

Ribosomal stalk proteins recruit translation elongation GTPases to the factor-binding center of the ribosome. Initiation factor 5B (eIF5B in eukaryotes and aIF5B in archaea) is a universally conserved GTPase that promotes the joining of the large and small ribosomal subunits during translation initiation.
KEYWORDS: IF2 GTPase, general control response, ribosome stalk complex, ribosomes, start codon, translation initiation, translation initiation factor
ABSTRACT
Ribosomal stalk proteins recruit translation elongation GTPases to the factor-binding center of the ribosome. Initiation factor 5B (eIF5B in eukaryotes and aIF5B in archaea) is a universally conserved GTPase that promotes the joining of the large and small ribosomal subunits during translation initiation. Here we show that aIF5B binds to the C-terminal tail of the stalk protein. In the cocrystal structure, the interaction occurs between the hydrophobic amino acids of the stalk C-terminal tail and a small hydrophobic pocket on the surface of the GTP-binding domain (domain I) of aIF5B. A substitution mutation altering the hydrophobic pocket of yeast eIF5B resulted in a marked reduction in ribosome-dependent eIF5B GTPase activity in vitro. In yeast cells, the eIF5B mutation affected growth and impaired GCN4 expression during amino acid starvation via a defect in start site selection for the first upstream open reading frame in GCN4 mRNA, as observed with the eIF5B deletion mutant. The deletion of two of the four stalk proteins diminished polyribosome levels (indicating defective translation initiation) and starvation-induced GCN4 expression, both of which were suppressible by eIF5B overexpression. Thus, the mutual interaction between a/eIF5B and the ribosomal stalk plays an important role in subunit joining during translation initiation in vivo.
INTRODUCTION
Translation of mRNA encompasses four major steps: initiation, elongation, termination, and ribosome recycling (1, 2). Upon the completion of the translation cycle, ribosomes dissociate into the small subunit (SSU) (30S in bacteria and archaea and 40S in eukaryotes) and the large subunit (LSU) (50S in bacteria and archaea and 60S in eukaryotes) to initiate the next round of translation. The universal translation initiation factor IF1A (in Archaea and Eukarya; IF1 in Bacteria), mRNA, and associated IFs bind the SSU. This results in the assembly of the SSU initiation complex (IC), which facilitates Met-tRNAi–start codon base pairing in the peptidyl tRNA-binding site (P-site) (3–5). Upon the formation of the SSU IC, IF5B (in Archaea and Eukarya; IF2 in Bacteria), which is a universal translation initiation factor with GTPase activity, promotes LSU:SSU joining (the subunit joining step). This results in the formation of the elongation-competent 80S ribosome (6–9).
During the elongation phase of translation, aminoacyl tRNAs (aa-tRNAs) are delivered to the aa-tRNA-binding site (A-site) of the ribosome by elongation factor 1A (EF1A in Archaea and Eukarya and EF-Tu in Bacteria) (10). The formation of the peptide bond is followed by the translocation of the ribosome, whereby peptidyl tRNA in the A-site and deacylated tRNA in the P-site are transferred to the P-site and the exit site (E-site), respectively. Translocation of the ribosome is facilitated by elongation factor 2 (EF2 in Archaea and Eukarya and EF-G in Bacteria). Both EF1A and EF2 exert GTPase activity. The rapid and accurate delivery of aa-tRNA during elongation is thought to be achieved via a high concentration of EF1A/EF-Tu relative to the ribosome (7:1 in bacteria) and facilitated, at least in part, by so-called ribosomal stalk proteins.
Ribosomal stalk proteins simultaneously associate with a base protein, which directly interacts with large rRNA, and elongation factors. The rRNA-binding domain of the base protein (L10 in Bacteria and P0 in Archaea and Eukarya) is universally conserved, and its C-terminal portion is folded into a long α-helix termed the spine. This region is bound by four to six stalk proteins as homodimers in Bacteria and Archaea or as heterodimers in Eukarya (11–13). Humans encode two stalk proteins, P1 and P2, which bind a molecule of P0 as two heterodimers, P1-P2 (14, 15). Given ribosome:EF1A/EF-Tu stoichiometry, every ribosome can be tethered to several stalk protein:EF complexes. These stalk complexes are therefore positioned to act as a molecular “rake,” which creates a gradient of distribution of elongation factors across the ribosomes. Moreover, the bacterial stalk proteins have been reported to directly promote GTP hydrolysis by EF-Tu and EF-G on the ribosome (16).
The bacterial stalk protein, L12/L7, contains the N-terminal domain (NTD) bound to the L10 spine, a long flexible linker, and the ∼70-amino-acid (aa)-long C-terminal domain (CTD), consisting of three α-helices and a three-stranded β-sheet (17). The L12-CTD directly binds the elongation factors EF-Tu and -G (18). In contrast, archaeal stalk P1 and eukaryal stalk P1/P2 proteins are homologous and yet are evolutionarily unrelated to the bacterial L12 protein. Thus, the archaeal and eukaryal stalk proteins comprise a distinct NTD that binds to the P0 spine, a flexible linker, and the small hydrophobic C-terminal tail involved in binding to EF1A and EF2 (19, 20). The C-terminal tails of both the stalk (archaeal P1 [aP1]/eukaryotic P1 [eP1]/eP2) and the base (a/eP0) proteins contain a common sequence that serves as a translational GTPase-binding site. Intriguingly, in humans, systemic lupus erythematosus (SLE), an autoimmune disease, is characterized by the production of anti-P antibody (21–23).
The role of stalk proteins does not appear to be limited to the elongation phase of translation, as the bacterial stalk proteins, L12, are also reported to bind initiation factor (IF2) and release factor (RF3) GTPases (18). Moreover, there is strong evidence that L12 binding to IF2 facilitates subunit joining during bacterial translation initiation (24). In the archaeon Pyrococcus horikoshii, a direct interaction between IF5B and the stalk protein P1 has been shown (19); however, the functional relevance of this interaction during initiation remains unknown. Extensive functional studies using the yeast Saccharomyces cerevisiae showed that most ribosomal proteins (RPs) are required for both ribosome biogenesis and function (25, 26). Accordingly, attenuation of the expression of a single RP decreases the abundance of the corresponding ribosomal subunit (25–27). Yeast encodes four copies of P1/P2 stalk proteins, P1α, P1β, P2α, and P2β (28, 29). Like other large RP (RPL) deletion mutants, stalk protein mutants expressing only one P1-P2 pair of the four possible stalk heterodimer combinations contain fewer 60S than 40S subunits (13). Since the severity of the ribosome biogenesis defect was greater with mutants deleted for P1α-P2α and P1β-P2β than with those deleted for P1α-P2β and P1β-P2α, P1α-P2β and P1β-P2α are considered “canonical” stalk protein heterodimers (13). In contrast, P1α-P2α and P1β-P2β are thought to be the “noncanonical heterodimers” (13).
The present study was motivated in part by the polyribosome (polysome) profiles of the above-mentioned stalk protein double-deletion mutants, displaying interesting phenotypes requiring further examination (13). For example, all four mutants appeared to show an increase in free 40S and 60S subunits, suggesting that ribosome association/dissociation is affected by the mutant stalk proteins. Moreover, the polysome abundance was lower in these mutants, which suggests that they exhibit a defect in translation initiation. This implicates the eukaryal stalk proteins in eIF5B-mediated ribosomal subunit joining. Here we show that the eukaryotic ribosome with only two stalk proteins is defective in translation initiation, which is likely mediated by the diminished interaction between stalk proteins and eIF5B. Furthermore, we identified the sites of binding between archaeal IF5B (aIF5B) domain I and the C-terminal tail of the aP1 stalk. An alanine substitution mutation in yeast eIF5B, which alters the predicted sites of binding to P1/P2, impairs ribosome-dependent GTP hydrolysis and results in phenotypes that closely resemble those exhibiting a subunit-joining defect. In conjunction with previous findings in Bacteria (24), our results emphasize the fundamental importance of the initiation factor-stalk interaction across all domains of life.
RESULTS
A yeast stalk mutant is defective in inducing the general control response, similar to an eIF5B mutant.
The yeast S. cerevisiae encodes four stalk proteins, P1α, P1β, P2α, and P2β. Four stalk protein deletion strains expressing only one of the four possible P1-P2 pairs displayed polysome phenotypes, suggesting defects in ribosome subunit assembly and translation initiation (13). To further characterize the phenotypes, we examined two strains, D56 (rpp1BΔ rpp2BΔ) and D57 (rpp1AΔ rpp2BΔ), as representatives expressing noncanonical (P1α-P2α) versus canonical (P1β-P2α) heterodimers, respectively (Fig. 1A).
FIG 1.

The deletion of the yeast P1α-P2β stalk dimer displays a general control phenotype. (A) Table summarizing strains used, their genotypes, stalk proteins expressed, 60S/40S abundance ratios, and 60S abundance levels computed as the ratio of the 60S/40S abundance compared to that of the wild type. The 60S/40S abundance ratio was measured by growing the indicated strains in a complex medium, yeast extract-peptone-dextrose (YPD), and analyzing ribosomes in their lysates through density gradient-velocity sedimentation, as described in Materials and Methods. The averages of data from two experiments are shown with standard deviations. (B) Model of GCN4 translational control and its deregulation by 60S subunit mutations. The schematics at the top describe the structure of GCN4 mRNA, with boxes indicating uORFs. The table below describes the uORF-dependent delayed-reinitiation model for GCN4 translation in the wild type (rows 1 and 2) or eIF5B (6) or 60S (8, 26, 27) mutants (rows 3 to 5). Ovals in the schematics represent ribosomes with 40S and 60S subunits or the subunit alone. Brown arrows indicate ribosome dissociation. See the text for details. For the vertical arrow, we propose that distinct general control phenotypes observed with 60S subunit mutants can be explained by a different severity of subunit-joining defects resulting in a corresponding degree of leaky scanning of start codons (see also Fig. S1 and the text in the supplemental material). (C) The transformants of the indicated strains carrying the GCN4-lacZ plasmid, p180, and an empty vector (to match growth media with experiments in Fig. 2C) were grown at 30°C in the absence (3AT−) (bars 1, 3, and 5) or presence (3AT+) (bars 2, 4, and 6) of 10 mM 3AT for 6 h, following preculturing for 2 h, and then assayed for β-galactosidase (β-gal) activity, as described in Materials and Methods. β-Galactosidase activity, in Miller units, is presented as averages and standard errors of six values obtained in duplicate from 3 independent experiments. Gcd, general control derepressed; Gcn, general control nonderepressible. Data for D57 were adapted from data shown in Fig. 2C, bars 3 and 4. (D and E) Complementation of phenotypes in D57 by P1α-P2β. Transformants of the indicated stains carrying empty vector (Vec) or YCpU-RPP1A and YCpW-RPP2B (P1α:P2β) were grown in synthetic complete (SC) medium lacking uracil and tryptophan and subjected to 3AT sensitivity (D) and ribosome subunit abundance (E) assays. (D) Fixed amounts (5 μl of culture diluted to an optical density at 600 nm [OD600] of 0.15) of the indicated transformants and their 10-fold serial dilutions were spotted onto SC medium plates lacking uracil, tryptophan, and histidine supplemented with 40 mM leucine with (+3AT) or without (−3AT) 40 mM 3AT and grown for 4 days at 30°C. Colonies are reddish due to the ade2 marker. (E) A254 profiles of ribosome subunit abundances are shown with 60S/40S ratios (n = 2 with standard deviations).
Because the 60S subunit joins the 40S IC at the last step of initiation, the sole consequence of 60S subunit mutation during translation initiation is likely to be a subunit-joining defect, which results in the bypass, or leaky scanning, of start codons. In order to test yeast phenotypes resulting from leaky scanning, we used GCN4 mRNA as the sensitive indicator of translation initiation defects (7). The GCN4 mRNA, encoding the transcription factor responsible for the general amino acid control (GAAC) response, has four upstream open reading frames (uORFs). The ribosome that has translated the first uORF (uORF1) remains linked to the mRNA and resumes scanning. GCN4 is normally not translated, because the scanning ribosome quickly translates one of uORFs 2 to 4 and then dissociates from the mRNA (Fig. 1B, row 1). Under amino acid starvation conditions, Gcn2p phosphorylates eIF2, reducing the amount of the eIF2–GTP–Met-tRNAi Met ternary complex (TC). This causes a delay in the scanning ribosome receiving the TC, allowing the bypass of uORFs 2 to 4, and thereby results in the translational induction of GCN4 (Fig. 1B, row 2).
A strong defect in subunit joining allows the ribosomes to bypass the uORF1 start codon, disrupting the delayed reinitiation mechanism required for GCN4 translation under starvation conditions (general control nonderepressible or Gcn− phenotype), as observed with mutants defective in eIF5B function (6) or rRNA interaction with eIF5B (8) (Fig. 1B, row 3). In contrast, a distinct, perhaps simply modest, defect (see below) in subunit joining would still allow a part of the ribosomes to translate uORF1 and resume scanning for start codons of uORFs 2 to 4. However, the defect allows a part of the reinitiating ribosomes to bypass even the latter, leading to GCN4 reinitiation in the absence of a starvation signal (general control derepressed or Gcd− phenotype). This was observed with an rpl16bΔ (27) or rpl33aΔ mutant displaying reduced 60S abundances or an rpl33a-G76R (gcd17-1) mutant defective in stabilizing Met-tRNAi Met in the 40S IC (26) (Fig. 1B, rows 4 and 5). We suggest that the contrasting phenotypes are due mainly to differences in the severities of the subunit joining defect; our simple simulation assuming an equal effect of a subunit joining defect on all start codons in GCN4 mRNA suggests that modest subunit joining defects (f, defined as the initiation rate at a given start codon by the fully loaded pre-IC, of 10 to 50%) lead to a Gcd− phenotype, while strong subunit joining defects (f of <5%) lead to a Gcn− phenotype (see Fig. S1 and the text in the supplemental material; also see Discussion).
We confirmed that the 60S subunit was more severely depleted in D56 expressing a noncanonical heterodimer than in D57 expressing a single canonical heterodimer (Fig. 1A), reinforcing the idea that canonical heterodimers normally bind the base protein P0 to form the stable stalk complex. As shown in Fig. 1C, the reporter assays with p180, the wild-type (WT) GCN4-lacZ plasmid (depicted in Fig. 1B), showed that D57 is defective in inducing GCN4 in response to 3-aminotrizole (3AT)-induced amino acid starvation (rows 5 and 6). In agreement with defective GAAC expression, D57 displayed 3AT-sensitive growth (Fig. 1D, row 2), in a manner complemented by the expression of the missing canonical heterodimer, P1α-P2β (row 3). Thus, D57 confers a weak Gcn− phenotype due to the stalk protein deletion. In contrast, D56, with a stronger 60S biogenesis defect (Fig. 1A), displays an elevated level of GCN4 and hence a Gcd− phenotype (Fig. 1C, rows 3 and 4). Based on our simulation, the frequencies of leaky scanning of a start codon within GCN4 mRNA (1 − f) are estimated to be ∼95% in D57 and ∼50 to 60% in D56 (see Fig. S1E and the text in the supplemental material). We chose to further characterize D57, because it shows a Gcn− phenotype considered to result from a strong defect in subunit joining, with the smallest effect on the 60S subunit abundance (Fig. 1A and E).
Our polysome analyses confirmed that D57 has a lower abundance of polysomes, as measured by the P (polysome)/M (monosome) ratio (P = 0.03) (Fig. S2A), and a higher abundance of free ribosomal subunits compared to the total ribosome abundance (P = 0.03) (Fig. S2B) than the wild type, in a manner complemented by P1α-P2β (Fig. S2). In addition, in D57, half-mer peaks representing polysomes associated with a 40S subunit were observed, but they disappeared when P1α and P2β were expressed (Fig. S2A), and the 60S/40S ratio was restored to the wild-type level (Fig. 1E).
eIF5B overexpression suppresses stalk mutant phenotypes related to translation initiation defects.
Next, we examined whether the Gcn− phenotype displayed by the stalk mutant D57 is suppressible by eIF5B overexpression by mass action. As shown in Fig. 2A and B, eIF5B overexpression significantly restored the lowered polysome abundance in D57, without eliminating the half-mer peaks (Fig. 2A, right). The latter observation suggests that eIF5B overexpression increases the polysome abundance without restoring the 60S biogenesis defect; rather, it does so through increasing stalk-eIF5B interactions by mass action. Furthermore, eIF5B overexpression rescued the Gcn− phenotype, i.e., the defect in GCN4 induction, as examined with p180 (Fig. 2C, bars 1 to 6).
FIG 2.
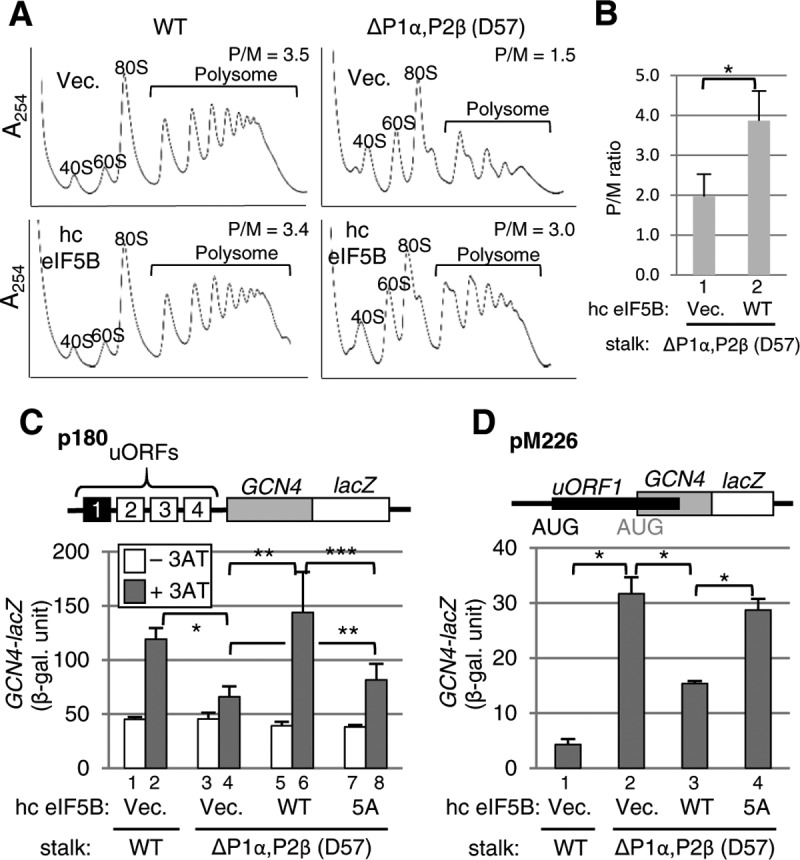
eIF5B overexpression suppresses translation initiation phenotypes caused by the P1α-P2β stalk dimer deletion. (A) Polysome profiles of the wild-type (left) and the P1α-P2β-deleted (D57) strains with (bottom) or without (top) eIF5B overexpression. Whole-cell extracts from these strains, grown in SC medium lacking tryptophan to an OD600 of ∼1.0 at 30°C, were layered onto a 5 to 45% (wt/vol) sucrose density gradient and resolved by ultracentrifugation. The gradients were scanned at 254 nm to visualize the indicated ribosome species. The polysome/monosome ratio (P/M ratio) for each experiment is presented at the top right. (B) Quantification of the P/M ratios for P1α-P2β-deleted (D57) strains. The results are the averages and standard deviations from three independent experiments. *, P = 0.009 by a t test. (C) Analysis of GCN4 expression by the β-galactosidase assay. Transformants of the wild-type (bars 1 and 2) and the P1α-P2β-deleted (D57) strains (bars 3 to 8) carrying p180, together with either a high-copy-number (hc) control (bars 1 to 4), eIF5B-WT (bars 5 and 6), or eIF5B-5A (bars 7 and 8) vector, were grown at 30°C in the absence (−3AT) (bars 1, 3, 5, and 7) or presence (+3AT) (bars 2, 4, 6, and 8) of 10 mM 3AT and assayed for β-galactosidase activity, as described in the legend of Fig. 1B. The data presented are the averages and standard errors of six values in duplicate from three independent experiments. *, P < 0.00001 by a t test; **, P < 0.05 by a t test; ***, P = 0.057 by a t test. (D) Analysis of leaky scanning of the uORF1 start codon. Transformants of the indicated strains carrying pM226, together with either a control (bars 1 and 2), eIF5B-WT (bar 3), or eIF5B-5A (bar 4) vector, were grown at 30°C and assayed for β-galactosidase activity. The data presented are the averages and standard errors of six values in duplicate from three independent experiments. *, P = 0.007 by a t test.
To directly examine whether the stalk heterodimer deletion in D57 leads to an increase in uORF1 bypass or leaky scanning, we used a mutant version of p180, named pM226, wherein a series of mutations, including a frameshift mutation, elongated uORF1, thereby strongly suppressing expression from GCN4-lacZ (Fig. 2D, bar 1, versus 2C, bar 1). As shown in Fig. 2D (bar 2), the stalk deletion increased the leaky scanning of uORF1, which would explain the inability of the mutant yeast to induce GCN4 (Fig. 1B, row 3). Importantly, eIF5B overexpression also suppressed this phenotype (Fig. 2D, bar 3). These results strongly suggest that the initiation-related phenotypes displayed by the stalk heterodimer P1α-P2β deletion mutant (D57) is due to a decreased interaction with eIF5B, in support of the functional interaction between eIF5B and the eukaryotic stalk proteins P1/P2.
Structure determination of the aIF5B·aP1 complex.
In order to identify the stalk protein-binding site in eIF5B, we next set out to solve the structure of the IF5B-P1 complex from an archaeon, Aeropyrum pernix. As shown in Fig. S3A in the supplemental material, aIF5B from A. pernix directly binds aP1 from the same organism. We then determined the regions in aIF5B and aP1 responsible for their binding interactions by using truncation mutants (see Fig. S3B to D and results in the supplemental material). Accordingly, we used the polypeptide fragment encompassing domains I and II of aIF5B (aIF5BI-II) and a synthetic peptide corresponding to the C-terminal 17 amino acids of aP1 (aP1CTD) for cocrystallization. Crystals of the aIF5BI-II-GDP-aP1CTD ternary complex were successfully obtained. The structure was solved by the molecular-replacement method and was refined at a 1.9-Å resolution to an R factor value of 17.1% and a free R factor value of 21.7%. The data collection and structure refinement statistics are summarized in Tables S1 and S2 in the supplemental material. The structure of aP1-free aIF5B was reported recently (30).
Figure 3 displays the aIF5BI-II-GDP-aP1CTD complex with aP1CTD adopting a type I β-turn (D101-L102-S103-G104) and a 310-helix (L105-S106-G107-M108) (Fig. 3A) (the model of the N-terminal part of the aP1 peptide [K95 to V100] could not be built, because of poor electron density). The structure also revealed that aP1 binds to a small hydrophobic pocket on the surface of aIF5B domain I, which is present on the opposite side (20 Å away) of the GTP/GDP-binding site (Fig. 3B). A comparison of the structures of aP1-bound and aP1-free aIF5B-GDP revealed the overall similarity in the structures of the two domains as well as their relative arrangement (Fig. 3C). The root mean square deviation (RMSD) between pairs of equivalent Cα atoms in domains I and II was 0.822 Å. These facts suggest that the binding of aP1 does not substantially affect the structure of the GDP-bound form of aIF5B.
FIG 3.

Crystal structure of the aIF5BI-II-GDP-aP1CTD complex. (A) Overall structure of the aIF5BI-II-GDP-aP1CTD complex. The structures are represented by ribbon models. aIF5B domains I and II and aP1CTD are shown in red, yellow, and blue, respectively. The bound GDP is shown by a stick model. The N and C termini of aP1CTD are indicated as N′ and C′, respectively. (B) Distance between aP1CTD (blue) and GDP (stick model) bound to domain 1 of aIF5B (red). (C) Superimposition of the aP1CTD-bound form of aIF5BI-II-GDP and its aP1CTD-free form. aIF5BI-II-GDP-aP1CTD is colored as described above for panel A, and the aP1CTD-free form of aIF5B-GDP is shown in gray.
The C-terminal region of aP1 bound to the binding pocket of domain I of aIF5B in a shape-complementary manner, burying its solvent-accessible surface area of 631 Å2 (Fig. 3A). The aP1-binding sites in aIF5B can be divided into four sites (sites 1 to 4) (Fig. 4A to E). At site 1 (a loop region forming a part of the aP1-binding pocket), the region of aP1 consisting of L105, M108, and F111 interacted with the region of aIF5B consisting of L70, V73, and I74, mainly by hydrophobic interactions (Fig. 4A and E). At site 2 (another loop on the rim opposite site 1), the region of aP1 consisting of D101, L105, S106, M108, and F109 interacted with the regions of aIF5B consisting of F193, T194, R195, K197, and F199, by numerous van der Waals contacts (Fig. 4B and E). In addition, the C-terminal carboxyl group of aP1 formed a hydrogen bond with the side chain of T194 of aIF5B in site 2 (Fig. 4B and E). Although site 2 includes negatively and positively charged amino acid residues in aP1 and aIF5B, respectively, no electrostatic interaction between these amino acid residues was observed. At site 3 (the base of the aP1-binding pocket), the region of aP1 consisting of L102, S103, L105, M108, and F111 interacted with the region of aIF5B consisting of I158, E218, A221, V222, L226, T229, and Y230, mainly by hydrophobic interactions (Fig. 4C and E). At site 4 (around the C terminus of aP1), the C-terminal carboxyl group of aP1 formed salt bridges with the side chains of R162 and R169 of aP1 (Fig. 4D and E).
FIG 4.

Atomic interactions between aIF5BI-II-GDP and aP1CTD. (A to D) Structure of the binding interface between aP1 and aIF5B. Hydrogen bonds are indicated by dashed lines in site 1 (A), site 2 (B), site 3 (C), and site 4 (D), as also depicted in panel E. (E) Schematic diagram of the interactions between aIF5B and aP1CTD. The van der Waals contacts and hydrogen bonds (<5 Å) between the side chains are represented by solid lines and dashed lines, respectively. (F) Induced fit of aIF5B by aP1 binding. The structure of the aP1CTD-free form of aIF5B is shown in gray. The structures of aIF5B and the aP1CTD complex are shown in red and light blue, respectively. Residues involved in the induced fit are indicated by stick models.
As described above, the overall structures of the aP1-bound and aP1-free forms of aIF5B are similar (Fig. 3C). However, some structural differences were detected in a limited region of the aP1-binding site. In comparison with the structure of aP1-free aIF5B, the loop including V73 and I74 of aIF5B in aP1-bound aIF5B moved toward the direction of opening the aP1-binding site (Fig. 4F). These amino acid residues display hydrophobic interactions with residues L105, M108, and F111 of aP1 in the aIF5B-aP1 complex (Fig. 4A and F). In addition, the side chains of R162 and R169 of aIF5B flipped toward aP1 in the complex and formed salt bridges with the C-terminal carboxyl group of aP1 (Fig. 4D and F). It seems likely that these conformational changes occur through an induced-fit mechanism for binding between aP1 and aIF5B.
Effect of amino acid substitutions on binding between aIF5B and aP1.
The structure of aP1-bound aIF5B revealed the amino acid residues involved in binding between aP1 and aIF5B. These amino acids are located in the C terminus of aP1 and a small hydrophobic pocket composed of four loop/helix sites (Fig. 3). To confirm the interactions detected in the structural data, we performed gel mobility shift assays using mutants of aP1 and aIF5B. We first focused on five hydrophobic amino acids in the C-terminal region of aP1 that interact with aIF5B (Fig. 5A). In this gel assay, the bands of the aP1-aIF5B complexes appeared in the top region of the gel, and accordingly, the free aP1 disappeared upon complex formation. Remarkably, binding was disrupted with the L105S (Fig. 5A, lane 10), M108S (lane 11), and F109S (lane 12) mutants, whereas the L102S (lane 9) mutation and the F111 deletion (lane 13) had minimal effects on binding. We next focused on the 12 amino acids of aIF5B interacting with L105, M108, and F109 (see Fig. S4 in the supplemental material). Again, binding was disrupted strongly with the L70A mutant of aIF5B. The other mutations, V73A, I74A, I158A, F193A, T194A, R195A, K197A, F199A, E218A, V222A, and L226A, also had some deleterious effects on binding (Fig. S4). Furthermore, we tested the effects of multiple amino acid substitution mutations in sites 1 to 3 of aIF5B on aP1 binding, i.e., site 1A mutations, including L70A, V73A, and I74A; site 2A mutations, including F193A, T194A, R195A, K197A, and F199A; and site 3A mutations, including I158A, E218A, V222A, and L226A. Since the deletion of the C-terminal amino acid residues of aP1 (ΔF111) did not affect binding (Fig. 5A), the assay was not performed for the site 4 residue mutation. The gel shift assays revealed that the aP1-aIF5B interaction was significantly reduced by site 1A (Fig. 5B, lane 7), site 2A (lane 8), and site 3A (lane 9) mutations, most strongly by the site 2A mutation. Thus, these biochemical binding data support the crystal structural evidence for aIF5BI-II-GDP-aP1CTD complex formation.
FIG 5.

Biochemical analyses of the interaction between IF5B and P1. (A) Gel electrophoretic analysis for binding of aP1 mutants to wild-type aIF5B. Wild-type aP1 (lanes 2 and 8) and individual aP1 variants (lanes 3 to 7 and 9 to 13) (100 pmol each) were incubated with (lanes 1 and 8 to 13) or without (lanes 2 to 7) 100 pmol of wild-type aIF5B and subjected to a gel mobility shift assay, as described in Materials and Methods. (B) Gel electrophoretic analysis for binding of aIF5B mutants to wild-type aP1. Wild-type aIF5B (lanes 1 and 6) and individual aIF5B variants (lanes 2 to 4 and 7 to 9) (100 pmol each) were incubated with (lanes 5 to 9) or without (lanes 1 to 4) 100 pmol of wild-type aP1 and subjected to a gel mobility shift assay. The site 1A mutations include L70A, V73A, and I74A; site 2A mutations include F193A, T194A, R195A, K197A, and F199A; and site 3A mutations include I158A, E218A, V222A, and L226A. (C) Structure comparison of the stalk-binding site in aIF5B (left) with the corresponding site in yeast eIF5B (right) (PDB ID 4NCF) (31). Both structures are shown in gray, but sites 1, 2, and 3 of aIF5B and their corresponding sites in yeast eIF5B are shown in green, red, and yellow, respectively. A. pernix aP1CTD is shown in blue (left). (D and E) Effect of mutations on the intrinsic and ribosome-dependent GTPase activity of eIF5B, respectively. Purified recombinant wild-type eIF5B (WT) (bars 1), the eIF5B-5A mutant (5A) (bars 2), or the eIF5B G489A mutant (G479A) (bars 3) was incubated with [γ-32P]GTP in the absence (D) or presence (E) of 80S ribosomes from S. cerevisiae, and their GTPase activity was then measured as described in Materials and Methods. The averages and standard deviations of data from three independent experiments are shown. The values in panel D were calculated from those in Fig. S6B in the supplemental material. *, P < 0.001 by a t test. (F) Effect of anti-P monoclonal antibody on the ribosome-dependent GTPase activity of eIF5B. A total of 2.5 pmol of the 80S ribosome from S. cerevisiae was preincubated without (lane 1) or with 32 pmol of anti-P monoclonal antibody (lane 2) or 32 pmol of nonimmune IgG (lane 3) for 1 h, and the ribosome-dependent GTPase activity was then measured in the presence of eIF5B, as described in Materials and Methods. Activity is shown as percent relative activity, with 100% being the activity without antibody. The averages and standard deviations of data from three independent experiments are shown. *, P < 0.001 by a t test.
The yeast eIF5B mutant altered within the stalk-binding site is defective in factor-dependent function in vitro and in vivo.
Ribosome-dependent GTP hydrolysis is a key step of translation initiation required for the release of eIF5B from the ribosome following subunit joining. To assess whether the interaction between eIF5B and the stalk protein mediates subunit joining and, hence, GTPase activity, we used the yeast assay system, taking advantage of the high level of conservation between the archaeal structural elements involved in aP1-aIF5B binding and the yeast counterparts found in P1/P2 stalk proteins (see Fig. S5A in the supplemental material) and eIF5B (Fig. 5C and Fig. S5B) (31). Because the aIF5B site 2A mutant showed the strongest effect on aP1-aIF5B binding (Fig. 5B), we constructed a corresponding mutant form of yeast eIF5B, termed eIF5B-5A, in which Y582, F583, Q584, K586, and M588 (F193, T194, R195, K197, and F199 in site 2 of archaeal IF5B) (Fig. S5B) are changed to alanine. This mutation alters an unstructured loop located 20 Å away from eIF5B-bound GDP and its ribosome-binding face (Fig. 3B). Therefore, we can disrupt the stalk-binding site alone by this mutation while avoiding the unexpected effects on GDP and ribosome binding.
The recombinant form of the eIF5B-5A mutant migrated similarly to WT eIF5B in a native gel (Fig. S6A) and displayed an affinity for GTP (10.5 ± 0.8 μM) equivalent to that of wild-type eIF5B (18.8 ± 2.5 μM), indicating that the eIF5B-5A mutant is folded properly and binds GTP. In agreement with this assertion, the mutant also displayed the same intrinsic GTPase activity as wild-type eIF5B, even though a G479A mutation, known to directly destabilize GTP binding (32), substantially reduced the activity (Fig. 5D and Fig. S6B). The addition of the ribosome to this reaction greatly stimulated the GTPase activity of WT eIF5B, signifying ribosome-dependent GTPase activity. Importantly, the eIF5B-5A mutation substantially reduced the activity, even though it did not affect intrinsic GTPase activity (Fig. 5D and E, bars 1 and 2). In contrast, the G479A mutation did not reduce the ribosome-dependent GTPase activity as strongly as it reduced the intrinsic activity (Fig. 5D, bar 3). These results strongly suggest that stalk binding to eIF5B plays an important role in mediating ribosome binding to eIF5B during this reaction.
Anti-P antibodies raised against the C-terminal sequence common to P1, P2, and P0 have been extensively used to inhibit translation (33, 34). As shown in Fig. 5F, the anti-P antibody raised against human P1/P2/P0, but not preimmune serum, significantly inhibited ribosome-dependent yeast eIF5B GTPase activity. These results agree with the high level of conversation of the P functional segment in eukaryotes (Fig. S4A) and endorse the idea that the conserved P protein C-terminal tail is directly involved in the eIF5B interaction with the ribosome.
In agreement with a defect in subunit joining in vivo, the eIF5B-5A mutation impaired the ability of overproduced eIF5B to suppress the phenotypes caused by the deletion of the stalk heterodimer P1α-P2β, as shown in Fig. 2: (i) eIF5B-7A decreased the magnitude of GCN4 induction by ∼2-fold in response to 3AT-induced amino acid starvation (Fig. 2C, bars 5 to 8), and (ii) it also increased the leaky scanning of uORF1 translation that had been suppressed by eIF5B overexpression (Fig. 2D, bars 3 and 4). Thus, the stalk-binding site of eIF5B is important for the genetic interaction between eIF5B and the stalk P1/P2.
Next, we examined the direct effect of the eIF5B-5A mutation on eIF5B function in vivo. Yeast deleted for eIF5B (Δfun12) grows very slowly, in agreement with eIF5B's stimulatory role in overall protein synthesis (6) (Fig. 6A). The eIF5B-5A mutation impaired the ability of eIF5B to confer rapid growth to Δfun12 strain J111 in vivo (Fig. 6A), without altering eIF5B expression (Fig. 6C). Thus, the stalk-binding site of eIF5B is important for yeast growth. To further investigate the effect of eIF5B-5A in vivo, we examined the yeast GAAC response dependent on the GCN4 gene. As shown in Fig. 6B, the 3AT sensitivity of the yeast Δfun12 strain (row 1), as described previously (6), was complemented by wild-type eIF5B (row 2) but not by the eIF5B-5A mutant (row 3). These results suggest that the eIF5B-5A mutation partially impairs GAAC induction. When measured directly with the GCN4-lacZ reporter (p180), GCN4 expression in the Δfun12 strain was elevated substantially during 3AT-induced starvation in the presence of WT eIF5B but not in the presence of eIF5B-5A (Fig. 6D, bars 3 to 6). Thus, the eIF5-5A mutant is partially Gcn−, signifying the involvement of the eIF5B stalk-binding site in translational GCN4 induction. To investigate whether eIF5B-5A impairs GCN4 induction through leaky scanning of the uORF1 start codon, we next used pM226, the frameshift mutant version of the GCN4-lacZ plasmid (Fig. 6E, top). As described above, the uORF1 bypass caused by ΔeIF5B results in high-level GCN4-lacZ expression from this plasmid (Fig. 6E, bar 1), which is complemented by WT eIF5B (6) (Fig. 6E, bar 2) but not by the eIF5B-5A mutant (Fig. 6E, bar 3). These results strongly suggest that the eIF5B mutation impairs the formation of the 80S IC at the initiation codon of uORF1, resulting in weaker GCN4 induction (Gcn− phenotype), because of a deficiency in the interaction between eIF5B and the ribosomal stalk in vivo.
FIG 6.
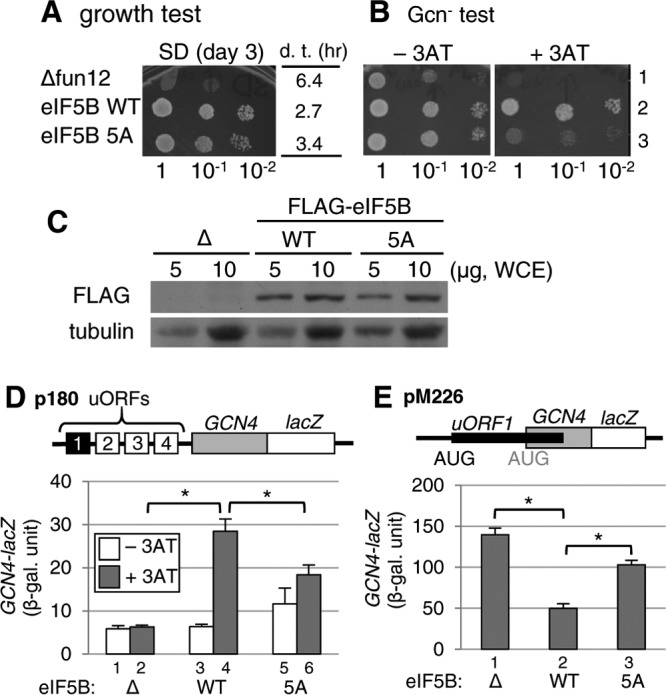
Effects of the eIF5B-5A mutation that disrupts the eIF5B·stalk interaction in yeast. (A and B) Growth and Gcn− phenotype tests. Fixed amounts (5 μl of culture diluted to an OD600 of 0.15) of Δfun12 strain J111 carrying the control vector (Δfun12) (row 1) or the single-copy plasmid pC1818 expressing FLAG-tagged eIF5B-WT (row 2) or eIF5B-5A (row 3) and their 10-fold serial dilutions were spotted onto synthetic dextrose (SD) (A) or SC (B) medium lacking histidine, with or without 4 mM 3AT, and then incubated for 3, 2, or 5 days at 30°C, respectively. In panel A, the table indicates the doubling time (d. t.) of each transformant in liquid medium. (C) Expression of plasmid-directed wild-type eIF5B and the eIF5B-5A mutant in yeast Δfun12 cells. Whole-cell extracts (WCE) from the Δfun12 strain and the same strains transformed with wild-type eIF5B and eIF5B-5A mutant genes with FLAG tags were subjected to immunoblot analyses using anti-FLAG and antitubulin antibodies. (D) Analysis of GCN4 expression by a β-galactosidase assay. Transformants of the Δfun12 strains carrying p180, together with the control (bars 1 and 2), eIF5B-WT (bars 3 and 4), or eIF5B-5A (bars 5 and 6) vector, were grown at 30°C in the absence (−3AT) (bars 1, 3, and 5) or presence (+3AT) (bars 2, 4, and 6) of 10 mM 3AT and assayed for β-galactosidase activity, as described in Materials and Methods. The graphs show averages and standard errors of six values in duplicate from three independent experiments. *, P < 0.001 by a t test. (E) Analysis of leaky scanning of the uORF1 start codon. Transformants of the Δfun12 strains carrying pM226, together with either the control (bar 1), eIF5B-WT (bar 2), or eIF5B-5A (bar 3) vector, were grown at 30°C and assayed for β-galactosidase activity. The data presented are the averages and standard errors of six values in duplicate from three independent experiments. *, P < 0.001 by a t test.
Finally, we reported previously that the 28S rRNA C2879U mutation, altering the LSU A-site (35), suppresses non-AUG initiation (Ssu− phenotype) caused by an eIF2β mutation (SUI3-2) (8). WT eIF5B overexpression alleviated the Ssu− phenotype, suggesting that eIF5B binding to the ribosomal A-site is responsible for stabilizing the 80S initiation complex on a UUG codon (8) (also see Fig. S7A, rows 1 to 6, in the supplemental material). Interestingly, eIF5B-5A overexpression did not alleviate the Ssu− phenotype (Fig. S7A, rows 7 and 8), even though it did not alter the eIF5B expression level (Fig. S7B). Thus, the eIF5B interaction with the stalk also appears to stimulate its interaction with the ribosomal complex initiating at the UUG codon. Together, these data led us to conclude that stalk binding to eIF5B is important for eIF5B function during subunit joining and the formation of the 80S initiation complex in vivo.
DISCUSSION
The present study was initiated by discovering leaky-scanning phenotypes displayed by a stalk protein deletion mutant termed D57 (Fig. 1C and D). This strain is deleted for P1β and P2α, two of the four stalk proteins that together form a canonical heterodimer. The strain displayed a decrease in the polysome abundance, suggesting a defect in translation initiation (Fig. 2A). Furthermore, it cannot efficiently translate uORF1 of GCN4 mRNA due to leaky scanning, or bypass, of its start codon (Fig. 2D). Since uORF1 is required for the delayed reinitiation mechanism of GCN4, the strain was not able to induce GCN4 under histidine starvation conditions (Gcn− phenotype). Because eIF5B overexpression suppressed all the polysomal, leaky-scanning, and GCN4-noninducing phenotypes (Fig. 2), it was concluded that the stalk complex missing one of the two heterodimers cannot bind efficiently to eIF5B and hence caused these phenotypes.
This result is in sharp contrast to that obtained with another stalk deletion mutant, D56, lacking P1β and P2β, which do not form a canonical heterodimer. D56 showed an increased level of GCN4 under unstressed conditions (Gcd− phenotype). Gcd− phenotypes were observed with two other 60S ribosomal protein deletion mutants bearing rpl16BΔ (27) or rpl33AΔ (26). In these strains, the 60S subunit abundances were reduced to 75% and 66%, respectively, compared to the wild type. Because only ∼10% of the ribosome is dissociated to initiate translation in growing yeast cells, while the majority (∼50 to 75%) resides in translating ribosomes (36), the minor (25 to 33%) decrease in the overall 60S subunit abundance can greatly impact the free 60S subunit abundance, to the extent that it causes a “modest” leaky-scanning defect, allowing a high-enough uORF1 initiation rate yet causing the bypass of the uORFs that follow (see Fig. S1 in the supplemental material). We propose that the 20% reduction in the 60S subunit abundance observed in D56 (Fig. 1A) is sufficient to cause this effect.
A conundrum in our observations is why D57 displayed an even stronger defect in subunit joining, and, hence, a Gcn− phenotype, although its 60S abundance was decreased by only 12%. One possibility is that D56, expressing only one pair of noncanonical heterodimers, is able to form a pentamer stalk complex due to the less specific binding of two noncanonical dimers to both of the two binding sites on the P0 spine. The formation of such a pentamer might be slow and inefficient, causing a greater defect in 60S biogenesis, but once formed, the 60S subunit would be more functional than that considered to carry only three stalks found in D57 (13). Another possibility is that the stalk complex is equally defective in D56 and D57. However, for the stalk complex to mediate eIF5B-dependent subunit joining, free 60S subunits must be present in a large-enough quantity (at least at the level found in rapidly growing wild-type yeast). Distinguishing these possibilities by further genetic and biochemical studies would be important for understanding the role of the stalk-eIF5B interaction during translation initiation. We believe that the application of the sophisticated kinetic model of GCN4 control (37) to the simplified leaky-scanning simulation (Fig. S1) greatly contributes to such studies.
Next, how does the stalk complex interact specifically with distinct translational GTPases? Here we showed that the C-terminal region of the stalk forms the type I β-turn and 310-helix when bound to aIF5B (Fig. 3 and 7A). In contrast, the C-terminal region of aP1 bound to aEF1A forms a long extended α-helix (Fig. 7B) (20), suggesting that this region of the stalk protein aP1 can adopt diverse conformations upon binding to different translational GTPases. In agreement with this idea, the C-terminal sequences of eukaryotic stalk proteins, P1/P2, homologous to aP1 (Fig. S5A) (38) can adopt an α-helix, a bent conformation, and an unstructured form in a ligand-free state, depending on the solution conditions (39–41). Moreover, the C-terminal region of the human stalk protein changes its structure to a unique, extended conformation upon antibody binding (39) or a conformation including a type II β-turn upon binding to trichosanthin, a ribosome-inactivating protein (42). Therefore, the C-terminal regions of both the archaeal and eukaryotic stalk proteins can likely adopt diverse conformations, in order to bind distinct GTPase ligands specifically.
FIG 7.

Structural comparisons of aIF5B and aEF1A bound to aP1CTD and aEF2. (A and B) Ribbon diagrams of aIF5BI-II-GDP-aP1CTD and aEF1A-GDP-aP1CTD (PDB ID 3WY9) (20) are shown in the same orientation, based on domain I. Domains I, II, and III of aEF1A and aP1CTD are shown in red, yellow, green, and light blue, respectively. (C and D) Ribbon diagrams of aIF5BI-II-GDP-aP1CTD and eEF2 (PDB ID 1N0V) (43) are shown in the same orientation, based on the domain I structure alignment. Domains I and II of aIF5B and eEF2 and the G′ domain of eEF2 are shown in red, yellow, and gray, respectively. The aP1 interaction region of aIF5B and the corresponding region of eEF2 are shown in magenta.
To account for the binding specificity governed by diverse conformations of the stalk C termini, distinct amino acids on the surface of aIF5B and aEF1A are involved in binding to aP1 (Fig. 7A to C). The structure of the P1 complex with EF2, belonging to the third translational GTPase superfamily, is not known. However, as shown in Fig. 7D, a/eEF2 possesses the G′ domain (in gray) (43) that appears to impede P1 binding in the manner observed with the archaeal IF5B complex (Fig. 7C). Furthermore, there is no discernible homology between the aP1-binding region of aIF5B and the corresponding region of aEF2 (Fig. S8). Therefore, the P1-binding sites seem to be different among all three translational GTPase families (also see the discussion in the supplemental material). It is likely that these diverse binding modes evolved independently to achieve an efficient recruitment of distinct translation factors to the factor-binding center of the ribosome (for bacterial stalk binding to various translational GTPases, see the discussion in the supplemental material).
It remains to be precisely determined how the a/eIF5B·stalk interaction revealed in this study mediates subunit joining in translation initiation. a/eIF5B binds first to the small subunit (SSU) in the initiation complex, before the large subunit (LSU) joins. Thus, the 60S stalk proteins are likely to play an important role in docking the 60S LSU to the SSU pre-IC bound to a/eIF5B. Alternatively, free a/eIF5B may be tethered to a free 60S LSU through the stalk, and subunit joining with the SSU IC is facilitated through the tethering effect. Regardless of these mechanisms, the C-terminal region of the P1 stalk should be able to bind a/eIF5B in a manner compatible with the LSU association with the SSU IC. To address this issue, we constructed a docking model, using the structures of the yeast 80S ribosome associated with eIF5B-GMPPCP (44) and the archaeal stalk complex (12) (Fig. 8A). Since there is no significant difference in the structures of the GDP-bound form of archaeal aIF5B (this study) and the GMPPCP-bound form of eIF5B (44), the stalk-binding site within a/eIF5B appears to be preserved in the 80S IC structure. In this docking model, the stalk-binding site of aIF5B faces away from the small subunit, and the C-terminal region of aP1 can access aIF5B on the small subunit through an unstructured hinge region, with no steric hindrance and without changing the binding orientation of the C-terminal region (Fig. 8A). The docking model therefore endorses the role for the interaction between a/eIF5B and the a/eP1 stalk in subunit joining before GTP hydrolysis (Fig. 8B).
FIG 8.
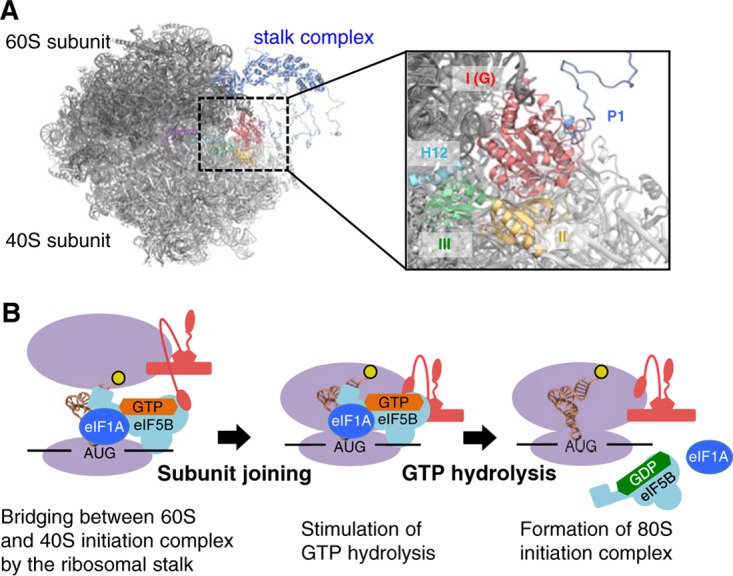
Model of subunit joining in translation initiation. (A) Model of the docking of the aIF5B-GDP-aP1 complex to the 80S initiation complex. The 60S and 40S eukaryotic ribosomal subunits are shown in black and light gray, respectively. The stalk complex, domain I, and domain II of aIF5B are shown in dark blue, red, and yellow, respectively, while domain III, α12 (H12), and domain IV of eIF5B are shown in green, light blue, and purple, respectively. In the docking model, aIF5BI-II-GDP-aP1CTD and the P. horikoshii stalk complex (PDB ID 3A1Y) (12) were superimposed onto the 80S initiation complex with eIF5B (PDB ID 4V8Y) (44), based on the structural alignment for domains I and II of eIF5B and the RNA-binding domain in eukaryotic P0, respectively. The aP0-aP1 stalk complex core and aP1CTD bound to aIF5B-GDP were connected by the flexible hinge region of aP1. Other hinge regions of aP1 were modeled arbitrarily. (B) Schematic representation of the interaction between IF5B and the stalk in the subunit-joining process. The ribosome subunits and the stalk complex are shown in purple and red, respectively. The N-terminal region bound to P0, the hinge region, and the C-terminal region of P1 are modeled as a pentagon, a curved line, and an ellipse, respectively.
We present ample in vivo evidence that the a/eIF5B-stalk interaction is functionally important in yeast (Fig. 2 and 6). The ribosome-dependent GTPase activity of eIF5B was significantly reduced by a mutation altering the five amino acids corresponding to aIF5B site 2 (Fig. 5E). Since the aP1-binding site in aIF5B is located 20 Å away from the bound GTP/GDP (Fig. 3B), it is unlikely that a/eP1 directly influences GTP hydrolysis. Instead, we presume that the a/eIF5B-stalk interaction contributes to the recruitment of the GTP-binding site of a/eIF5B to the sarcin/ricin loop of the large subunit, the generally accepted catalytic site for GTPase activation (45, 46), thereby enhancing GTP hydrolysis, as illustrated in Fig. 8B. GTP hydrolysis and the concomitant dissociation of initiation factors, including a/eIF5B itself, then promote the formation of the 80S IC to start translation elongation (Fig. 8B).
In conclusion, the identification of IF2/IF5B-binding sites in evolutionarily distinct stalk proteins, bacterial L12 (18) and archaeal/eukaryotic aP1/eP1/eP2 (this study), emphasizes the functional importance of the initiation factor·stalk interaction across diverse life forms. Relevant to our findings, 15 to 35% of sera from SLE patients (23) or 42% of sera from patients with active disease (47) contain antibodies against the common C-terminal tails of P1, P2, and P0. Previous studies suggested that these peptides are phosphorylated (21), which, in light of our study, might regulate stalk binding to translational GTPases. Presumably, translational regulation is altered in SLE patients, leading to the efficient cell surface presentation of the stalk peptides and, hence, anti-P antibody production. How this occurs at the level of the ribosome will be an important subject for future investigation.
MATERIALS AND METHODS
Plasmid construction.
The gene encoding full-length aIF5B from the crenarchaeon Aeropyrum pernix was inserted into the pET-21b vector (Novagen) to create a fusion protein with a C-terminal hexahistidine tag. The gene fragments encoding full-length A. pernix aP1 and S. cerevisiae eIF5B with a truncation of the N-terminal 396 residues were cloned into the pET-47b vector (Novagen) to generate proteins with an N-terminal hexahistidine tag. The plasmids encoding mutants of aIF5B, eIF5B, and aP1 were constructed by PCR using the above-described respective vectors.
In yeast, the FUN12 gene encodes eIF5B. For expression in yeast, we used plasmid pC1818 (LEU2 FL-FUN12), a single-copy plasmid derived from YCplac111, expressing N-terminally truncated S. cerevisiae eIF5B. pC1818 contains the natural promoter and terminator of FUN12, encoding eIF5B, and the sequence encoding amino acid residues 28 to 396 of eIF5B was replaced with the FLAG tag sequence. Plasmid pC1818-5A (p1673), encoding an eIF5B mutant (eIF5B-5A) in which 5 amino acid residues involved in stalk binding were replaced with alanines, was prepared by QuikChange PCR mutagenesis using pC1818 as the template. Plasmid YEpW-FUN12 (TRP1 FUN12) (p1656) encoding full-length eIF5B was generated by transferring the BamHI-SalI FUN12 fragment of pC565-3 (URA3 FUN12) (48) to a high-copy-number vector, YEplac112. The five-alanine substation mutation altering the stalk-binding site was introduced by using the same oligonucleotides as the ones employed to prepare pC1818-5A, generating YEpW-FUN12-5A (p1657). Plasmids p180 and pM226, for the GCN4-lacZ expression assay, were described previously (36).
For yeast complementation assays of P1 and P2, we produced YCpU-RPP1A (p1841) and YCpW-RPP2B (p1843) by subcloning the RPP1A and RPP2B gene fragments generated by PCR using oligonucleotides 5′ RPP1A-Sac (5′-gggagctCCCTCATGATTACCGCTCG-3′) and 3′ RPP1A-Hd (5′-CGAAGCTTAAACGGGAAGCCATCAC-3′) and oligonucleotides 5′ RPP2B-Ec (5′-ccgaaTTCCCTATTTCTTTTACACATACCC-3′) and 3′ PRR2B-Hd (5′-AGAAGCTTCTGAAGATGATGATATC-3′) into the SacI-HindIII sites of YCplac33 (Ura+) and the EcoRI-HindIII sites of YCplac22 (Trp+), respectively (in the oligonucleotide sequences listed above, uppercase type indicates those derived from the S. cerevisiae genome, lowercase types indicates those added while designing the oligonucleotides, and underlining indicates restriction enzyme sites).
In vivo phenotypic assays in yeast.
Plasmids and yeast strains used for phenotypic assays in yeast are listed in Table 1. The analyses of GCN4 expression and cell growth were performed with transformants of yeast strains D56 (rpp1BΔ rpp2BΔ), D57 (rpp1AΔ rpp2BΔ) (13), and J111 (fun12Δ) (48), as described previously (8, 36). 3-Aminotrizole (3AT) is used to induce a general amino acid control response (49, 50). To verify the expression of wild-type eIF5B and its mutant, whole-cell extracts prepared from the transformants were subjected to immunoblot analyses, using anti-FLAG (Sigma), anti-yeast eIF5B (a gift of Thomas E. Dever), antitubulin (Sigma), or anti-yeast PGK (3-phosphoglyceric phosphokinase) (catalog number ABIN459152; Antibodies-online, Inc.). Polysome profiles and 40S/60S abundance profiles were obtained by sucrose density gradient-velocity sedimentation, as described previously (36, 51, 52). To evaluate polysome abundance in vivo, the P/M ratio was defined as the ratio of polysome abundance to that of free 40S, 60S, and 80S subunits. It is normally defined as the polysome-to-monosome (80S) ratio. However, because the subunit dissociation observed in stalk deletion mutants (see Fig. S2 in the supplemental material) would underestimate the abundance of vacant 80S ribosomes, we included free 40S and 60S subunits in the calculation of the P/M ratio.
TABLE 1.
Plasmids and yeast strains used in this study
| Plasmid or yeast strain | Genotypea | Reference or source |
|---|---|---|
| Plasmids | ||
| pC1818 | sc LEU2 FL-FUN12 (Δ28–396) | 6 |
| pC1818-5A | sc LEU2 FL-FUN12-5A (Δ28–396) | This study |
| YEpW-FUN12 | hc TRP1 FUN12 | This study |
| YEpW-FUN12-5A | hc TRP1 FUN12-5A | This study |
| YCpU-RPP1A | sc URA3 RPP1A | This study |
| YCpW-RPP2B | sc TRP1 RPP2B | This study |
| Yeast strains | ||
| W303a | MATa leu2-3,112 trp1-1 his3-11,15 ura3-1 ade2-1 can1-100 | 13 |
| D56 | MATa rpl1BΔ::TRP1 rpp2BΔ::HIS3 ura3-1 leu2-3,112 can1-100 | 13 |
| D57 | MATa rpp1AΔ::LEU2 rpp2BΔ::HIS3 ura3-1 trp1-1 can1-100 | 13 |
| J111 | MATa ura3-52 leu2-3 leu2-112 fun12::hisG | 48 |
| KAY761 | MATa ade2-1 his3-11 leu2-3,112 ura3-1 trp1-1 can1-100 rdnΔΔ::HIS3 pNOY373 [2μ RDN LEU2] | 52 |
| KAY968 | MATa ade2-1 his3-11 leu2-3,112 ura3-1 trp1-1 can1-100 rdnΔΔ::HIS3 pNOY373-40 [2μ rdn-40 LEU2] | 35 |
sc, single copy; hc, high copy number.
Simulation of the effect of leaky scanning on GCN4 translational control.
To evaluate the effect of a subunit-joining defect on GCN4 translational control, we used the following formula to compute A, representing the frequency of translation initiation from the GCN4 start codon: A = (1 − f)4f + 0.5f2 [a2 + (1 − a2)(1 − f)][a3 + (1 − a3)(1 − f)][a4 + (1 − a4)(1 − f)], where f is the frequency of initiation by the fully loaded pre-IC from a given start codon within GCN4 mRNA (e.g., f = 1 for wild-type yeast and f < 1 for mutant yeast defective in subunit joining) and an is the frequency of bypass of uORFn (n = ∼2 to 4) by the ribosome that has resumed scanning after uORF1 translation but not yet received the eIF2–GTP–Met-tRNAiMet ternary complex (see Fig. S1 and the text in the supplemental material for details). Based on data reported previously (8, 27, 52, 53), we obtained the following values for an (Fig. S1B and C): (i) an a2 value of 0.5, an a3 value of 0.1, and an a4 value of 0.05 (unstressed conditions); (ii) an a2 value of 0.9, an a3 value of 0.6, and an a4 value of 0.7 (starvation conditions in yeast S288c strains); and (iii) an a2 value of 0.8, an a3 value of 0.5, and an a4 value of 0.4 (starvation conditions in yeast W303 strains). For each 60S subunit mutant, f is estimated based on A obtained under unstressed and starvation conditions (Fig. S1D to G).
Preparation of proteins and the aP1 peptide.
The pET vectors expressing A. pernix aIF5B and S. cerevisiae eIF5B were used to transform Escherichia coli Rosetta(DE3) cells (Novagen). The aIF5B proteins were purified by heat treatment, nickel affinity chromatography, ion-exchange chromatography, and gel filtration chromatography and then dialyzed against buffer A (10 mM HEPES-KOH [pH 7.6], 100 mM KCl, 10 mM MgCl2, and 7 mM 2-mercaptoethanol). The aP1 proteins were purified by heat treatment and nickel affinity chromatography and then treated with HRV3C protease to remove the hexahistidine tag. After digestion of the purification tag, the aP1 proteins were dialyzed against buffer containing 10 mM HEPES-KOH (pH 7.6) and 7 mM 2-mercaptoethanol. N-terminally truncated eIF5Bs were purified by nickel affinity chromatography, ion-exchange chromatography, and gel filtration chromatography. After purification, the eIF5B proteins were dialyzed against buffer containing 20 mM Tris-HCl (pH 7.6), 100 mM NaCl, 10% glycerol, and 7 mM 2-mercaptoethanol. After dialysis, all proteins were concentrated with an Amicon Ultra-15 centrifugal filter unit (Merck Millipore), quantified with a Micro BCA protein assay reagent kit (Thermo Scientific), and stored at −80°C.
The peptide for crystallization, composed of the C-terminal 17 amino acid residues of A. pernix aP1 (residues 95 to 111) (here referred to as aP1CTD) with four lysine residues fused to the N terminus of the peptide to increase its solubility, was synthesized and purified by Hokkaido System Science Co., Ltd.
Crystallization.
Prior to crystallization, the protein solution for crystallization was incubated at 70°C for 10 min. The crystals of aIF5B domains I and II (residues 1 to 358) (here referred to as aIF5BI-II) complexed with GDP and the aP1CTD peptide were obtained by the sitting-drop vapor diffusion method at 20°C in 1 week, by mixing 1 μl of protein solution (17.8 mg ml−1 aIF5BI-II, 10.8 mg/ml aP1CTD, and 4.5 mM GDP in buffer A) and an equal volume of reservoir solution (0.1 M citric acid [pH 6.5], 35% [wt/vol] polyethylene glycol 600 [PEG 600]).
Data collection, structure determination, and refinement.
X-ray diffraction data for aIF5BI-II-GDP-aP1CTD were collected at 95 K with a Quantum 315r charge-coupled-device (CCD) detector (Area Detector Systems Corporation) on beamline BL5A at KEK PF (Tsukuba, Japan). The data sets were processed with the program XDS (54).
The initial model of the aIF5BI-II-GDP-aP1CTD complex was obtained by molecular replacement with the program MOLREP (55), using the structure of aIF5B-GDP from A. pernix as the search model (Protein Data Bank [PDB] ID 5FG3) (30). The models were then manually refined, using the programs COOT (56) and REFMAC5 (57). The resultant data collection and refinement statistics are summarized in Tables S1 and S2 in the supplemental material.
Structure and sequence analysis.
All molecular graphics were prepared by using the program CueMol2 (http://www.cuemol.org/). The multiple-sequence alignment, based on the structural information, was generated with the programs PROMALS3D (58) and ESPript (59). The calculations of the root mean square deviations (RMSDs) between pairs of equivalent Cα atoms were executed by using the program SUPERPOSE (60).
Pulldown assay.
A total of 1,000 pmol of aIF5B and equal amounts of maltose binding protein (MBP) or MBP-aP1C15 were incubated with 20 μl of amylose resin (New England BioLabs) in 100 μl of buffer B (20 mM Tris-HCl [pH 7.6], 200 mM NaCl, 10 mM MgCl2, and 7 mM 2-mercaptoethanol) for 1 h at 4°C with gentle mixing. The beads were washed two times with 500 μl of buffer B, and the bound proteins were then eluted with buffer B containing 50 mM maltose. The eluates were subjected to SDS-PAGE and stained with Coomassie brilliant blue G-250.
Gel mobility shift assay.
The aP1 protein (100 pmol) was mixed with 100 pmol aIF5B in 10 μl of a solution containing 20 mM Tris-HCl (pH 7.6) and 300 mM KCl and then incubated at 70°C for 10 min. After the preincubation step, each sample was subjected to electrophoresis on a 6% polyacrylamide gel at pH 8.8, as described previously (19).
Measurement of the ribosome-dependent GTPase activity of eIF5B.
The high-KCl/puromycin-treated 80S ribosomes from S. cerevisiae were prepared as described previously (61). The 20-μl reaction mixture, containing 2.5 pmol yeast 80S ribosome, 10 pmol eIF5B, 3 nmol [γ-32P]GTP, 20 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 50 mM NH4Cl, and 5 mM 2-mercaptoethanol, was incubated at 30°C for 30 min. After mixing with charcoal, the 32Pi released was recovered and quantitated, as described previously (62).
Measurement of the intrinsic GTPase activity of eIF5B.
The intrinsic GTPase activity of eIF5B was measured as described previously (63), with some modifications. Briefly, 20 μM eIF5B was mixed with 300 μM [γ-32P]GTP in a buffer containing 20 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 1 M NaCl, and 5 mM 2-mercaptoethanol and incubated at 30°C. At various times after incubation, aliquots were taken and mixed with charcoal. The 32Pi released was recovered and quantitated, as described previously (62).
Measurement of Kd values for GTP binding to eIF5B.
Kd (dissociation constant) values for GTP binding to eIF5B were measured by a N-methylanthraniloyl–GDP competition experiment as described previously (32). In our experiment, fluorescence intensities were measured with a Fluorolog-3 spectrofluorometer (Horiba Jobin Yvon). The data were analyzed using a one-site-binding nonlinear regression model with Prism 6 (GraphPad Software, Inc.).
Acidic native PAGE.
A total of 100 pmol of eIF5B with a volume of 10 μl was mixed with 2 μl of a dye solution containing 47.5 mM acetate-KOH (pH 6.8), 40% glycerol, and 0.02% xylene cyanol. The samples were subjected to a gel comprised of a 10% separating gel layer (acrylamide-bisacrylamide at a ratio of 39:1, 386 mM acetate-KOH [pH 4.3], and 10% [vol/vol] glycerol) and a 3% stacking gel layer (acrylamide-bisacrylamide at a ratio of 39:1 and 63 mM acetate-KOH [pH 6.8]). Electrophoresis was performed at 4°C for 2 h at 11.8 V/cm with an acidic electrophoresis buffer (350 mM 6-aminocaproic acid and 175 mM acetate, with the pH adjusted to pH 4.3 with HCl). The direction of protein migration was toward the cathode. The gel was stained with Coomassie brilliant blue G-250.
Accession number(s).
Coordinates and structure factors have been deposited in the RCSB Protein Data Bank with accession number 5YT0 for the aIF5BI-II-GDP-aP1CTD complex.
Supplementary Material
ACKNOWLEDGMENTS
We are deeply indebted to Thomas E. Dever (National Institutes of Health) for timely gifts of material, discussions, and advice. We also thank the beamline staffs of the KEK PF and PF-AR (High Energy Accelerator Research Organization) for technical assistance during data collection, Miguel Remacha and Juan P. G. Ballesta (Consejo Superior de Investigaciones Científicas and Universidad Autónoma de Madrid) for providing the yeast P1/P2 strains, Kathrin Schrick (Kansas State University) for comments, and Brian Ackley (University of Kansas, Lawrence) and Ivan Topisirovic (McGill University) for proofreading.
This work was supported in part by a grant-in-aid for JSPS fellows (15J04972 to R.M.), a grant-in-aid for scientific research (B) (16H04741 to T.U.), a grant-in-aid for young scientists (B) (21770108 to K.I.), and a grant-in-aid for scientific research (C) (15K06964 to K.I.) from the Japan Society for the Promotion of Science; a student exchange support program from the Japan Student Services Organization (to R.M.); a grant from the Uchida Energy Science Promotion Foundation (22-1-10 to K.I.); grants from the National Institutes of Health (R01 GM64781 and R15 GM124671 to K.A.); a pilot grant from University of Kansas COBRE-PSF (P30 GM110761 to K.A.); a bridging grant from the Kansas IDeA Network of Biomedical Research Excellence (P20 GM103418 to K.A.); and an innovative award from the KSU Terry Johnson Cancer Center (to K.A.).
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00067-18.
REFERENCES
- 1.Merrick WC, Hershey JWB. 1996. The pathway and mechanism of eukaryotic protein synthesis, p 31–69. In Hershey JWB, Matthews MB, Sonenberg N (ed), Translational control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 2.Asano K. 2013. Translational control, p 2278–2282. In Dubitzky W, Wolkenhauser O, Cho K-H, Yokota H (ed), Encyclopedia of systems biology. Springer, New York, NY. [Google Scholar]
- 3.Asano K. 2014. Why is start codon selection so precise in eukaryotes? Translation 2:e28387. doi: 10.4161/trla.28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asano K, Sachs MS. 2007. Translation factor control of ribosome conformation during start codon selection. Genes Dev 21:1280–1287. doi: 10.1101/gad.1562707. [DOI] [PubMed] [Google Scholar]
- 5.Milón P, Maracci C, Filonava L, Gualerzi CO, Rodnina MV. 2012. Real-time assembly landscape of bacterial 30S translation initiation complex. Nat Struct Mol Biol 19:609–615. doi: 10.1038/nsmb.2285. [DOI] [PubMed] [Google Scholar]
- 6.Shin BS, Maag D, Roll-Mecak A, Arefin MS, Burley SK, Lorsch JR, Dever TE. 2002. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111:1015–1025. doi: 10.1016/S0092-8674(02)01171-6. [DOI] [PubMed] [Google Scholar]
- 7.Hinnebusch AG, Dever TE, Asano K. 2007. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, p 225–268. In Mathews MB, Sonenberg N, Hershey JWB (ed), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 8.Hiraishi H, Shin B, Udagawa T, Nemoto N, Chowdhury W, Graham J, Cox C, Reid M, Brown SJ, Asano K. 2013. Interaction between 25S rRNA A loop and eIF5B promotes subunit joining and ensures stringent AUG selection. Mol Cell Biol 33:3540–3548. doi: 10.1128/MCB.00771-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto H, Unbehaun A, Loerke J, Behrmann E, Collier M, Bürger J, Mielke T, Spahn CMT. 2014. Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat Struct Mol Biol 21:721–727. doi: 10.1038/nsmb.2859. [DOI] [PubMed] [Google Scholar]
- 10.Asano K, Ito K. 2013. Translation elongation, p 2259–2263. In Dubitzky W, Wolkenhauser O, Cho K-H, Yokota H (ed), Encyclopedia of systems biology. Springer, New York, NY. [Google Scholar]
- 11.Diaconu M, Kothe U, Schlünzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. 2005. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Naganuma T, Nomura N, Yao M, Mochizuki M, Uchiumi T, Tanaka I. 2010. Structural basis for translation factor recruitment to the eukaryotic/archaeal ribosomes. J Biol Chem 285:4747–4756. doi: 10.1074/jbc.M109.068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardenas D, Revuelta-Cervantes J, Jimenez-Díaz A, Camargo H, Remacha M, Ballesta JPG. 2012. P1 and P2 protein heterodimer binding to the P0 protein of Saccharomyces cerevisiae is relatively non-specific and a source of ribosomal heterogeneity. Nucleic Acids Res 40:4520–4529. doi: 10.1093/nar/gks036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yusupova G, Yusupov M. 2014. High-resolution structure of the eukaryotic 80S ribosome. Annu Rev Biochem 83:467–486. doi: 10.1146/annurev-biochem-060713-035445. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Azorin F, Remacha M, Ballesta JPG. 2008. Functional characterization of ribosomal P1/P2 proteins in human cells. Biochem J 413:527–534. doi: 10.1042/BJ20080049. [DOI] [PubMed] [Google Scholar]
- 16.Mohr D, Wintermeyer W, Rodnina MV. 2002. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry 41:12520–12528. doi: 10.1021/bi026301y. [DOI] [PubMed] [Google Scholar]
- 17.Wahl MC, Bourenkov GP, Bartunik HD, Huber R. 2000. Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J 19:174–186. doi: 10.1093/emboj/19.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helgstrand M, Mandava CS, Mulder FAA, Liljas A, Sanyal S, Akke M. 2007. The ribosomal stalk binds to translation factors IF2, EF-Tu, EF-G and RF3 via a conserved region of the L12 C-terminal domain. J Mol Biol 365:468–479. doi: 10.1016/j.jmb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 19.Nomura N, Honda T, Baba K, Naganuma T, Tanzawa T, Arisaka F, Noda M, Uchiyama S, Tanaka I, Yao M, Uchiumi T. 2012. Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc Natl Acad Sci U S A 109:3748–3753. doi: 10.1073/pnas.1112934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito K, Honda T, Suzuki T, Miyoshi T, Murakami R, Yao M, Uchiumi T. 2014. Molecular insights into the interaction of the ribosomal stalk protein with elongation factor 1α. Nucleic Acids Res 42:14042–14052. doi: 10.1093/nar/gku1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elkon K, Skelly S, Parnassa A, Moller W, Danho W, Weissbach H, Brot N. 1986. Identification and chemical synthesis of a ribosomal protein antigenic determinant in systemic lupus erythematosus. Proc Natl Acad Sci U S A 83:7419–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toubi E, Shoenfeld Y. 2007. Clinical and biological aspects of anti-P-ribosomal protein autoantibodies. Autoimmun Rev 6:119–125. doi: 10.1016/j.autrev.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Viana VT, Durcan L, Bonfa E, Elkon KB. 2017. Ribosomal P antibody: 30 years on the road. Lupus 26:453–462. doi: 10.1177/0961203317690243. [DOI] [PubMed] [Google Scholar]
- 24.Huang C, Mandava CS, Sanyal S. 2010. The ribosomal stalk plays a key role in IF2-mediated association of the ribosomal subunits. J Mol Biol 399:145–153. doi: 10.1016/j.jmb.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira-Cerca S, Poll G, Gleizes PE, Tschochner H, Milkereit P. 2005. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol Cell 20:263–275. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Martin-Marcos P, Hinnebusch AG, Tamame M. 2007. Ribosomal protein L33 is required for ribosome biogenesis, subunit joining, and repression of GCN4 translation. Mol Cell Biol 27:5968–5985. doi: 10.1128/MCB.00019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foiani M, Cigan AM, Paddon CJ, Harashima S, Hinnebusch AG. 1991. GCD2, a translational repressor of the GCN4 gene, has a general function in the initiation of protein synthesis in Saccharomyces cerevisiae. Mol Cell Biol 11:3203–3216. doi: 10.1128/MCB.11.6.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remacha M, Jimenez-Diaz A, Bermejo B, Rodriguez-Gabriel MA, Guarinos E, Ballesta JPG. 1995. Ribosomal acidic phosphoproteins P1 and P2 are not required for cell viability but regulate the pattern of protein expression in Saccharomyces cerevisiae. Mol Cell Biol 15:4754–4762. doi: 10.1128/MCB.15.9.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos C, Ballesta JPG. 1995. The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J Biol Chem 270:20608–20614. doi: 10.1074/jbc.270.35.20608. [DOI] [PubMed] [Google Scholar]
- 30.Murakami R, Miyoshi T, Uchiumi T, Ito K. 2016. Crystal structure of translation initiation factor 5B from the crenarchaeon Aeropyrum pernix. Proteins 84:712–717. doi: 10.1002/prot.25009. [DOI] [PubMed] [Google Scholar]
- 31.Kuhle B, Ficner R. 2014. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J 33:1177–1191. doi: 10.1002/embj.201387344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin B-S, Acker MG, Maag D, Kim J-R, Lorsch JR, Dever TE. 2007. Intragenic suppressor mutations restore GTPase and translation functions of a eukaryotic initiation factor 5B switch II mutant. Mol Cell Biol 27:1677–1685. doi: 10.1128/MCB.01258-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchiumi T, Traut RR, Kominami R. 1990. Monoclonal antibodies against acidic phosphoproteins P0, P1, and P2 of eukaryotic ribosomes as functional probes. J Biol Chem 265:89–95. [PubMed] [Google Scholar]
- 34.Sato T, Uchiumi T, Arakawa M, Kominami R. 1994. Serological association of lupus autoantibodies to a limited functional domain of 28S ribosomal RNA and to the ribosomal proteins bound to the domain. Clin Exp Immunol 98:35–39. doi: 10.1111/j.1365-2249.1994.tb06603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nemoto N, Udagawa T, Chowdhury D, Kitabatake M, Hiraishi H, Wang S, Singh CR, Brown SJ, Ohno M, Asano K. 2013. Random mutagenesis of yeast 25S rRNA identify bases critical for 60S subunit structural integrity and function. Translation 1:e26402. doi: 10.4161/trla.26402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee B, Udagawa T, Singh CS, Asano K. 2007. Yeast phenotypic assays on translational control. Methods Enzymol 429:139–161. doi: 10.1016/S0076-6879(07)29007-X. [DOI] [PubMed] [Google Scholar]
- 37.You T, Stansfield I, Romano MC, Brown AJP, Coghil GM. 2011. Analysing GCN4 translational control in yeast by stochastic chemical kinetics modelling and simulation. BMC Syst Biol 5:131. doi: 10.1186/1752-0509-5-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grela P, Bernadó P, Svergun D, Kwiatowski J, Abramczyk D, Grankowski N, Tchórzewski M. 2008. Structural relationships among the ribosomal stalk proteins from the three domains of life. J Mol Evol 67:154–167. doi: 10.1007/s00239-008-9132-2. [DOI] [PubMed] [Google Scholar]
- 39.Soares MR, Bisch PM, Campos de Carvalho AC, Valente AP, Almeida FCL. 2004. Correlation between conformation and antibody binding: NMR structure of cross-reactive peptides from T. cruzi, human and L. braziliensis. FEBS Lett 560:134–140. doi: 10.1016/S0014-5793(04)00088-2. [DOI] [PubMed] [Google Scholar]
- 40.Kessenbrock K, Fritzler MJ, Groves M, Eissfeller P, von Mühlen CA, Höpfl P, Mahler M. 2007. Diverse humoral autoimmunity to the ribosomal P proteins in systemic lupus erythematosus and hepatitis C virus infection. J Mol Med (Berl) 85:953–959. doi: 10.1007/s00109-007-0239-5. [DOI] [PubMed] [Google Scholar]
- 41.Lee KM, Yusa K, Chu LO, Yu CW, Oono M, Miyoshi T, Ito K, Shaw PC, Wong KB, Uchiumi T. 2013. Solution structure of human P1·P2 heterodimer provides insights into the role of eukaryotic stalk in recruiting the ribosome-inactivating protein trichosanthin to the ribosome. Nucleic Acids Res 41:8776–8787. doi: 10.1093/nar/gkt636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Too PH, Ma MK, Mak AN, Wong YT, Tung CK, Zhu G, Au SW, Wong KB, Shaw PC. 2009. The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res 37:602–610. doi: 10.1093/nar/gkn922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jørgensen R, Ortiz PA, Carr-Schmid A, Nissen P, Kinzy TG, Andersen GR. 2003. Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase. Nat Struct Biol 10:379–385. doi: 10.1038/nsb923. [DOI] [PubMed] [Google Scholar]
- 44.Fernández IS, Bai XC, Hussain T, Kelley AC, Lorsch JR, Ramakrishnan V, Scheres SH. 2013. Molecular architecture of a eukaryotic translational initiation complex. Science 342:1240585. doi: 10.1126/science.1240585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liljas A, Ehrenberg M. 2013. Structural aspects of protein synthesis, 2nd ed, p 71–113. World Scientific, Singapore. [Google Scholar]
- 46.Moazed D, Robertson JM, Noller HF. 1988. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 334:362–364. doi: 10.1038/334362a0. [DOI] [PubMed] [Google Scholar]
- 47.Sato T, Uchiumi T, Ozawa T, Kikuchi M, Nakano M, Kominami R, Arakawa M. 1991. Autoantibodies against ribosomal proteins found with high frequency in patients with systemic lupus erythematosus with active disease. J Rheumatol 18:1681–1684. [PubMed] [Google Scholar]
- 48.Choi SK, Olsen SD, Roll-Mecak A, Martung A, Remo KL, Burley SK, Hinnebusch AG, Dever TE. 2000. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol Cell Biol 20:7183–7191. doi: 10.1128/MCB.20.19.7183-7191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. 2001. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 21:4347–4368. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Udagawa T, Nemoto N, Wilkinson C, Narashimhan J, Watt S, Jiang L, Zook A, Jones N, Wek RC, Bähler J, Asano K. 2008. Int6/eIF3e promotes general translation and Atf1 abundance to modulate Sty1 MAP kinase-dependent stress response in fission yeast. J Biol Chem 283:22063–22075. doi: 10.1074/jbc.M710017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asano K, Clayton J, Shalev A, Hinnebusch AG. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev 14:2534–2546. doi: 10.1101/gad.831800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nemoto N, Udagawa T, Singh CR, Wang S, Thorson E, Winter ZA, Ohira T, Brown SJ, Asano K. 2010. Yeast 18S rRNA is directly involved in the ribosomal response to stringent AUG selection during translation initiation. J Biol Chem 285:32200–32212. doi: 10.1074/jbc.M110.146662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mueller PP, Hinnebusch AG. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201–207. doi: 10.1016/0092-8674(86)90384-3. [DOI] [PubMed] [Google Scholar]
- 54.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vagin A, Teplyakov A. 1997. MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30:1022–1025. doi: 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 56.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pei J, Kim BH, Grishin NV. 2008. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 61.Shin B-S, Dever TE. 2007. Molecular genetic structure-function analysis of translation initiation factor eIF5B. Methods Enzymol 429:185–201. doi: 10.1016/S0076-6879(07)29009-3. [DOI] [PubMed] [Google Scholar]
- 62.Frolova L, Le Goff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L. 1996. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA 2:334–341. [PMC free article] [PubMed] [Google Scholar]
- 63.Kuhle B, Ficner R. 2014. A monovalent cation acts as structural and catalytic cofactor in translational GTPases. EMBO J 33:2547–2563. doi: 10.15252/embj.201488517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.