enhance numerous functional outcomes [24, 34]. Treating co-cultures of rodent RTECs and undifferentiated monocytes with GM-CSF stimulated monocytes to convert to M2 macrophages by elevating STAT5 activity [24]. In vivo inhibition of GM-CSF improved RTEC proliferation in post-AKI mice but the study did not examine effects of the treatment on longer-term pathological outcomes (i.e. fibrosis and progression to chronic kidney disease). Another pathway implicated in macrophage polarization is retinoic acid (RA) signaling, a critical pathway for kidney development and in AKI for reducing injury and fibrosis [34]. Chiba et al. demonstrated that locally synthesized RA simultaneously reduces M1 macrophages and activates RA signaling in RTECs. When RA signaling is genetically blocked in RTECs, M2 macrophage markers significantly decreased, suggesting that RA is part of an RTEC/macrophage crosstalk pathway that regulates macrophage polarization. Other studies have shown erythropoietin (EPO), a hormone widely known for its role in hematopoietic differentiation, suppressed inflammatory cytokine expression in rodent kidneys [35, 36]. EPO decreases CCL7 expression in macrophages, thereby limiting RTEC apoptosis. EPO also influenced macrophage polarization by increasing JAK2 and STAT3 activity, resulting in a significantly higher number of peritubular M2 macrophages [36]. Studies have focused on utilizing bone-marrow derived mesenchymal stem cells (MSC) to affect macrophage polarization. It is speculated that MSCs increase M2 macrophage polarization both in vivo and in vitro via MSC-secreted trophic factors, particularly IL-10 [37, 38].
Table 1.
Summary of macrophage polarization studies in kidney injury
| Factor tested | Injury type | Major findings | Effect on macrophages | Ref |
|---|---|---|---|---|
| Relaxin | UUO AKI | Relaxin administration lowers TLR4 expression Increased renal function and attenuated injury and fibrosis |
Increased M2 polarization | [74] |
| PRDX6 | LPS AKI | PRDX6 overexpression decreased mortality and renal injury by lowering ROS via decreasing p38 MAPK and JNK. | Overall lower macrophage infiltration | [75] |
| Gpnmb | IR AKI | Gpnmb is highly expressed in M2. Knocking down Gpnmb results in higher IL-1B and TNFa. Gpmb induces M2 activation via IL-4/STAT6 |
Required for IL-4/STAT6 dependent M2 activation | [76] |
| IL-4/IL-13 | DT AKI; IR AKI | IL-4/IL-13 activate JAK3/STAT6 Deletion of IL-4/13 results in worsened fibrosis |
Required for M2a activation | [77] |
| EPO | RI AKI | EPO administration increases macrophage polarization reduces macrophage recruitment, increased phenotype switch to M2, repressed M1 and increases Jak2/STAT3/STAT6 pathway | Overall lower macrophage infiltration; higher ratio of M2 activation | [36] |
| RA | I/R AKI | RA synthesis (Raldh3) is activated in recruited renal macrophages All-Trans Retinoic Acid reduces post-AKI fibrosis RTEC-specific RAR knockdown results reduces M2 polarization in mice |
RTEC-specific RA required for M2 activation | [34] |
| EPO | I/R AKI | EPO administration resulted in increased function Increased expression of Wnt7b, B-Cat, downregulation of miR-21, −214,−210, −199a |
EPO lowers renal macrophage infiltration | [78] |
| CSF-1 | I/R AKI | CSF-1 depletion results in reduced M2 polarization, delayed functional, structural recovery, increased fibrosis | Required for M2 polarization | [79] |
| GM-CSF | I/R AKI | GM-CSF secreted by RTECs increase STAT5 activation and increases RTEC proliferation | Required for M2 polarization | [24] |
| SOCS-3 | RI AKI; I/R AKI | SOCS-3 KO in RTECs resulted in increased proliferation and attenuated injury | Inhibits M2 polarization | [80] |
| MSC | RI AKI | Increased renal function and injury attenuation function and injury attenuation. | Increases M2 polarization | [37] |
UUO - unilateral uretral obstruction
LPS - Lipopolysaccharide
DT - diphtheria toxin
RI - Rhabdomyolysis induced
I/R - Ischemia/reperfusion
PRDX6 - Peroxiredoxin 6
Gpnmb - Glycoprotein non-metastatic melanoma protein b
EPO - Erythropoietin
RA - Retinoic Acid
CSF-1 - Colony Stimulating Factor-1
GM-CSF - Granulocyte-macrophage colony-stimulating factor
SOCS-3 - suppressor of cytokine signaling 3
MSC - Mesenchymal Stem Cells
Macrophage polarization in human AKI
To translate rodent AKI studies to clinical therapies, the pathways responsible for macrophage polarization need to be conserved in human AKI. It has been reported that an increase in the number of macrophages is seen in human patients with AKI [39, 40]. Analysis of human kidney biopsies with acute tubular injury during the early repair phase showed that 75% of the recruited macrophages expressed the M2 macrophage marker, CD163. Further structural analysis demonstrated that CD163+ macrophages closely adhered to the basement membrane of damaged tubular cells whereas the M1 or CD163− macrophages did not show specific cellular contact [41]. However, the effect of M1 and M2 macrophages in patients with AKI is complex. In patients with macroscopic hematuria induced-AKI, the incomplete recovery group had increased M2 (CD163+) macrophages in the kidney. However, patients with leptospirosis-induced AKI showed an increased number of M1 (HLA-DR+) macrophages (biopsied at week4) [39]. The opposing results can possibly be attributed to lack of biopsied samples and the inherent variability in injury severity and outcome. Other human studies have conducted prophylactic administration of EPO and Pentoxifylline in patients undergoing cardiac surgery, a procedure with a high risk of inducing AKI [42–46]. In these studies, positive outcomes were associated with decreased inflammatory markers, including lower urinary NGAL, TNF α, IL-6, and C-reactive protein [42, 46]. This was accompanied by a significant decrease in the number of leukocytes, though M1/M2 polarization changes were not investigated. Overall, these studies suggest that suppression of the early inflammatory response is beneficial to high-risk patients.
To date, only a handful of human clinical trials have focused on chemically manipulating macrophage infiltration or polarization to enhance repair of acute injuries. Recombinant human (rh) GM-CSF, a pro-polarization cytokine, has been utilized for promoting an increased M2 population in humans. Severe burn victims treated with rhGM-CSF displayed significantly improved healing rates [47, 48]. However, the studies did not follow up with histological analysis to demonstrate that increased repair is a direct result of increased M2 polarization. Others have used drugs that target pathways important for macrophage recruitment to reduce fibrosis. Angiotensin converting enzyme (ACE) inhibitors have been shown to lower Monocyte chemotactic protein-1 (MCP-1), minimizing fibrosis and improving renal function in a mouse model of diabetic nephropathy [49] [50]. In a human clinical study, ACE inhibitor treatment in patients with diabetic nephropathy resulted in decreased urinary levels of MCP-1 as well as increased renal function [51]. Aside from amelioration of acute injuries, macrophage polarization can be manipulated to achieve other clinical goals, such as immunosuppression in transplant patients. Mercalli et al. demonstrated that Rapamycin, normally administered to transplantation patients to suppress T helper cells, simultaneously drives changes in macrophage polarization dynamics both in vitro and in vivo. [52].
Zebrafish as a model organism for AKI and the innate immune response
It is well established that early kidney development and function are conserved between zebrafish and mammals [53, 54]. Zebrafish models also recapitulate the pathophysiology of injury and repair typically observed in mammalian models of AKI [55, 56]. At the onset of injury, both zebrafish and mammalian RTECs loses cell polarity, increase expression of injury markers (i.e. KIM-1), and undergo apoptosis and necrosis [34, 57–59]. Additionally, the larval zebrafish appears to possess a relatively robust capacity for renal repair. A nephrotoxic model of AKI in larval zebrafish responds to injury with increased proliferation and reactivation of developmental genes, as evidenced by increased RA signaling and Pax2a positive RTECs [34, 57, 60, 61]. With conserved mechanisms in development and repair, paired with the power of high-throughput screening, zebrafish studies have elucidated various signaling pathways and identified therapeutic compounds with the potential to improve repair [57, 62–64].
Aside from conserved kidney function, zebrafish leukocyte differentiation and function are also comparable to mammalian models. Mammalian and zebrafish HSCs share conserved mechanism of myelopoesis during embryonic development. As in mammals, there are two waves of HSC migration and differentiation in the zebrafish, termed primitive and definitive myelopoiesis. Primitive myelopoiesis occurs between 12–48 hours post-fertilization (hpf), when precursor cells initiate differentiation and produce mature cells from mesodermal tissue along the notochord [65]. Definitive myelopoiesis occurs when HSCs migrate and seed the anterior segment of the pronephros. The migrated HSCs remain as the definitive pool of self-renewing cells that continue to produce mature leukocytes from 96 hpf to adulthood [65, 66]. There are key regulatory factors that are conserved between mammalian and zebrafish macrophage differentiation. For example, Pu.1, Spi-b, and Irf8 are required for macrophage development and differentiation in both models [67]. While conservation of mammalian and zebrafish myelopoiesis is well outlined, zebrafish macrophage polarization is relatively a new area of study. To date, few in situ studies for zebrafish M1 and M2 macrophage markers have been conducted.
The field has generated several versatile transgenic tools to observe neutrophil and macrophage response. For example, Tg(lyzC:egfp) and Tg(mpeg1:egfp) allows visualization of neutrophil and macrophages in vivo, respectively [68, 69]. Tg(mpeg1:dendra2) allows photo-conversion of macrophages to trace their fate at desired time points, and Tg(mpeg1:gal4/UAS:ntr-mcherry) allows genetic ablation of macrophages [70]. These lines have been used to address the roles of macrophages in repair after fin and liver regeneration [71, 72]. A recent study has shown live imaging of macrophage polarization in fin amputation, using transgenic reporters for tnfa (M1 marker) and macrophage expressed 1 (mpeg1) [73]. Nguyen-Chi et al. visualized by in vivo imaging activation and subsequent repression of TNFα in macrophages recruited to the site of injury. TNFα+ macrophage recruitment was observed as early as 1 hour post amputation, while TNFα-macrophage recruitment occurred at approximately 25 hours post amputation. The convenience of generating transgenic lines paired with imaging tools resulted in tracking macrophages in vivo, enabling characterization of M1/M2 velocity, contact frequency, and morphological changes [73]. Taken together, the zebrafish larva has the potential to serve as a good model organism to elucidate the effect of macrophage polarization during AKI [34].
Towards the future
For the past several decades, the field has elucidated the pathways required to polarize macrophages. However, among the multiple pathways outlined throughout this review, it is still unclear how the identified pathways interact with one another to ultimately drive polarization in injury settings. One reason for the shortcoming may be that specific cellular mechanisms driving macrophage polarization could vary in each organ or injury model. In order to implement macrophage polarization as a clinical immunotherapy, the effects of macrophages in different injury types must be specifically outlined as follows: 1. What specific pathways are required for macrophage polarization in each injury model and tissue; 2. What signaling molecules are secreted by M1 and M2 under each condition; 3. What are the downstream effects of M1 and M2 signaling molecules in various cell types that reside in the organ; and 4. Will pro-polarization drugs have any off-target effects? With a more comprehensive understanding of the immune response the field of regenerative medicine may be able to take advantage of reparative powers of the immune system.
Acknowledgments
Dr. Hukriede’s laboratory is supported by the National Institutes of Health (NIH) National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) grants 2R01DK069403, 1R01DK112652, 1P30DK079307, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) grant 2R01HD053287, and the Department of Defense DoD-W81XWH-17-1-0610. Dr. Davidson’s laboratory is support by the Health Research Council of New Zealand (grants 15/057 & 17/425)
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11:88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 2.Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93:875–881. doi: 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33:643–675. doi: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 4.Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144:541–548. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mescher AL. Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration (Oxf) 2017;4:39–53. doi: 10.1002/reg2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang H, Xu F, Wen XJ, Liu HZ, Wang HB, Zhong JY, Yang CX, Zhang B. Interleukin-33 signaling contributes to renal fibrosis following ischemia reperfusion. Eur J Pharmacol. 2017;812:18–27. doi: 10.1016/j.ejphar.2017.06.031. [DOI] [PubMed] [Google Scholar]
- 7.Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, Lee J, Christmann RB, Lafyatis R. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther. 2014;16:R136. doi: 10.1186/ar4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. 1972;46:845–852. [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson AJ, Zon LI. The ‘definitive’ (and ‘primitive’) guide to zebrafish hematopoiesis. Oncogene. 2004;23:7233–7246. doi: 10.1038/sj.onc.1207943. [DOI] [PubMed] [Google Scholar]
- 10.Gentek RMK, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. 2014;262:56–73. doi: 10.1111/imr.12224. [DOI] [PubMed] [Google Scholar]
- 11.Conger J. Hemodynamic factors in acute renal failure. Adv Ren Replace Ther. 1997;4:25–37. [PubMed] [Google Scholar]
- 12.Brooks DP. Role of endothelin in renal function and dysfunction. Clin Exp Pharmacol Physiol. 1996;23:345–348. doi: 10.1111/j.1440-1681.1996.tb02835.x. [DOI] [PubMed] [Google Scholar]
- 13.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonavia A, Singbartl K. A review of the role of immune cells in acute kidney injury. Pediatr Nephrol. 2017 doi: 10.1007/s00467-017-3774-5. [DOI] [PubMed] [Google Scholar]
- 15.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 16.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014 doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chauhan P, Sodhi A, Shrivastava A. Cisplatin primes murine peritoneal macrophages for enhanced expression of nitric oxide, proinflammatory cytokines, TLRs, transcription factors and activation of MAP kinases upon co-incubation with L929 cells. Immunobiology. 2009;214:197–209. doi: 10.1016/j.imbio.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012;122:4519–4532. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2011;16:233–238. doi: 10.1177/1074248411409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Huen SC, Huynh L, Marlier A, Lee Y, Moeckel GW, Cantley LG. GM-CSF Promotes Macrophage Alternative Activation after Renal Ischemia/Reperfusion Injury. J Am Soc Nephrol. 2015;26:1334–1345. doi: 10.1681/ASN.2014060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Gaetano M, Crean D, Barry M, Belton O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front Immunol. 2016;7:275. doi: 10.3389/fimmu.2016.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klinkert K, Whelan D, Clover AJP, Leblond AL, Kumar AHS, Caplice NM. Selective M2 Macrophage Depletion Leads to Prolonged Inflammation in Surgical Wounds. Eur Surg Res. 2017;58:109–120. doi: 10.1159/000451078. [DOI] [PubMed] [Google Scholar]
- 27.Melgar-Lesmes P, Edelman ER. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J Hepatol. 2015;63:917–925. doi: 10.1016/j.jhep.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H, Liao JJ, Graeler M, Huang MC, Goetzl EJ. Lysophospholipid regulation of mononuclear phagocytes. Biochim Biophys Acta. 2002;1582:175–177. doi: 10.1016/s1388-1981(02)00153-1. [DOI] [PubMed] [Google Scholar]
- 29.Roszer T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015 doi: 10.1155/2015/816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saha S, Aranda E, Hayakawa Y, Bhanja P, Atay S, Brodin NP, Li J, Asfaha S, Liu L, Tailor Y, Zhang J, Godwin AK, Tome WA, Wang TC, Guha C, Pollard JW. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun. 2016;7:13096. doi: 10.1038/ncomms13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long ME, Eddy WE, Gong KQ, Lovelace-Macon LL, McMahan RS, Charron J, Liles WC, Manicone AM. MEK1/2 Inhibition Promotes Macrophage Reparative Properties. J Immunol. 2017;198:862–872. doi: 10.4049/jimmunol.1601059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, Pei H, Chen L, Zheng JJ, Carroll TJ, Pollard JW, McMahon AP, Lang RA, Duffield JS. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lech M, Grobmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, Susanti HE, Kobayashi KS, Flavell RA, Anders HJ. Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J Am Soc Nephrol. 2014;25:292–304. doi: 10.1681/ASN.2013020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiba T, Skrypnyk NI, Skvarca LB, Penchev R, Zhang KX, Rochon ER, Fall JL, Paueksakon P, Yang H, Alford CE, Roman BL, Zhang MZ, Harris R, Hukriede NA, de Caestecker MP. Retinoic Acid Signaling Coordinates Macrophage-Dependent Injury and Repair after AKI. J Am Soc Nephrol. 2016;27:495–508. doi: 10.1681/ASN.2014111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagnis C, Beaufils H, Jacquiaud C, Adabra Y, Jouanneau C, Le Nahour G, Jaudon MC, Bourbouze R, Jacobs C, Deray G. Erythropoietin enhances recovery after cisplatin-induced acute renal failure in the rat. Nephrol Dial Transplant. 2001;16:932–938. doi: 10.1093/ndt/16.5.932. [DOI] [PubMed] [Google Scholar]
- 36.Wang S, Zhang C, Li J, Niyazi S, Zheng L, Xu M, Rong R, Yang C, Zhu T. Erythropoietin protects against rhabdomyolysis-induced acute kidney injury by modulating macrophage polarization. Cell Death Dis. 2017;8:e2725. doi: 10.1038/cddis.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geng Y, Zhang L, Fu B, Zhang J, Hong Q, Hu J, Li D, Luo C, Cui S, Zhu F, Chen X. Mesenchymal stem cells ameliorate rhabdomyolysis-induced acute kidney injury via the activation of M2 macrophages. Stem Cell Res Ther. 2014;5:80. doi: 10.1186/scrt469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eirin A, Zhu XY, Puranik AS, Tang H, McGurren KA, van Wijnen AJ, Lerman A, Lerman LO. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017;92:114–124. doi: 10.1016/j.kint.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka K, Tanabe K, Nishii N, Takiue K, Sugiyama H, Wada J. Sustained Tubulointerstitial Inflammation in Kidney with Severe Leptospirosis. Intern Med. 2017;56:1179–1184. doi: 10.2169/internalmedicine.56.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubio-Navarro A, Carril M, Padro D, Guerrero-Hue M, Tarin C, Samaniego R, Cannata P, Cano A, Villalobos JM, Sevillano AM, Yuste C, Gutierrez E, Praga M, Egido J, Moreno JA. CD163-Macrophages Are Involved in Rhabdomyolysis-Induced Kidney Injury and May Be Detected by MRI with Targeted Gold-Coated Iron Oxide Nanoparticles. Theranostics. 2016;6:896–914. doi: 10.7150/thno.14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutierrez E, Egido J, Rubio-Navarro A, Buendia I, Blanco Colio LM, Toldos O, Manzarbeitia F, de Lorenzo A, Sanchez R, Ortiz A, Praga M, Moreno JA. Oxidative stress, macrophage infiltration and CD163 expression are determinants of long-term renal outcome in macrohematuria-induced acute kidney injury of IgA nephropathy. Nephron Clin Pract. 2012;121:c42–53. doi: 10.1159/000342385. [DOI] [PubMed] [Google Scholar]
- 42.Barkhordari K, Karimi A, Shafiee A, Soltaninia H, Khatami MR, Abbasi K, Yousefshahi F, Haghighat B, Brown V. Effect of pentoxifylline on preventing acute kidney injury after cardiac surgery by measuring urinary neutrophil gelatinase - associated lipocalin. J Cardiothorac Surg. 2011;19:6–8. doi: 10.1186/1749-8090-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tasanarong A, Duangchana S, Sumransurp S, Homvises B, Satdhabudha O. Prophylaxis with erythropoietin versus placebo reduces acute kidney injury and neutrophil gelatinase-associated lipocalin in patients undergoing cardiac surgery: a randomized, double-blind controlled trial. BMC Nephrol. 2013;14:136. doi: 10.1186/1471-2369-14-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh SW, Chin HJ, Chae DW, Na KY. Erythropoietin improves long-term outcomes in patients with acute kidney injury after coronary artery bypass grafting. J Korean Med Sci. 2012;27:506–511. doi: 10.3346/jkms.2012.27.5.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JE, Song SW, Kim JY, Lee HJ, Chung KH, Shim YH. Effect of a Single Bolus of Erythropoietin on Renoprotection in Patients Undergoing Thoracic Aortic Surgery With Moderate Hypothermic Circulatory Arrest. Ann Thorac Surg. 2016;101:690–696. doi: 10.1016/j.athoracsur.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Cagli K, Ulas MM, Ozisik K, Kale A, Bakuy V, Emir M, Balci M, Topbas M, Sener E, Tasdemir O. The intraoperative effect of pentoxifylline on the inflammatory process and leukocytes in cardiac surgery patients undergoing cardiopulmonary bypass. Perfusion. 2005;20:45–51. doi: 10.1191/0267659105pf779oa. [DOI] [PubMed] [Google Scholar]
- 47.Wang ZY, Zhang Q, Liao ZJ, Han CM, Lv GZ, Luo CQ, Chen J, Yang SX, Yang XD, Liu Q. Effect of recombinant human granulocyte-macrophage colony stimulating factor on wound healing in patients with deep partial thickness burn. Zhonghua Shao Shang Za Zhi. 2008;24:107–110. [PubMed] [Google Scholar]
- 48.Italiani P, Boraschi D. New Insights Into Tissue Macrophages: From Their Origin to the Development of Memory. Immune Netw. 2015;15:167–176. doi: 10.4110/in.2015.15.4.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.el Nahas AM. The role of growth hormone and insulin-like growth factor-I in experimental renal growth and scarring. Am J Kidney Dis. 1991;17:677–679. doi: 10.1016/s0272-6386(12)80350-7. [DOI] [PubMed] [Google Scholar]
- 50.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amann B, Tinzmann R, Angelkort B. ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care. 2003;26:2421–2425. doi: 10.2337/diacare.26.8.2421. [DOI] [PubMed] [Google Scholar]
- 52.Mercalli A, Calavita I, Dugnani E, Citro A, Cantarelli E, Nano R, Melzi R, Maffi P, Secchi A, Sordi V, Piemonti L. Rapamycin unbalances the polarization of human macrophages to M1. Immunology. 2013;140:179–190. doi: 10.1111/imm.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wingert RA, Davidson AJ. The zebrafish pronephros: a model to study nephron segmentation. Kidney Int. 2008;73:1120–1127. doi: 10.1038/ki.2008.37. [DOI] [PubMed] [Google Scholar]
- 54.Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16:299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- 55.Cianciolo Cosentino C, Roman BL, Drummond IA, Hukriede NA. Intravenous microinjections of zebrafish larvae to study acute kidney injury. J Vis Exp. 2010 doi: 10.3791/2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diep CQ, Peng Z, Ukah TK, Kelly PM, Daigle RV, Davidson AJ. Development of the zebrafish mesonephros. Genesis. 2015;53:257–269. doi: 10.1002/dvg.22846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, West J, Korotchenko VN, McDermott L, Day BW, Davidson AJ, Harris RC, de Caestecker MP, Hukriede NA. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol. 2013;24:943–953. doi: 10.1681/ASN.2012111055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humphreys BD, Czerniak S, DiRocco DP, Hasnain W, Cheema R, Bonventre JV. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A. 2011;108:9226–9231. doi: 10.1073/pnas.1100629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin W, Naini SM, Chen G, Hentschel DM, Humphreys BD, Bonventre JV. Mammalian Target of Rapamycin Mediates Kidney Injury Molecule 1-Dependent Tubule Injury in a Surrogate Model. J Am Soc Nephrol. 2016;27:1943–1957. doi: 10.1681/ASN.2015050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cirio MC, de Groh ED, de Caestecker MP, Davidson AJ, Hukriede NA. Kidney regeneration: common themes from the embryo to the adult. Pediatr Nephrol. 2014;29:553–564. doi: 10.1007/s00467-013-2597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hentschel DM, Park KM, Cilenti L, Zervos AS, Drummond I, Bonventre JV. Acute renal failure in zebrafish: a novel system to study a complex disease. Am J Physiol Renal Physiol. 2005;288:F923–929. doi: 10.1152/ajprenal.00386.2004. [DOI] [PubMed] [Google Scholar]
- 62.deGroh ED, Swanhart LM, Cosentino CC, Jackson RL, Dai W, Kitchens CA, Day BW, Smithgall TE, Hukriede NA. Inhibition of Histone Deacetylase Expands the Renal Progenitor Cell Population. J Am Soc Nephrol. 2010;21:794–802. doi: 10.1681/ASN.2009080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Skrypnyk NI, Sanker S, Brilli-Skvarca L, Novitskaya T, Woods C, Chiba T, Patel K, Goldberg ND, McDermott L, Vinson PN, Calcutt MW, Huryn DM, Vernetti LA, Vogt A, Hukriede N, de Caestecker MP. Delayed treatment with PTBA analogs reduces post injury renal fibrosis after kidney injury. Am J Physiol Renal Physiol. 2015;310:F705–F716. doi: 10.1152/ajprenal.00503.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanker S, Cirio MC, Vollmer LL, Goldberg ND, McDermott LA, Hukriede NA, Vogt A. Development of high-content assays for kidney progenitor cell expansion in transgenic zebrafish. J Biomol Screen. 2013;18:1193–1202. doi: 10.1177/1087057113495296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ellett F, Lieschke GJ. Zebrafish as a model for vertebrate hematopoiesis. Curr Opin Pharmacol. 2010;10:563–570. doi: 10.1016/j.coph.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 66.Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin HF, Handin RI, Herbomel P. Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity. 2006;25:963–975. doi: 10.1016/j.immuni.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 67.Yu T, Guo W, Tian Y, Xu J, Chen J, Li L, Wen Z. Distinct regulatory networks control the development of macrophages of different origins in zebrafish. Blood. 2017;129:509–519. doi: 10.1182/blood-2016-07-727651. [DOI] [PubMed] [Google Scholar]
- 68.Henry KM, Loynes CA, Whyte MK, Renshaw SA. Zebrafish as a model for the study of neutrophil biology. J Leukoc Biol. 2013;94:633–642. doi: 10.1189/jlb.1112594. [DOI] [PubMed] [Google Scholar]
- 69.Hall C, Flores MV, Storm T, Crosier K, Crosier P. The zebrafish lysozyme C promoter drives myeloid-specific expression in transgenic fish. BMC Dev Biol. 2007;7:42. doi: 10.1186/1471-213X-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood. 2011;117:e49–56. doi: 10.1182/blood-2010-10-314120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Petrie TA, Strand NS, Yang CT, Rabinowitz JS, Moon RT. Macrophages modulate adult zebrafish tail fin regeneration. Development. 2014;141:2581–2591. doi: 10.1242/dev.098459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu J, Choi TY, Shin D. tomm22 Knockdown-Mediated Hepatocyte Damages Elicit Both the Formation of Hybrid Hepatocytes and Biliary Conversion to Hepatocytes in Zebrafish Larvae. Gene Expr. 2017;17:237–249. doi: 10.3727/105221617X695195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen-Chi M, Laplace-Builhe B, Travnickova J, Luz-Crawford P, Tejedor G, Phan QT, Duroux-Richard I, Levraud JP, Kissa K, Lutfalla G, Jorgensen C, Djouad F. Identification of polarized macrophage subsets in zebrafish. 2015 doi: 10.7554/eLife.07288. [DOI] [PMC free article] [PubMed] [Google Scholar]