

Using Sos1 knockout (Sos1-KO), Sos2-KO, and Sos1/2 double-knockout (Sos1/2-DKO) mice, we assessed the functional role of Sos1 and Sos2 in skin homeostasis under physiological and/or pathological conditions. Sos1 depletion resulted in significant alterations of skin homeostasis, including reduced keratinocyte proliferation, altered hair follicle and blood vessel integrity in dermis, and reduced adipose tissue in hypodermis.
KEYWORDS: DMBA/TPA, RasGEFs, skin tumors, Sos1, Sos2, wound repair
ABSTRACT
Using Sos1 knockout (Sos1-KO), Sos2-KO, and Sos1/2 double-knockout (Sos1/2-DKO) mice, we assessed the functional role of Sos1 and Sos2 in skin homeostasis under physiological and/or pathological conditions. Sos1 depletion resulted in significant alterations of skin homeostasis, including reduced keratinocyte proliferation, altered hair follicle and blood vessel integrity in dermis, and reduced adipose tissue in hypodermis. These defects worsened significantly when both Sos1 and Sos2 were absent. Simultaneous Sos1/2 disruption led to severe impairment of the ability to repair skin wounds, as well as to almost complete ablation of the neutrophil-mediated inflammatory response in the injury site. Furthermore, Sos1 disruption delayed the onset of tumor initiation, decreased tumor growth, and prevented malignant progression of papillomas in a DMBA (7,12-dimethylbenz[α]anthracene)/TPA (12-O-tetradecanoylphorbol-13-acetate)-induced skin carcinogenesis model. Finally, Sos1 depletion in preexisting chemically induced papillomas resulted also in decreased tumor growth, probably linked to significantly reduced underlying keratinocyte proliferation. Our data unveil novel, distinctive mechanistic roles of Sos 1 and Sos2 in physiological control of skin homeostasis and wound repair, as well as in pathological development of chemically induced skin tumors. These observations underscore the essential role of Sos proteins in cellular proliferation and migration and support the consideration of these RasGEFs as potential biomarkers/therapy targets in Ras-driven epidermal tumors.
INTRODUCTION
Ras proteins are signal transduction regulators that regulate biological processes, including cell proliferation, differentiation, migration, and survival. These proteins continuously cycle between inactive and active conformations in a process modulated by negative (Ras-GAPs) and positive (Ras-GEFs) regulators (1–4). Among the main Ras-GEF families, the Sos proteins are the most widely expressed and functionally relevant with regard to Ras and Rac activation by upstream cellular signals (5, 6).
Ras activation by point mutation leads to a variety of pathological alterations, including multiple tumor types and various developmental syndromes (3, 7). It is also known that hyperactivation of certain Ras-GEFs or dysregulation of other components of Ras-mediated signaling may be associated with different pathological alterations (8–13).
The Sos family of RasGEFs encompasses two ubiquitously expressed, homologous members, Sos1 and Sos2, although their specific functional properties have not yet been clearly ascertained (6). Our prior work showed that constitutive Sos1-null animals die during gestation (14), whereas adult Sos2 knockout (Sos2-KO) mice are viable (15). The generation of conditional Sos1-null mutants has also made it possible to bypass the lethality of Sos1-null mutations and to address unanswered questions regarding the viability of adult mice expressing Sos1-null alleles or the functional specificity/redundancy of Sos proteins in specific cell lineages and tissues (16, 17). Interestingly, Sos1/2 double-knockout (Sos1/2-DKO) animals died precociously, whereas single Sos1- or Sos2-deficient mice were viable, suggesting functional redundancy between Sos proteins in organism homeostasis and survival (17). We also demonstrated that Sos1-null mouse embryonic fibroblasts (MEFs), but not Sos2-null MEFs, showed decreased cell proliferation and migration (18). However, the specific role of Sos1/2 proteins in other different cell lineages and tissues, under physiological and/or pathological conditions, has not been yet examined.
Ras activation and Ras-mediated signaling pathways are critical for epidermal development and carcinogenesis (19–21). In this regard, Sos1 upregulation results in development of skin papillomas with 100% penetrance (22, 23). Although these studies suggest that Sos proteins must be involved in skin homeostasis and/or in response to skin-related pathologies, little is known about the specific role of these proteins in these biological contexts.
Here, we used our viable Sos1-KO and Sos1/2-DKO mutants with an aim at evaluating the role of Sos1/2 proteins in maintenance of skin homeostasis and the healing processes upon injury. In addition, the effects of Sos1/2 disruption on initiation and/or progression of skin tumors was also investigated by using the DMBA (7,12-dimethylbenz[α]anthracene) and TPA (12-O-tetradecanoylphorbol-13-acetate)-induced skin tumorigenesis (24).
Unraveling the functional peculiarities of Sos1 and Sos2 will further improve our understanding of the relevance of Ras/Sos-dependent cellular signaling mechanisms and pathways in skin physiological and tumoral processes. Ascertaining the exact mechanistic contributions of these Ras-GEF proteins in those processes may also provide a basis for their use as diagnostic markers or therapy targets in pathological contexts.
RESULTS
Sos1 is functionally prevalent over Sos2 for maintenance of skin architecture.
We examined whether Sos1/2 depletion, individually or in combination, altered skin homeostasis. PCR assays using specific primers documented that disruption of the floxed Sos1 allele was 100% complete in our animals after 12 days of tamoxifen (TAM) feeding (Fig. 1a, left panel). Western blot (WB) analysis confirmed the disappearance of the Sos1 and Sos2 proteins in skin of mice treated under these experimental conditions and confirmed our previous observations in MEFs (18), indicating that there are no compensatory changes of either Sos1 or Sos2 protein expression when the other form is absent (Fig. 1a, right panel). Histological examination after that time revealed no differences in the overall skin architecture of Sos2-KO mutants compared to wild-type (WT) counterparts (Fig. 1b). The thicknesses of the three skin layers were almost identical for both genotypes (Fig. 1g to i), with similar keratinocyte organizations and hair follicle morphologies and distributions in the epidermis and comparable organizations in the dermis and hypodermis (Fig. 1c to e). In contrast, single Sos1 deletion resulted in reduction of all layers in comparison to WT and Sos2-KO counterparts (Fig. 1). Moreover, dual Sos1/2 absence caused a severe overall disruption of skin architecture. Epidermal thickness was affected in Sos1/2-DKO mice, exhibiting a reduction compared to the rest of groups (Fig. 1b, c, and g). We also observed distorted hair follicles and sebaceous glands whose number and size appeared diminished compared to WT counterparts (Fig. 1f). Dermal thickness was reduced in relation to WT and Sos2-KO mice, although with similar values compared to the Sos1-KO group (Fig. 1b, d, and h). Similarly to Sos1-KO mice, the cellularity in the dermis of Sos1/2-DKO mice was decreased in comparison with WT and Sos2-KO mice (Fig. 1d). Combined Sos1/2 deletion also resulted in a dramatic reduction of hypodermis with respect to the rest of the genotypes (Fig. 1b, e, and i).
FIG 1.
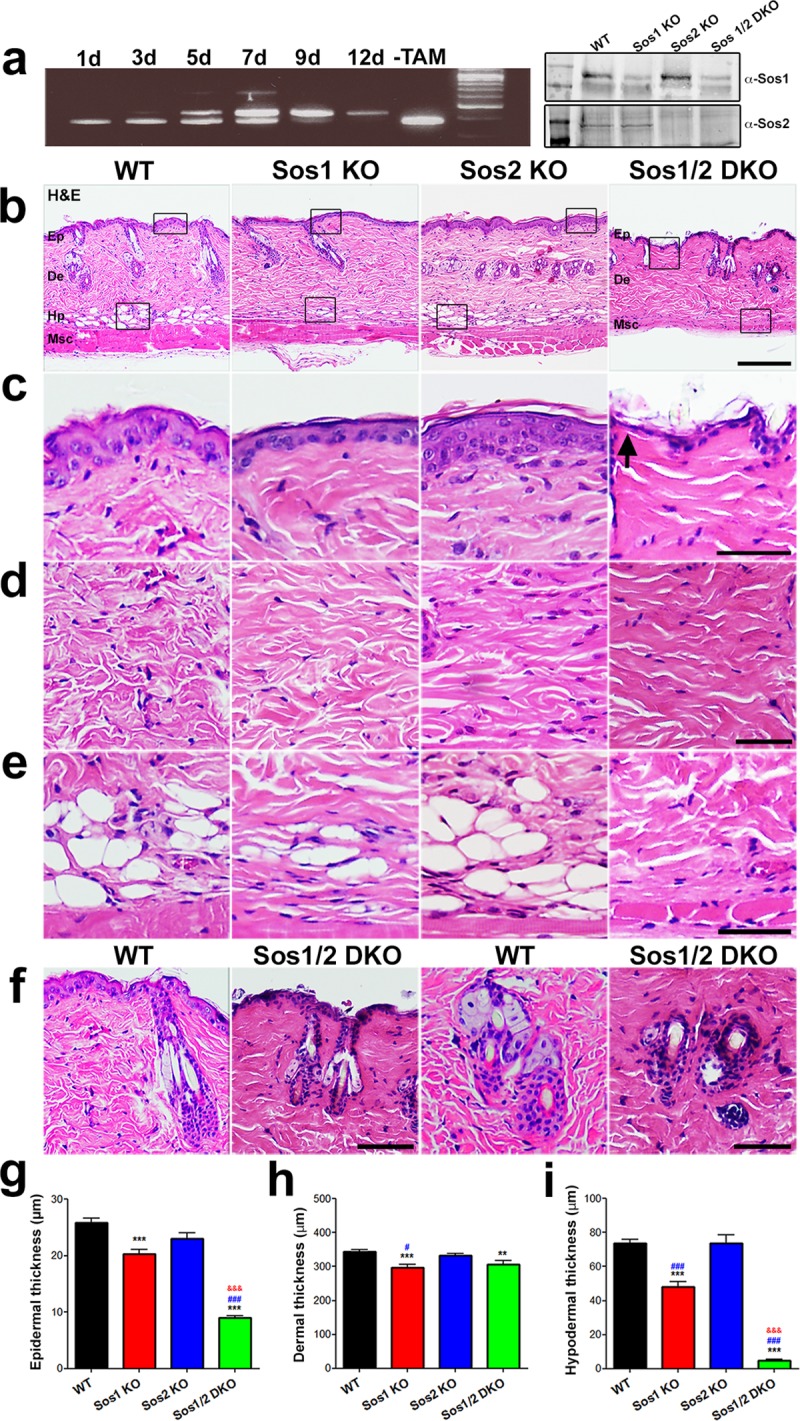
Histological analysis of overall skin architecture in WT and Sos1/2-KO mice. (a) The left panel shows PCR amplification of DNA from the dorsal skin of Sos1fl/fl mice treated with TAM for the indicated times (days). Progressive excision of Sos1 exon 10 (revealed by the upper band) was complete after 12 days TAM treatment. No excision occurred in the untreated (−TAM) Sos1fl/fl mice. In the right panel, a Western immunoblot shows Sos1 and Sos2 protein depletion in the dorsal skin of mice after 12 days of TAM treatment. (b) H&E-stained, paraffin-embedded sections of skin from WT, Sos1-KO, Sos2-KO, and Sos1/2-DKO mice that were previously fed with TAM-containing diet for 12 days (for complete disruption of the floxed Sos1 allele). Scale bar, 200 μm. Ep, epidermis; De, dermis; Hp, hipodermis; Msc, muscle. (c to e) Higher magnification of boxed areas marked in panel b, focusing on the epidermis (c), dermis (d), and hypodermis (e) of each of the indicated genotypes. Scale bars, 25 μm. (f) Higher magnification of areas containing hair follicles (left panels; scale bar, 100 μm) and/or sebaceous cells (right panels; scale bar, 50 μm) in skin sections of the indicated Sos genotypes, documenting the alteration and/or loss of integrity of these structures in the skin of Sos1/2-DKO mice compared to similarly TAM-treated WT mice. (g to i) Bar plots quantitating thickness of the epidermis (g), dermis (h), and hypodermis (i) layers of the skin of mice of the indicated genotypes. Data are expressed as means ± the standard errors of the mean (SEM). n = 3 per genotype (100 measurements per mouse). #, P < 0.05 versus Sos2-KO; **, P < 0.01 versus WT; ***, &&&, and ###, P < 0.001 versus WT, Sos1-KO, and Sos2-KO, respectively.
Differential contribution of Sos1 and Sos2 to control of keratinocyte proliferation and survival in the skin.
We next tested whether the loss of epidermal thickness observed in Sos1-KO and Sos1/2-DKO mice was a consequence of altered keratinocyte proliferation and/or increased of cell apoptosis in epidermis. Single Sos2 depletion did not alter the proliferative ability of keratinocytes respect to the WT (Fig. 2a and e). However, Sos1-KO mice exhibited a reduction of proliferating keratinocytes with respect to WT counterparts (Fig. 2a and e) that became more pronounced in the Sos1/2-DKO mice (Fig. 2a and e). Consistent with the reduction of proliferation in the epidermis of Sos1-deficient mice, a diminution of phospho-extracellular signal-regulated kinase (pERK) expression was detected in the epidermis of Sos1-depleted groups (Fig. 2b). A slight reduction of the basal level of Rac-GTP was also apparent by means of pulldown assays in Sos1-depleted samples (Fig. 2h).
FIG 2.

Evaluation of keratinocyte proliferation and survival in the skin of WT and Sos1/2-KO mice. (a) Representative confocal microscopy images of skin sections of the four relevant genotypes after costaining for BrdU (green) and DAPI (4′,6′-diamidino-2-phenylindole; blue). Scale bar, 25 μm. (b) Representative immunostaining pictures of pERK detected in paraffin-embedded skin sections of the four relevant Sos genotypes. Scale bar, 50 μm. (c) Cleaved-caspase-3 (green) and DAPI (blue) colabeling performed in paraffin-embedded sections of skin from the four experimental groups. Arrows point to cleaved-caspase-3-positive cells in hair follicles. Note the nonspecific staining in the upper layers of the epithelium. Scale bar, 50 μm. (d) TUNEL (green) and DAPI (blue) colabeling performed in paraffin-embedded sections of skin from the four experimental groups. Arrows point to TUNEL-positive cells. Scale bar, 50 μm. (e) Percentage of BrdU-positive keratinocytes in skin of the indicated Sos genotypes per field. n = 4 per genotype (four microscopy fields per mouse). (f) Quantitation of percentage of cleaved-caspase-3-positive cells counted in the skin of the indicated Sos genotypes. n = 4 per genotype (four microscopy fields per mouse). (g) Quantitation of percentage of apoptotic, TUNEL-positive cells in skin of the indicated Sos genotypes. n = 4 per genotype (four microscopy fields per mouse). (h) Representative image of Rac-GTP pulldown assays. Normalized Rac-GTP/Total Rac ratios were as follows: WT = 1 ± 0.17, Sos1-KO = 0.77 ± 0.11, Sos2-KO = 0.98 ± 0.08, and Sos1/2-DKO = 0.61 ± 0.15. n = 3 per genotype. Data are expressed as means ± the SEM. (a to c) n = 3 per genotype (three sections per animal). (d) WT, n = 6; Sos1-KO, n = 4; Sos2-KO, n = 3; and Sos1/2-DKO, n = 7. & and #, P < 0.05 versus Sos1-KO and Sos2-KO, respectively; **, &&, and ##, P < 0.01 versus WT and Sos2-KO; ***, P < 0.001 versus WT. Ep, epidermis; De, dermis.
The percentage of cleaved-caspase-3-positive (Fig. 2c and f) or TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling)-positive (Fig. 2d and g) cells found in the skin of WT, Sos1-KO, or Sos2-KO mice was very limited. In contrast, although the absolute number of dying cells was still quite low, the percentage of apoptotic cells was higher in the skin of Sos1/2-DKO mice (Fig. 2f and g), thus raising the question of the potential relevance of these minor apoptotic changes with regard to the severe changes of skin homeostasis observed in this experimental group.
Sos1 disruption is associated with decreased vascularization and angiogenesis in the skin.
Using CD31 immunostaining, we observed that Sos1 depletion resulted in reduced skin vascularization in the dermis compared to the WT and Sos2-KO counterparts (Fig. 3a and c), an observation consistent with the reduction of dermis seen in Sos1-depleted mice (Fig. 1). The potential contribution of Sos GEFs to angiogenesis was also analyzed using aortic ring assays (Fig. 3b). Our data showed that single absence of either Sos1 or Sos2 resulted in minor reduction of the number of new sprouts arising from the aortic rings, whereas the formation of these new sprouts was almost completely abolished in the Sos1/2-DKO samples (Fig. 3d). In contrast, the length of the sprouts was only diminished under simultaneous Sos1/2 deletion (Fig. 3e).
FIG 3.

In vivo and ex vivo analysis of vascularization and angiogenesis in WT and Sos1/2-KO mice. (a) Representative confocal microscopy images of skin sections of the four relevant genotypes (WT, Sos1-KO, Sos2-KO, and Sos1/2-DKO) costained for CD31 (green) and DAPI (blue). Scale bar, 100 μm. (b) Representative images of aortic ring explant assays after the indicated times (1, 3, 5, and 8 days) in culture. Scale bars: 200 μm for day 1 to day 8 columns and 500 μm for the rightmost column, where the boxes enclose the areas magnified in the corresponding pictures under the day 8 column. (c) Quantitation of the CD31-positive immunolabeling area in the dermis of the four relevant Sos genotypes. n = 3 per genotype. (d) Quantitation of the number of sprouts counted in aortic ring assays performed on samples of the four relevant Sos genotypes at the indicated postexplant times (days) in culture. (e) Graphic representation of the average length of the sprouts generated by aortic rings of the four relevant Sos genotypes after 8 days of explant culture. n = 5 per genotype. Data are expressed as means ± the SEM. * and &, P < 0.05 versus WT; **, &&, and ##, P < 0.01 versus WT and Sos1-KO; ***, &&&, and ###, P < 0.001 versus WT, Sos1-KO, and Sos2-KO, respectively.
Concomitant Sos1/2 depletion impairs tissue remodeling in wounded skin.
The sequence of events occurring after cutaneous injury includes transitory changes in the balance between cell proliferation and differentiation and the activation of the immune system (25). Here, we evaluated the effect of Sos1/2 depletion during healing of round full-thickness excisions performed in the dorsal skin of our TAM-treated mice (Fig. 4).
FIG 4.

Wound healing assays in the skin of WT and Sos1/2-KO mice. (a) Graph depicting the kinetics of wound repair after injury to the skin of mice of the four indicated Sos genotypes. WT, Sos1-KO, and Sos2-KO mice underwent 12 days of TAM treatment before initiating the experiment, whereas, because of their premature lethality, Sos1/2-DKO mice were TAM treated for only 4 days prior to starting the wound healing procedure. TAM feeding was then kept in this group for the whole duration of the assay. Data are expressed as means ± the SEM. n = 4 per genotype. *, P < 0.05 versus WT; ** and ##, P < 0.01 versus WT and Sos2-KO; ***, &&&, and ###, P < 0.001 versus WT, Sos1-KO, and Sos2-KO, respectively. (b) Representative pictures of the wound region on the backs of mice of the four relevant Sos genotypes at the endpoint (10 days) of the wound repair assays. Scale bar, 4 mm. (c) Representative images of the wound closure area in injured skin of WT and Sos1/2-DKO mice at day 1 and day 10 of our wound repair assays. Scale bar, 4 mm. (d) H&E staining of paraffin-embedded sections of the wound area in skin from injured WT and Sos1/2-DKO mice after 3 days of the wound. Notice that the density of the blood vessels (red arrows) in the skin of WT mice is significantly higher than in Sos1/2-DKO mice. Scale bar, 100 μm. (e) Paraffin-embedded, skin sections of the injured area in mice of the indicated genotypes, after costaining for BrdU (green) and DAPI (blue). Scale bar, 100 μm. (f) Higher magnification of areas boxed in panels d and e focusing on epidermal hyperplasia (red arrows), cellularity of the dermis (blue arrows), and blood vessels adjacent to the injury (green arrows). Scale bars: H&E (250 μm) and BrdU (100 μm). (g) Paraffin-embedded sections of the wound area in WT, Sos1-KO, and Sos1/2-DKO injured skin (10 days after injury), costained for K14 (green) and vimentin (red) and counterstained with DAPI (blue). Scale bar, 100 μm. (h) Paraffin-embedded sections of the injured wound area in mice of the four relevant genotypes after immunostaining with the macrophage marker F4/M80. Scale bar, 50 μm.
We observed that complete healing in the WT group took about 10 days (Fig. 4a and b). Single Sos2 absence did not alter the kinetics of the wound repair process in comparison to WT counterparts (Fig. 4a and b). In contrast, single Sos1 depletion slightly delayed wound closure kinetics in comparison to WT and Sos2-KO groups, and simultaneous Sos1/2 depletion strongly affected the reepithelialization process (Fig. 4a and b). During the initial phases of healing (days 1 to 3), closure of the wounds was very similar in Sos1/2-DKO mice compared to the rest of groups (Fig. 4a). Of note, during that time frame, Sos1 depletion was not yet complete. However, after 4 days and onward, the repaired area of the wound remained basically unchanged in Sos1/2-DKO mice, with about 40% of the wound still persisting open (Fig. 4a to c).
During skin repair, fibroblasts migrate to the injury site, and the mechanical forces created by their movement through the wounded tissue generate mechanical tension which promotes wound closure (25). Whereas the levels of contraction of the tissue in the proximity of the wound were similar in WT, Sos1-KO, and Sos2-KO mice (not shown), we did not visualize this phenomenon in Sos1/2-DKO counterparts (Fig. 4c).
We also analyzed inflammatory and proliferative phases of healing. Consistent with proliferative and migratory keratinocyte responses, we observed epidermal hyperplasia at the wound edges in WT mice (Fig. 4d). In contrast, Sos1/2-DKO mice did not exhibit any hyperplasia in the epidermis at the wound edge (Fig. 4d). Bromodeoxyuridine (BrdU) immunolabeling confirmed a profound reduction of proliferating keratinocytes at the wound edge in Sos1/2-DKO compared to WT mice (Fig. 4e and f). In addition, increased cellularity and blood vessel density was detected in the dermis of the WT mice, whereas, in contrast, no significant increment of either cellularity or blood vessel density was observed in response to injury in the Sos1/2-DKO samples (Fig. 4d and f). Vimentin immunostaining also confirmed the reduction of fibroblast migration at the wound edge in Sos1-depleted mice compared to WT counterparts (Fig. 4g). Finally, the level of macrophage infiltration in the injury zone was also reduced in the absence of Sos1 (Fig. 4h).
Sos1 depletion impairs the initiation, promotion, and malignant progression of DMBA/TPA-induced skin tumors.
We next evaluated the role of Sos1 and Sos2 during the initiation and promotion phases of DMBA/TPA-induced skin carcinogenesis. WT, Sos1-KO and Sos2-KO mice were TAM treated (12 days) before undergoing DMBA/TPA “protocol 1” (see Materials and Methods) (Fig. 5a). The application of this protocol to Sos1/2-DKO mice was not feasible due to the premature death of these mice (17), and the animals of this genotype only received TAM treatment at 28 weeks after start of the carcinogenic protocol [Sos1/2-DKO(−TAM/+TAM)] (Fig. 5b).
FIG 5.

DMBA/TPA-induced skin tumor initiation and progression in WT and Sos1/2-KO mice. (a) Representative images of TAM-treated, WT, Sos1-KO, and Sos2-KO mice and the tumors generated in each of those genotypes after undergoing 30 weeks of DMBA/TPA-induced carcinogenic treatment under protocol 1, as described in Materials and Methods. (b, left) Sos1/2-DKO(−TAM). Representative images are shown of a Sos1/2-DKO mouse and the dorsal skin tumors developed after 28 weeks of the DMBA/TPA carcinogenic treatment of mice of this particular genotype that were kept for this complete time period without the TAM treatment needed for Sos1 removal. (b, right) Sos1/2-DKO(+TAM). A representative image is shown of a mouse of the same Sos1/2-DKO genotype and the skin tumors observed in it at the 30-week time point mark of the DMBA/TPA carcinogenic protocol in Sos1/2-DKO mice that were only administered TAM (for Sos1 depletion) after previously undergoing 28 weeks of the carcinogenic protocol. (c) Percentage of mice showing development of tumors until week 28 of application of the carcinogenesis protocol to mice of the experimental groups depicted in panel a (TAM treated, WT, Sos1-KO, and Sos2-KO) or panel b (TAM untreated, Sos1/2-DKO). (d and e) Graphic representation of the number of tumors per individual mouse that appeared up to week 30 of our carcinogenic protocol in the indicated Sos genotypic groups. Notice that whereas WT and individual Sos1-KO and Sos2-KO mice were pretreated with TAM before undergoing the carcinogenic protocol, the DKO mice were only treated with TAM (for Sos1 depletion) after week 28 of the protocol. The arrow marks the start of TAM treatment for Sos1/2-DKO mice. The box in panel d encircles the time period amplified in more detail in panel e. (f and g) Evolution of the size of tumors arising in each experimental genotypic group. Data are expressed as the average area (mm2) of all tumors detected in the indicated experimental time points along the carcinogenic protocol. The arrow marks the time point where TAM treatment started for the group of Sos1/2-DKO mice. The box in panel f encloses the specific time period that is amplified to show data in more detail in panel g. (h) Percentage of the progressive histopathological grades identified in chemically induced tumors arising in mice of the indicated Sos genotypes. Tumor pathology stages were morphologically identified here as papilloma (P), sebaceous adenoma (A), keratoacanthoma (K), and squamous cell carcinoma (SSC). (i) Quantitative distribution of grades of histopathological progression within the benign tumors (adenomas and papillomas) counted in each of the four Sos experimental groups. Pathology progression stages were morphologically identified here as low-grade papilloma (P1), medium/high-grade papilloma (P2-P3), low-grade sebaceous adenoma (A1), and medium/high-grade sebaceous adenoma (A2-A3). Data are expressed as means ± the SEM. n = 13 for WT and Sos1/2-DKO, n = 11 for Sos1-KO, and n = 15 for Sos2-KO. * and #, P < 0.05 versus WT and Sos2-KO; ** and &&, P < 0.01 versus WT and Sos1-KO; ***, ###, and &&&, P < 0.001 versus WT, Sos1-KO, and Sos2-KO, respectively. TAM, tamoxifen.
Sos1 depletion delayed tumor development compared to WT group (Fig. 5c). In contrast, single Sos2 absence had no impact on the kinetics of DMBA/TPA-induced carcinogenesis relative to WT counterparts (Fig. 5c). Of note, tumors appeared earlier in Sos1/2-DKO(−TAM) samples than in the rest of groups (Fig. 5c). Because this group lacked TAM treatment during the initial stages of analysis, this may suggest a partial off-target effect of TAM retarding tumor initiation. Both the number of tumors per mouse and the tumor size were reduced in Sos1-KO mice compared to the WT group (Fig. 5d to g). In contrast, Sos2-depleted mice showed similar numbers and sizes of tumors compared to their WT counterparts (Fig. 5d to g). A noticeable reduction was observed in both the numbers and sizes of tumors in the Sos1/2-DKO(−TAM) group upon TAM administration at week 28 of the carcinogenic protocol (Fig. 5b and d to g).
The influence of the Sos1/2 proteins on the histopathological grades reached by the DMBA/TPA-induced tumors was investigated by quantifying the percentages of benign tumors, premalignant lesions, and malignant carcinomas generated in each group (Fig. 5h and i). About 60% of the DMBA/TPA-induced tumors in WT mice were papillomas, whereas about 30% of the remainder tumors were catalogued as sebaceous adenomas (Fig. 5h). Although with low frequency, tumors with a higher grade of malignancy, including keratoacanthomas (K) and squamous cell carcinomas (SCC), were also detected in WT mice (Fig. 5h). In all Sos-KO genotypes, the chemically induced tumors were mostly identified as papillomas (∼85%) and sebaceous adenomas (15 to 20%). In contrast to the observations in WT mice, no malignant SCC were detected in any of the Sos-null-mutant groups (Fig. 5h). Furthermore, the pathological progression of the benign papillomas and adenomas developed in WT mice was higher than those observed in Sos-depleted mutants (Fig. 5i). Lower number of the more aggressive tumors, such as SCC, would be expected in the Sos1/2-DKO group since TAM treatment (to eliminate Sos1) in these mice only started after week 28.5 because of their inherent lethality problems (17). Notice that these mice were originally already Sos2-KO and then became Sos1 depleted upon TAM administration. It was also noticed that single Sos2 absence appeared to prevent formation of malignant tumors (Fig. 5h and i), suggesting that the depletion of Sos2 in the DKO group could by itself contribute to attenuating progression to malignancy in the DMBA/TPA-induced carcinogenic assays.
These observations were consistent with the reduction of BrdU-positive keratinocytes and pERK activation detected in Sos1-depleted tumors compared to WT and Sos2-KO tumors (Fig. 6a, b, and e). The very low rates of cell death measured in Sos1-KO and Sos2-KO samples suggested that single Sos1 or Sos2 depletion did not significantly prime skin tumor cells for apoptosis, However, combined Sos1/2 depletion slightly increased the percentage of apoptosis in tumors compared to WT, Sos1-KO, and Sos2-KO mice (Fig. 6c, d, f, and g), and therefore we cannot discard the possibility that this increment of cell death rate plays a role, at least in part, in the reduction of tumor size observed in the DKO mice (Fig. 5f and g).
FIG 6.

Analysis of cell proliferation, pERK distribution, and cell survival in skin tumors of WT and Sos1/2-KO mice. (a) Representative confocal images of paraffin-embedded skin sections of mice of the four relevant Sos genotypes subjected to the DMBA/TPA protocol that were immunostained for cytokeratin K14 (red) and BrdU (green). Scale bar, 50 μm. (b) Representative images of pERK immunolabeling of skin sections corresponding to the same genotypes and carcinogenic treatments as in panel a. Scale bar, 100 μm. (c) Representative confocal images of skin sections, corresponding to the same genotypes and treatments as in panels a and b, that were costained for cleaved caspase-3 (green) and DAPI (blue). Boxes in the upper row enclose areas amplified in the pictures of the lower row. Scale bars, 100 μm (upper row) and 25 μm (lower row). (d) Representative confocal images of skin sections corresponding to same genotypes and treatments as in panels a, b, and c that were colabeled for TUNEL assay (green) and DAPI (blue). Boxes indicate TUNEL-positive cells. Note TUNEL unspecific labeling in the hair follicles (asterisks). Scale bar, 100 μm. (e) Quantitation of the percentage of BrdU-positive cells detected in the skin of mice of the indicated genotypes after undergoing the DMBA/TPA carcinogenic protocol. Data are expressed as means ± the SEM. n = 5 for all genotypes (four microscopy fields per mouse). #, P < 0.05 versus Sos2-KO; **, P < 0.01 versus WT. (f) Quantitation of the percentage of caspase-3-positive, apoptotic cells detected mice of the indicated genotypes that were subjected to the DMBA/TPA carcinogenesis protocol. Data are expressed as means ± the SEM. n = 4 for all genotypes (four microscopy fields per mouse). *, P < 0.05 versus WT. (g) Quantitation of the percentage of TUNEL-positive cells detected inmice of the indicated genotypes that were subjected to the DMBA/TPA carcinogenesis protocol. n = 4 for all genotypes (four microscopy fields per mouse).
Sos1 depletion reduces TPA-mediated hyperproliferation.
We analyzed further the events taking place during the initiation and early promotion phases of the carcinogenesis by performing specific short-term experiments inducing hyperproliferative, inflammatory, and proapoptotic responses in the skin (Fig. 7). These assays additionally allowed us to evaluate the effect of concomitant Sos1/2 absence during the early phases of DMBA/TPA-induced cancer.
FIG 7.

Analysis of proliferative, inflammatory, and apoptotic responses in the skin of WT and Sos1/2-KO mice after short-term TPA and DMBA treatments. (a) Representative confocal immunofluorescence images detecting BrdU (green), K-14 (red), and DAPI (blue) in TPA-treated skin sections from the four indicated genotypes. Scale bar, 100 μm. (b) Higher magnification of the areas enclosed by boxes in panel a. Scale bar, 5 μm. (c) Representative confocal images of TPA-treated skin sections of the indicated genotypes, immunostained with anti-neutrophil elastase (green) and counterstained with DAPI (blue). Scale bar, 100 μm. (d) Representative confocal images of skin sections from DMBA-treated mice of the indicated genotypes, immunolabeled for cleaved caspase-3 (green) and counterstained with DAPI (blue). Arrows point to apoptotic keratinocytes. Scale bar, 25 μm. (e) Quantitation of epidermal thickness measured in mice topically treated on the dorsal skin with four consecutive doses (one dose/day) of TPA of the four relevant Sos genotypes. n = 3 per genotype (60 measurements per mouse). (f) Quantitation of percentage of BrdU-positive cells present in the epidermis of mice of the indicated genotypes that were subjected to short-term TPA treatment. n = 3 per genotype (four microscopy fields per mouse). (g) Quantitation of number of neutrophils per 1,000 μm2 of tissue detected in skin of mice of the four indicated genotypes after undergoing four consecutive doses of TPA treatment. n = 3 per genotype. (h) Quantitation of the percentage of cleaved-caspase-3-positive cells counted in the epidermis of mice of the indicated genotypes after undergoing a single exposure to DMBA. n = 3 per genotype (four microscopy fields per mouse). In panels e to h, data are expressed as means ± the SEM. *, &, and #, P < 0.05 versus WT, Sos1-KO, and Sos2-KO, respectively; ** and ##, P < 0.01 versus WT and Sos2-KO, respectively; ***, &&&, and ###, P < 0.001 versus WT, Sos1-KO, and Sos2-KO, respectively. Ep, epidermis; De, dermis; Hp, hypodermis.
TPA-induced hyperproliferation caused gain of epidermal thickness in all groups relative to untreated conditions (Fig. 1b and 7a). In particular, the gain of epidermal thickness in single Sos2-KO mice was similar to that in WT mice (Fig. 7a, b, and e). In contrast, single Sos1 depletion caused a slight reduction of the epidermis in comparison to WT animals, and this reduction became much more dramatic under concomitant Sos1/2 depletion (Fig. 7a, b, and e).
Consistently, the number of BrdU-positive cells detected in skin of mice of the four groups after TPA application was also increased in comparison to untreated animals of their respective genotypes (Fig. 2 and 7). Indeed, consistent with the epidermal reduction detected in TPA-treated, Sos1-depleted mice, the number of BrdU-positive keratinocytes was diminished in Sos1-KO and Sos1/2-DKO mice compared to WT and Sos2-KO counterparts (Fig. 7f).
Short-term TPA administration also allowed us to investigate the role of Sos proteins on neutrophil-mediated inflammatory response upon treatment with this compound. Our observations evidenced a reduction of dermis in Sos1-KO mice compared to the WT and Sos2-KO groups. This reduction was more evident in the Sos1/2-DKO mice (Fig. 7a and c). On the other hand, the density of neutrophils infiltrated in the dermis of TPA-treated Sos1-KO mice was similar to WT counterparts, whereas the Sos2-KO mice displayed very significant increments of neutrophil infiltration compared to the rest of groups (Fig. 7c and g). In contrast, combined Sos1/2 depletion resulted in almost total ablation of neutrophil response to the inflammatory stimulus (Fig. 7c and g).
Finally, we studied the effect of Sos1/2 depletion on DMBA-induced apoptosis after single-dose application of this compound to the dorsal skin of our mice. An increased apoptotic rate was observed in the skin of all experimental groups compared to untreated mice of their respective genotype (Fig. 2 and 7). In particular, only concomitant Sos1/2 absence appeared to slightly sensitize keratinocytes to DMBA-induced apoptosis by producing an increment of the number of cleaved-caspase-3-positive cells in the skin of Sos1/2-DKO, DMBA-treated mice (Fig. 7d and h). In contrast, individual depletion of Sos1 or Sos2 did not result in significant alteration of the rate of apoptosis in skin cells in comparison to WT mice (Fig. 7d and h).
Sos1 disruption reduces growth of preexisting papillomas.
In view of the prevalence of Sos1 over Sos2 uncovered above for most functional aspects of the carcinogenic events triggered by DMBA/TPA, we also checked whether Sos1 depletion could effectively alter or reduce growth in preexisting, DMBA/TPA-induced tumors by using our carcinogenesis “protocol 2” (see Materials and Methods) (Fig. 8).
FIG 8.
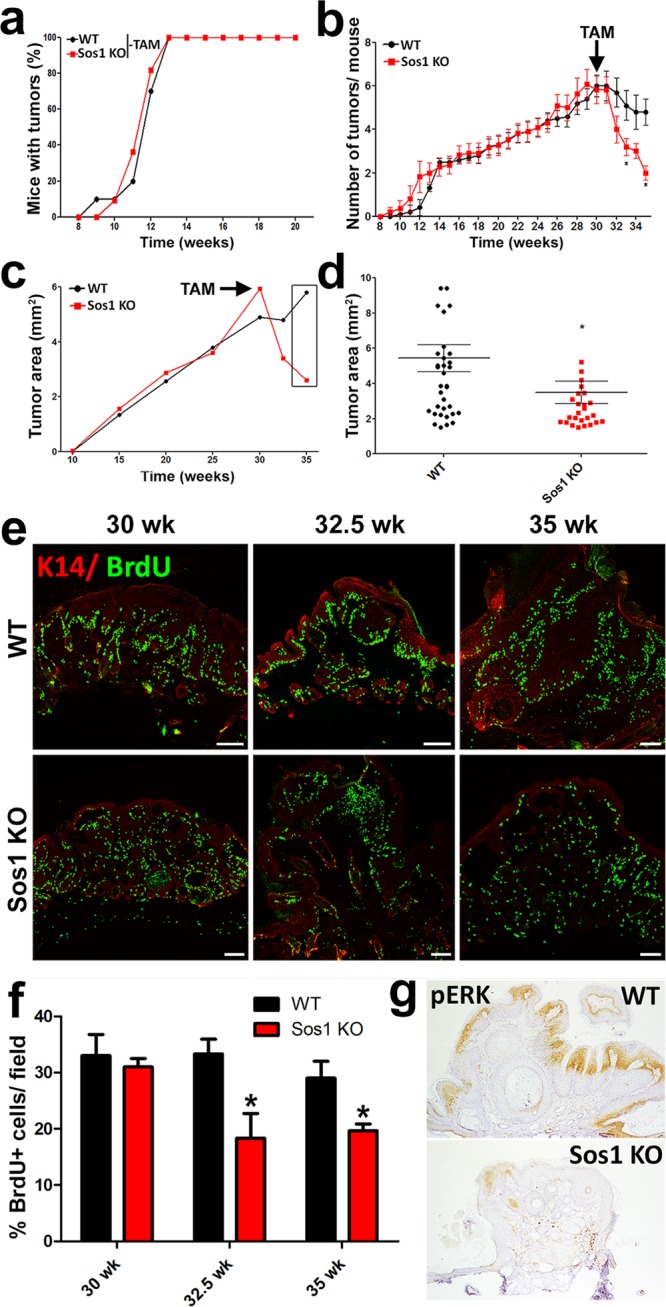
Effect of Sos1 depletion on the number and size of preformed, DMBA/TPA-induced skin tumors. (a) Similar kinetics of tumor initiation and development after DMBA/TPA carcinogenic induction of WT and Sos1-KO mice that were kept untreated (−TAM) with TAM (needed for Sos1 depletion) for the indicated period of carcinogenic treatment. (b) Graphic representation of the number of DMBA/TPA-induced tumors per individual mouse appearing in WT and Sos1-KO mice before and after administration of TAM (arrow) to both experimental groups at the 28.5-week time point. (c and d) Graphic representation of evolution of the average size of tumors arising in the same experimental groups of WT and Sos1-KO mice described for panel b. The arrow marks the time point of TAM administration in both experimental groups. The box in panel c encircles the specific, final time points shown in more detail in panel d. Data for panels a to d are expressed as means ± the SEM. n = 11 for WT and n = 13 for Sos1-KO. *, P < 0.05 versus WT. (e) Representative images of paraffin-embedded tumor sections immunostained for K14 (red) and BrdU (green) that were collected from WT and Sos1-KO mice subjected to the same carcinogenic protocol as in panels b and c. Samples correspond to the beginning (30 weeks), midpoint (32.5 weeks), and end (35 weeks) of the TAM treatment period. Scale bars, 100 μm. (f) Bar chart quantitating the percentage of BrdU-positive, proliferating cells per field in comparable tumors from WT and Sos1-KO mice at the different time points of the same carcinogenic protocol applied in panels b and c. Data are expressed as means ± the SEM. n = 3 per genotype and condition (four microscopy fields per mouse). *, P < 0.05 versus WT. (g) Representative images of paraffin-embedded sections from WT and Sos1-KO tumors that were immunostained for pERK at the endpoint of the experiment (35 weeks).
Interestingly, our results documented a reduction of both the number and size of preexisting tumors in Sos1-KO mice upon TAM application (Fig. 8a to d). In addition, we also noted a slight reduction in the number of tumors after TAM treatment in WT mice, probably reflecting a partial, off-target effect of TAM (Fig. 8b). Consistent with our previous data, the tumor reduction observed in Sos1-KO mice appeared to directly correlate with a progressive decrease in keratinocyte proliferation and ERK phosphorylation levels specifically detected in tumors of Sos1-KO mice upon TAM administration compared to WT counterparts (Fig. 8e to g).
DISCUSSION
We have documented here the functional relevance of Sos1/2 proteins for the maintenance of skin homeostasis under physiological conditions, as well as for the mechanisms involved in wound repair and chemically induced skin carcinogenesis. While Ras-mediated signaling was previously reported to be critical for skin development (19, 20), the specific involvement of the Sos family members (Sos1 and Sos2) of Ras-GEFs during these processes remained poorly understood.
Our data have established a direct mechanistic correlation between Sos1 depletion and the generation of specific alterations of skin homeostasis under physiological conditions, including a profound reduction of skin thickness correlated with decreased keratinocyte proliferation and pERK activation, defects in hair follicle and sebaceous gland integrity and diminution of cellularity and blood vessel density, and strong affectation of the hypodermis. These phenotypes were always much more pronounced in Sos1/2-DKO mice, indicating that Sos2 is able to play a partial, contributing role in the maintenance of skin architecture when Sos1 is absent.
It is unclear at this time whether the observed phenotypic alterations linked to Sos1/2 depletion in the skin originate from specific functional differences between the Sos proteins or because of differences in their expression levels. Whereas WB analysis of Sos protein disappearance in the skin of our KO mice (Fig. 1a) suggested that Sos1 is more highly expressed than Sos2, quantitation of the RNA transcripts for Sos1 and Sos2 in large sets of microarray hybridization data from normal mouse skin (e.g., a data set originated from 196 independent mouse skin samples [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?targ=self&form=html&view=brief&acc=GSE52524]) clearly showed that the level of Sos1 transcripts is lower than that of Sos2 transcripts in the mouse skin, raising the possibility that the different intensities of WB signals observed for Sos1 and Sos2 proteins are merely due to different affinities of the commercial antibodies (sc-256 for Sos1 and sc-15358 for Sos2) used in the immunoblot assays. It is apparent that only future, detailed mass spectrometric quantitation of the Sos1 or Sos2 proteins (focusing on peptides specific for each of these proteins) in the skin may offer a definite answer to this relevant question.
Despite the marked alterations of skin homeostasis caused by Sos1 or Sos1/2 disruption, it is relevant to mention that we detected a small, but statistically significant, increase in apoptosis in the skin of Sos1-KO and Sos1/2-DKO mice. Our observations are consistent with previous reports demonstrating that deletion of the three canonical Ras targets of Sos GEFs blocked keratinocyte or MEF proliferation but did not significantly affect cell survival (19, 26). Consistently, transgenic mice expressing a dominant Sos1 isoform in the skin showed a hyperproliferative response in keratinocytes in vitro and also in vivo, leading to tissue hyperplasia (22). On the other hand, different bioinformatic tools predict that Sos1 is a putative target gene for miR-181a (27), which is also known to reduce proliferation in primary keratinocytes (28). In this regard, upon Ras elimination, keratinocytes cease proliferation and enter into senescence without any signs of apoptosis induction (19). The key role of Sos1 in control of cell proliferation has also been widely documented in other cell lineages, tissues, and organs (5, 17, 18, 29, 30).
The disrupted hair follicle and sebaceous gland organization observed in Sos1/2-DKO mice is also consistent with a previous report showing that deregulation of epidermal growth factor receptor (EGFR)-mediated Ras signaling affects hair follicle integrity (21). Consistent with the notion that Sos1 coordinates the activation of both Ras and Rac (31, 32), it has been shown that Rac1 plays a critical role in sustaining the hair follicle and sebaceous gland integrity although it is not essential for maintenance of the epidermis (33–35). In this regard, our results here have also shown a slight reduction of Rac-GTP levels in the skin of Sos1-depleted mice.
Interestingly, CD31 immunostaining showed only significant reduction of in vivo, steady-state density of blood vessels in the dermis of Sos1/2-DKO mice. On the other hand, aortic ring assays probing neoformation of microvessels showed that single Sos1 or Sos2 depletion resulted in slight decrease (<20%) in number, but not length, of the newly generated sprouts, whereas more than 90% inhibition of both parameters was measured in the Sos1/2-DKO samples. Although the mechanistic details for these processes remain unclear, these data suggest an important functional involvement of Sos1 and Sos2 in the regulation of angiogenesis and the integrity of blood vessels. The data are also consistent with reports supporting a critical involvement of small GTPases in angiogenic processes, endothelial migration, and the regulation of the integrity and functionality of blood vessels (36–38). Our observations suggest that the functional relevance of Sos1/2 proteins for angiogenesis and neovascularization is mechanistically linked to their function activating small GTPases essential for those processes.
The integrity of the hypodermis, composed mainly of adipose tissue, was altered in Sos1-KO and almost completely disappeared in Sos1/2-DKO mice. This is consistent with our previous work describing dramatic disappearance of adipose tissue in mice after simultaneous Sos1/2 disruption (17). Reduction of the hypodermis might also be a secondary, indirect effect since hair follicle cycle alteration is known to affect the thickness of the underlying fat layer via cytokine production (39). Our observations indicate that Sos proteins, particularly Sos1, importantly contribute to adipocytic homeostasis in the skin, but more detailed studies will be needed to precisely understand the specific role of Sos1/2 in this particular cell type.
Cutaneous remodeling after damage involves a number of well-coordinated biological events (40). In contrast to the dominant role of Sos1 over Sos2 in cell proliferation, our data support functional redundancy between both Sos isoforms regarding the events controlling tissue remodeling. During the healing process, different cell types, including keratinocytes and fibroblasts, migrate to the injury site and proliferate. Consistently, our analyses under physiological conditions first documented the functional relevance of Sos1 (predominant) and Sos2 for normal skin cell proliferation, and our skin wound repair studies further demonstrated a reduction of cell proliferation and migration of fibroblasts to the wounded area in Sos1/2-DKO mice. Other reports have also demonstrated a significant blockade of migration in both Ras- and Sos-depleted MEFs after wounding (18, 26).
Wound closure also involves inflammatory steps, including macrophage recruitment to the injury site. No alterations in the distribution of the macrophages were detected in our studies of the skin of Sos-deficient mice under physiological conditions. Consistently, our previous analysis did not show any differences in the number of blood circulating macrophages between Sos-depleted and WT mice (17). However, we observed that Sos1 disruption affected the recruitment of macrophages upon skin injury, suggesting a defect in the proliferation and/or migration of these cells upon inflammatory stimuli. Since Sos proteins are known to participate in macrophage proliferation and migration (41, 42), we suggest that Sos absence affects the capability of macrophages to proliferate, although additional Ras- or Rac-mediated alterations of the migratory capacity of macrophages should not be discarded (42). Later stages of healing involve an angiogenic process (43). As we uncovered a significant contribution of both Sos proteins during angiogenesis in ex vivo aortic ring assays, defects in angiogenesis during skin wound repair in Sos-depleted mice should also be expected.
Overall, we demonstrated here defective skin reepithelialization in Sos1/2-DKO mice that is linked to specific functional alterations during tissue remodeling, including decreased keratinocyte proliferation and migration of different cell types, and deficient inflammatory and angiogenic processes. Consistently, previous studies documented the importance of the EGFR-Ras-Raf pathway during the healing process, controlling cell proliferation, migration, and adhesion (44–46). Nevertheless, we should also note that EGFR activation is important but not necessary to complete healing (47) and that Rac, which can also be activated by Sos1, appears to be required during tissue remodeling (35).
Sos1/2 ablation after DMBA and/or TPA treatments allowed us to evaluate the relevance/contribution of Sos proteins to chemically induced skin carcinogenesis and their possible usefulness as potential therapy targets in cutaneous tumors. We showed that Sos1 depletion, but not Sos2 absence, impaired the initiation, promotion, and progression of DMBA/TPA-induced tumors. Our analyses also suggested that Sos1 depletion triggers those antitumorogenic effects by acting preferentially through a reduction of keratinocyte proliferation and ERK phosphorylation in the tumors, rather than through the modest increases of cell death rates observed in the tumor samples. However, we do not discard the potential impact of apoptosis in the decrease in tumor progression in Sos1/2-DKO mice. Consistently, previous reports indicate that components of Ras signaling pathways play pivotal roles in epidermal tumorigenesis (20, 21, 48, 49) and, in particular, that the expression of a dominant Sos1 isoform in basal keratinocytes leads to development of skin papillomas (22). We also documented a complete absence of malignant SCC in Sos-depleted mice. In this regard, it was reported that ERK activation occurs at the transition from papilloma to SCC (50). In addition, Sos upregulation leads to cisplatin resistance in SCC cells through mitogen-activated protein kinase (MAPK)/ERK activation (51), and belinostat blocks Sos-mediated MAPK activation, exerting an antitumor effect in SCC (52). It has also been reported that the absence of Rac activators such as Tiam1 or Vav2/3 has a potential therapeutic effect in DMBA/TPA-induced skin tumors (53, 54). Based on our detection of slightly reduced levels of Rac-GTP in the absence of Sos1, it is conceivable that our observed Sos depletion-related reduction of skin tumorigenesis may be mediated through the downregulation of both Ras- and/or Rac-mediated signaling.
Strikingly, the neutrophil infiltration triggered by acute short-term TPA-treatment did not change in Sos1-KO mice compared to WT mice, but single Sos2 depletion clearly incremented neutrophil infiltration relative to WT. However, concomitant Sos1/2 deletion almost completely abrogated the recruitment of these cells. Since neutrophils are not proliferative, migration or adhesion to tumor matrix components may play a role in controlling the presence of neutrophils in skin tumors of the different Sos genotypes. Interestingly, we reported previously that Sos2-KO MEFs exhibit enhanced cell adhesion ability in comparison to WT controls (18) whereas other functional parameters remained unchanged in the same Sos2-KO MEFs fibroblasts; however, Sos1/2-DKO MEFs significantly lost their ability to adhere to culture surfaces (15–18). We suggest that possible alterations in the capacity of adhesion of neutrophils from the different groups of Sos-KO mice analyzed here may play a significant role in the response of these cells after injury stimuli. Further studies will be needed to ascertain the level of functional specificity/redundancy displayed by Sos1 and/or Sos2 in all of these biological processes.
Our observations clearly showed that Sos1 deletion impairs skin tumor initiation and promotion, and it decreases tumor growth in preexisting papillomas by reducing proliferation in the tumor. It is generally accepted that DMBA/TPA-induced tumors are mostly driven by oncogenic mutations in the H-Ras locus (downstream of Sos proteins), and therefore it could be argued that tumorigenesis should be, in theory, unaffected by the disruption of Ras upstream activators. However, recent reports have shown that normal Ras gene products can also contribute to generation of the transformed phenotype. Indeed, it has been shown that oncogenic Ras promotes the activation of wild-type Ras through a mechanism mediated by allosteric stimulation of Sos that confers a growth advantage to cancer cells harboring oncogenic Ras (55). A second possibility consistent with our observations is that EGFR-mediated signals, upstream of Sos, act as a survival factor in oncogenic transformation, and therefore Sos depletion could lead to attenuated EGFR-mediated signaling under our experimental oncogenic conditions.
Overall, our data unveiled previously unknown functional roles of the Sos proteins during various phases of skin tumor development, thus making it possible to postulate the Sos1/2 isoforms as potential markers or therapeutic targets for prevention and/or treatment of epidermal cancers.
MATERIALS AND METHODS
Generation of tamoxifen-inducible, Sos1-null mice.
We generated the mice of the four genotypes of interest (WT, Sos1-KO, Sos2-KO, and Sos1/2-DKO) as previously described (17). The genotypes of animals and Sos1 deletion in the skin after TAM feeding were monitored by PCR (17). Mice were kept, handled, and sacrificed in the NUCLEUS animal facility of the University of Salamanca in accordance with current European and Spanish legislations. All experiments were approved by the Bioethics Committee of our research center.
Western blot and pulldown assays.
Mice of the four relevant Sos genotypes (n = 3/genotype) were TAM treated for 12 days, and dorsal skin tissue samples were collected and quickly frozen before tissue homogenization in radioimmunoprecipitation assay buffer using a GentleMacs dissociator. A 90-μg portion of protein was loaded in electrophoresis gels, and immunoblotting was performed, as previously described (18). Rabbit anti-Sos1 (sc-256; Santa Cruz Biotechnology), rabbit anti-Sos2 (sc-15358; Santa Cruz Biotechnology), and mouse antitubulin (T5293; Sigma) antibodies were used.
For pulldown assays, dorsal skin of 12 days-TAM-treated mice was homogenized in MLB buffer using a GentleMacs dissociator. Rac-GTP levels were measured as previously described (18) using a mouse anti-Rac1 antibody (610651; BD).
Role of Sos1/2 isoforms under physiological conditions.
To examine the functional role of Sos proteins in skin homeostasis, 6- to 8-week-old mice of the four genotypes received a TAM-containing diet for 12 days. The dorsal skin was shaved 48 h prior to the end of TAM treatment. At the end of the experiment, mice were intraperitoneally injected with BrdU (100 μg/g [body weight]; Sigma) 1.5 h before sacrifice. After euthanasia, the skin was collected, fixed in 4% PFA overnight at 4°C, and paraffin embedded.
Wound healing assays.
Two round excisional wounds (4 mm in diameter) were performed in previously shaved skin of anesthetized, TAM-treated (12 days), 6- to 8-week-old, WT, Sos1-KO, and Sos2-KO mice. Because of lethality among Sos1/2-DKO mice (17), animal groups of this genotype were TAM treated for only 4 days prior to starting the wound healing procedure, and TAM feeding was maintained for 8 additional days. The wound area was measured daily for 10 days after injury. After the last measurement, the skin was isolated, fixed, and paraffin embedded. For more detailed analyses, A set of animals of the four genotypes were killed 3 days after the injury, and the skin was collected and handled as indicated above.
TPA-induced hyperproliferation and inflammation and DMBA-induced apoptosis.
For short-term studies of hyperproliferation and apoptosis, 6- to 8-week-old, TAM-treated (12 days) mice from the four experimental groups were used. TPA (6.8 nmol in 200 μl of acetone; Sigma) was applied to shaved skin for either one or four consecutive doses (one dose/day). A single dose of DMBA (6.4 μg/200 μl of acetone; Sigma, D3254) was applied to the dorsal skin to analyze DMBA-induced apoptosis. At 24 h after the last treatment, mice were intraperitoneally injected with BrdU. After euthanasia, the skin was isolated, fixed, and paraffin embedded.
DMBA/TPA-induced skin carcinogenesis.
Two different procedures were used to induce DMBA/TPA carcinogenesis. The role of Sos1/2 proteins in tumor initiation and progression was analyzed using “protocol 1.” WT (n = 13), Sos1-KO (n = 11), and Sos2-KO (n = 15) mice (6 to 8 weeks old, treated for 12 days with TAM) were treated with DMBA/TPA according to the standard protocol (24). A single DMBA dose (25 μg/200 μl of acetone) was topically applied. After 3 days, TPA (10−4 M in acetone; Sigma) was applied twice a week for 30 weeks. Sos1/2-DKO mice (n = 13) were only treated with TAM after 28.5 weeks past the start of the DMBA/TPA protocol.
The role of Sos proteins in established tumors was investigated by using “protocol 2.” Non-TAM-treated, 6- to 8-week-old WT (n = 11) and Sos1-KO (n = 13) mice were subjected to the DMBA/TPA protocol as described above for 28.5 weeks. A TAM-containing chow diet was then administered for the following 2.5 to 5 weeks. In both protocols, the number, size, and incidence of papillomas were measured weekly. At the conclusion of the experiments, mice were intraperitoneally injected with BrdU, and the tumors were collected, fixed, and paraffin embedded.
Histology and immunostaining.
Paraffin-embedded skin samples were cut and stained with hematoxylin and eosin (H&E) according to standard procedures. Pathological classification of tumor samples ranged from benign lesions (papilloma, sebaceous adenoma, and keratoacanthoma) to SCC (malignant stage).
For immunohistochemistry, sections were dewaxed, microwaved in citrate buffer (pH 6), and incubated overnight with anti-pERK1/2 (Cell Signaling) or anti-F4/80 (Life Technologies) at 4°C. Sections were then incubated with biotin-conjugated secondary antibodies, followed by Vectastain Elite ABC reagent. The reaction product was visualized by incubating the sections in 0.025% 3,3′-diaminobenzidine and 0.003% H2O2 in phosphate-buffered saline.
For immunofluorescence, sections were treated as described above. After incubation of the primary antibodies, the fluorochrome-conjugated secondary antibodies were applied for 1 h at room temperature. The following primary antibodies were used: anti-BrdU (Accurate Chemical), anti-CD31 (BD), anti-cleaved-caspase-3 (Cell Signaling), anti-keratin 14 (Covance), anti-neutrophil elastase (Abcam), and antivimentin (Sigma). Late apoptosis was analyzed using an in situ cell death fluorescein kit (Sigma). The percentages of BrdU-, cleaved-caspase-3-, and TUNEL-positive cells were quantified in the same area (160,000 μm2) corresponding to four different microscopy fields per animal by using ImageJ software.
Aortic ring assay.
Aortic ring assay was performed as described previously (56) in 8-week-old, TAM-treated (12 days) mice of the four genotypes. The aortic rings were maintained at 37°C and 5% CO2 for 8 days. Vascular sprouts were scored at 1, 3, 5, and 8 days. The numbers of sprouts per ring and the lengths of the sprouts were measured by using ImageJ software.
Statistical analysis.
Statistical analyses were performed with SPSS software (SPSS, Inc., Chicago, IL) using one-way analysis of variance and the Bonferroni and Student t tests.
ACKNOWLEDGMENTS
This study was supported by grants FIS PI16/02137 from ISCIII (MINECO), SA043U16 (UIC 076) from JCyL, and AECC Spain (to E.S.); by MINECO grant SAF2015-66015-R; and by MSyC grants ISCIII-RETIC RD12/0036/0009, PIE 15/00076, and CB/16/00228 (to J.M.P.). This research was cofinanced by FEDER funds.
P.L.-B. performed experimental work. D.J. and C.G. did the statistical analysis. M.M.-M., L.F.L.-M., and X.R.B. helped in designing DMBA/TPA experiments. N.C. managed the mouse colony. R.G.-N. and R.F.-M. performed Rac pulldown assays. D.J., J.M.P., and C.S. performed various immunolabeling studies. F.C.B. and E.S. designed and supervised the study and wrote the article. All authors reviewed the manuscript.
REFERENCES
- 1.Buday L, Downward J. 2008. Many faces of Ras activation. Biochim Biophys Acta 1786:178–187. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Hennig A, Markwart R, Esparza-Franco MA, Ladds G, Rubio I. 2015. Ras activation revisited: role of GEF and GAP systems. Biol Chem 396:831–848. doi: 10.1515/hsz-2014-0257. [DOI] [PubMed] [Google Scholar]
- 3.Castellano E, Santos E. 2011. Functional specificity of Ras isoforms: so similar but so different. Genes Cancer 2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherfils J, Zeghouf M. 2013. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 5.Pierre S, Bats A-S, Chevallier A, Bui L-C, Ambolet-Camoit A, Garlatti M, Aggerbeck M, Barouki R, Coumoul X. 2011. Induction of the Ras activator Son of Sevenless 1 by environmental pollutants mediates their effects on cellular proliferation. Biochem Pharmacol 81:304–313. doi: 10.1016/j.bcp.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Rojas JM, Oliva JL, Santos E. 2011. Mammalian son of sevenless guanine nucleotide exchange factors: old concepts and new perspectives. Genes Cancer 2:298–305. doi: 10.1177/1947601911408078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Medarde A, Santos E. 2011. Ras in cancer and developmental diseases. Genes Cancer 2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, Imielinski M, Hu X, Ling S, Akbani R, Rosenberg M, Cibulskis C, Ramachandran A, Collisson EA, Kwiatkowski DJ, Lawrence MS, Weinstein JN, Verhaak RGW, Wu CJ, Hammerman PS, Cherniack AD, Getz G, Artyomov MN, Schreiber R, Govindan R, Meyerson M, Meyerson M. 2016. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernández-Medarde A, Santos E. 2011. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim Biophys Acta Rev Cancer 1815:170–188. doi: 10.1016/j.bbcan.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JPL, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. 2007. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 11.Easton AC, Rotter A, Lourdusamy A, Desrivières S, Fernández-Medarde A, Biermann T, Fernandes C, Santos E, Kornhuber J, Schumann G, Müller CP. 2014. Rasgrf2 controls dopaminergic adaptations to alcohol in mice. Brain Res Bull 109:143–150. doi: 10.1016/j.brainresbull.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Jimeno D, Gómez C, Calzada N, de la Villa P, Lillo C, Santos E. 2016. RASGRF2 controls nuclear migration in postnatal retinal cone photoreceptors. J Cell Sci 129:729–742. doi: 10.1242/jcs.180919. [DOI] [PubMed] [Google Scholar]
- 13.Cordeddu V, Yin JC, Gunnarsson C, Virtanen C, Drunat S, Lepri F, De Luca A, Rossi C, Ciolfi A, Pugh TJ, Bruselles A, Priest JR, Pennacchio LA, Lu Z, Danesh A, Quevedo R, Hamid A, Martinelli S, Pantaleoni F, Gnazzo M, Daniele P, Lissewski C, Bocchinfuso G, Stella L, Odent S, Philip N, Faivre L, Vlckova M, Seemanova E, Digilio C, Zenker M, Zampino G, Verloes A, Dallapiccola B, Roberts AE, Cavé H, Gelb BD, Neel BG, Tartaglia M. 2015. Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan syndrome. Hum Mutat 36:1080–1087. doi: 10.1002/humu.22834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian X, Esteban L, Vass WC, Upadhyaya C, Papageorge AG, Yienger K, Ward JM, Lowy DR, Santos E. 2000. The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J 19:642–654. doi: 10.1093/emboj/19.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esteban LM, Fernández-Medarde A, López E, Yienger K, Guerrero C, Ward JM, Tessarollo L, Santos E. 2000. Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol Cell Biol 20:6410–6413. doi: 10.1128/MCB.20.17.6410-6413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kortum RL, Sommers CL, Alexander CP, Pinski JM, Li W, Grinberg A, Lee J, Love PE, Samelson LE. 2011. Targeted Sos1 deletion reveals its critical role in early T-cell development. Proc Natl Acad Sci U S A 108:12407–12412. doi: 10.1073/pnas.1104295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baltanás FC, Pérez-Andrés M, Ginel-Picardo A, Diaz D, Jimeno D, Liceras-Boillos P, Kortum RL, Samelson LE, Orfao A, Santos E. 2013. Functional redundancy of Sos1 and Sos2 for lymphopoiesis and organismal homeostasis and survival. Mol Cell Biol 33:4562–4578. doi: 10.1128/MCB.01026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liceras-Boillos P, García-Navas R, Ginel-Picardo A, Anta B, Pérez-Andrés M, Lillo C, Gómez C, Jimeno D, Fernández-Medarde A, Baltanás FC, Santos E. 2016. Sos1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene 35:6389–6402. doi: 10.1038/onc.2016.169. [DOI] [PubMed] [Google Scholar]
- 19.Drosten M, Lechuga CG, Barbacid M. 2014. Ras signaling is essential for skin development. Oncogene 33:2857–2865. doi: 10.1038/onc.2013.254. [DOI] [PubMed] [Google Scholar]
- 20.Kern F, Niault T, Baccarini M. 2011. Ras and Raf pathways in epidermis development and carcinogenesis. Br J Cancer 104:229–234. doi: 10.1038/sj.bjc.6606009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doma E, Rupp C, Baccarini M. 2013. EGFR-Ras-Raf signaling in epidermal stem cells: roles in hair follicle development, regeneration, tissue remodeling and epidermal cancers. Int J Mol Sci 14:19361–19384. doi: 10.3390/ijms141019361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, Schlessinger J, Wagner EF. 2000. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell 102:211–220. doi: 10.1016/S0092-8674(00)00026-X. [DOI] [PubMed] [Google Scholar]
- 23.Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. 2010. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 140:268–279. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Losada J, Balmain A. 2003. Stem-cell hierarchy in skin cancer. Nat Rev Cancer 3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 25.Laurens N, Koolwijk P, de Maat MP. 2006. Fibrin structure and wound healing. J Thromb Haemost 4:932–939. [DOI] [PubMed] [Google Scholar]
- 26.Drosten M, Dhawahir A, Sum EYM, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. 2010. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29:1091–1104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakraborty C, Sharma AR, Patra BC, Bhattacharya M, Sharma G, Lee S-S. 2016. MicroRNAs mediated regulation of MAPK signaling pathways in chronic myeloid leukemia. Oncotarget 7:42683–42697. doi: 10.18632/oncotarget.7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neu J, Dziunycz PJ, Dzung A, Lefort K, Falke M, Denzler R, Freiberger SN, Iotzova-Weiss G, Kuzmanov A, Levesque MP, Dotto G-P, Hofbauer GFL. 2017. miR-181a decelerates proliferation in cutaneous squamous cell carcinoma by targeting the proto-oncogene KRAS. PLoS One 12:e0185028. doi: 10.1371/journal.pone.0185028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Depeille P, Henricks LM, van de Ven RA, Lemmens E, Wang CY, Matli M, Werb Z, Haigis KM, Donner D, Warren R, Roose JP. 2015. RasGRP1 opposes proliferative EGFR-SOS1-Ras signals and restricts intestinal epithelial cell growth. Nat Cell Biol 17:804–815. doi: 10.1038/ncb3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen P-C, Wakimoto H, Conner D, Araki T, Yuan T, Roberts A, Seidman CE, Bronson R, Neel BG, Seidman JG, Kucherlapati R. 2010. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest 120:4353–4365. doi: 10.1172/JCI43910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scita G, Tenca P, Areces LB, Tocchetti A, Frittoli E, Giardina G, Ponzanelli I, Sini P, Innocenti M, Di Fiore PP. 2001. An effector region in Eps8 is responsible for the activation of the Rac-specific GEF activity of Sos-1 and for the proper localization of the Rac-based actin-polymerizing machine. J Cell Biol 154:1031–1044. doi: 10.1083/jcb.200103146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Innocenti M, Tenca P, Frittoli E, Faretta M, Tocchetti A, Di Fiore PP, Scita G. 2002. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol 156:125–136. doi: 10.1083/jcb.200108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benitah SA, Frye M, Glogauer M, Watt FM. 2005. Stem cell depletion through epidermal deletion of Rac1. Science 309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- 34.Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C. 2006. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol 26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castilho RM, Squarize CH, Leelahavanichkul K, Zheng Y, Bugge T, Gutkind JS. 2010. Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PLoS One 5:e10503. doi: 10.1371/journal.pone.0010503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chrzanowska-Wodnicka M, Kraus AE, Gale D, White GC, Vansluys J. 2008. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 111:2647–2656. doi: 10.1182/blood-2007-08-109710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, Li DY, Ruoslahti E, Komatsu M. 2012. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 22:235–249. doi: 10.1016/j.ccr.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J. 2014. RAS interaction with PI3K p110α is required for tumor-induced angiogenesis. J Clin Invest 124:3601–3611. doi: 10.1172/JCI74134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruglikov IL, Scherer PE. 2016. Dermal adipocytes and hair cycling: is spatial heterogeneity a characteristic feature of the dermal adipose tissue depot? Exp Dermatol 25:258–262. doi: 10.1111/exd.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinke JM, Sorg H. 2012. Wound repair regeneration. Eur Surg Res 49:35–43. doi: 10.1159/000339613. [DOI] [PubMed] [Google Scholar]
- 41.Lanfrancone L, Pelicci G, Brizzi MF, Aronica MG, Casciari C, Giuli S, Pegoraro L, Pawson T, Pelicci PG, Arouica MG. 1995. Overexpression of Shc proteins potentiates the proliferative response to the granulocyte-macrophage colony-stimulating factor and recruitment of Grb2/SoS and Grb2/p140 complexes to the beta receptor subunit. Oncogene 10:907–917. [PubMed] [Google Scholar]
- 42.Baruzzi A, Remelli S, Lorenzetto E, Sega M, Chignola R, Berton G. 2015. Sos1 regulates macrophage podosome assembly and macrophage invasive capacity. J Immunol 195:4900–4912. doi: 10.4049/jimmunol.1500579. [DOI] [PubMed] [Google Scholar]
- 43.Park JE, Barbul A. 2004. Understanding the role of immune regulation in wound healing. Am J Surg 187(5A):11S–16S. [DOI] [PubMed] [Google Scholar]
- 44.Marikovsky M, Breuing K, Liu PY, Eriksson E, Higashiyama S, Farber P, Abraham J, Klagsbrun M. 1993. Appearance of heparin-binding EGF-like growth factor in wound fluid as a response to injury. Proc Natl Acad Sci U S A 90:3889–3893. doi: 10.1073/pnas.90.9.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shirakata Y, Kimura R, Nanba D, Iwamoto R, Tokumaru S, Morimoto C, Yokota K, Nakamura M, Sayama K, Mekada E, Higashiyama S, Hashimoto K. 2005. Heparin-binding EGF-like growth factor accelerates keratinocyte migration and skin wound healing. J Cell Sci 118:2363–2370. doi: 10.1242/jcs.02346. [DOI] [PubMed] [Google Scholar]
- 46.Yoshioka R, Shiraishi A, Kobayashi T, Morita S-I, Hayashi Y, Higashiyama S, Ohashi Y. 2010. Corneal epithelial wound healing impaired in keratinocyte-specific HB-EGF-deficient mice in vivo and in vitro. Invest Ophthalmol Vis Sci 51:5630–5639. doi: 10.1167/iovs.10-5158. [DOI] [PubMed] [Google Scholar]
- 47.Repertinger SK, Campagnaro E, Fuhrman J, El-Abaseri T, Yuspa SH, Hansen LA. 2004. EGFR enhances early healing after cutaneous incisional wounding. J Investig Dermatol 123:982–989. doi: 10.1111/j.0022-202X.2004.23478.x. [DOI] [PubMed] [Google Scholar]
- 48.May U, Prince S, Vähätupa M, Laitinen AM, Nieminen K, Uusitalo-Järvinen H, Järvinen TAH. 2015. Resistance of R-Ras knockout mice to skin tumour induction. Sci Rep 5:11663. doi: 10.1038/srep11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Menacho-Márquez M, García-Escudero R, Ojeda V, Abad A, Delgado P, Costa C, Ruiz S, Alarcón B, Paramio JM, Bustelo XR. 2013. The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol 11:e1001615. doi: 10.1371/journal.pbio.1001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Segrelles C, Ruiz S, Perez P, Murga C, Santos M, Budunova IV, Martínez J, Larcher F, Slaga TJ, Gutkind JS, Jorcano JL, Paramio JM. 2002. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene 21:53–64. doi: 10.1038/sj.onc.1205032. [DOI] [PubMed] [Google Scholar]
- 51.Kong LR, Chua KN, Sim WJ, Ng HC, Bi C, Ho J, Nga ME, Pang YH, Ong WR, Soo RA, Huynh H, Chng WJ, Thiery J-P, Goh BC. 2015. MEK Inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol Cancer Ther 14:1750–1760. doi: 10.1158/1535-7163.MCT-15-0062. [DOI] [PubMed] [Google Scholar]
- 52.Kong LR, Tan TZ, Ong WR, Bi C, Huynh H, Lee SC, Chng WJ, Eichhorn PJA, Goh BC. 2017. Belinostat exerts antitumor cytotoxicity through the ubiquitin-proteasome pathway in lung squamous cell carcinoma. Mol Oncol 11:965–980. doi: 10.1002/1878-0261.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. 2002. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 417:867–871. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- 54.Menacho-Márquez M, García-Escudero R, Ojeda V, Abad A, Delgado P, Costa C, Ruiz S, Alarcón B, Paramio JM, Bustelo XR. 2013. The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol 11:e1001615. doi: 10.1371/journal.pbio.1001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeng H-H, Taylor LJ, Bar-Sagi D. 2012. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun 3:1168. doi: 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D'Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K. 2011. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc 7:89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]