

Abstract
Exposure to low-dose lipopolysaccharide (LPS) before cerebral ischemia is neuroprotective in stroke models, a phenomenon termed preconditioning (PC). Although it is well established that LPS-PC induces central and peripheral immune responses, the cellular mechanisms modulating ischemic injury remain unclear. Here, we investigated the role of immune cells in the brain protection afforded by PC and tested whether monocytes may be reprogrammed by ex vivo LPS exposure, thus modulating inflammatory injury after cerebral ischemia in male mice. We found that systemic injection of low-dose LPS induces a Ly6Chi monocyte response that protects the brain after transient middle cerebral artery occlusion (MCAO) in mice. Remarkably, adoptive transfer of monocytes isolated from preconditioned mice into naive mice 7 h after transient MCAO reduced brain injury. Gene expression and functional studies showed that IL-10, inducible nitric oxide synthase, and CCR2 in monocytes are essential for neuroprotection. This protective activity was elicited even if mouse or human monocytes were exposed ex vivo to LPS and then injected into male mice after stroke. Cell-tracking studies showed that protective monocytes are mobilized from the spleen and reach the brain and meninges, where they suppress postischemic inflammation and neutrophil influx into the brain parenchyma. Our findings unveil a previously unrecognized subpopulation of splenic monocytes capable of protecting the brain with an extended therapeutic window and provide the rationale for cell therapies based on the delivery of autologous or allogeneic protective monocytes in patients after ischemic stroke.
SIGNIFICANCE STATEMENT Inflammation is a key component of the pathophysiology of the brain in stroke, a leading cause of death and disability with limited therapeutic options. Here, we investigate endogenous mechanisms of protection against cerebral ischemia. Using lipopolysaccharide (LPS) preconditioning (PC) as an approach to induce ischemic tolerance in mice, we found generation of neuroprotective monocytes within the spleen, from which they traffic to the brain and meninges, suppressing postischemic inflammation. Importantly, systemic LPS-PC can be mimicked by adoptive transfer of in vitro-preconditioned mouse or human monocytes at translational relevant time points after stroke. This model of neuroprotection may facilitate clinical efforts to increase the efficacy of BM mononuclear cell treatments in acute neurological diseases such as cerebral ischemia.
Keywords: adoptive transfer, cerebral ischemia, lipopolysaccharide, monocytes, neuroprotection
Introduction
Induction of ischemic tolerance by the exposure to subinjurious stressors, also known as preconditioning (PC), is a powerful mechanism to evoke endogenous neuroprotective programs (Iadecola and Anrather, 2011). In the brain, PC can be achieved by a variety of agents and stressors, including ischemia, inflammatory mediators, metabolic blockers, anesthetics, cortical spreading depression, and seizures (Kirino, 2002). The immune system plays a key role in the establishment of ischemic tolerance. For example, toll-like receptors (TLRs), key innate immunity receptors, are potent mediators of cerebral PC (Marsh et al., 2009b; Pradillo et al., 2009; Wang et al., 2010; Garcia-Bonilla et al., 2014a) and activation of TLR4 by systemic administration of lipopolysaccharide (LPS) is a potent PC stimulus (Tasaki et al., 1997; Ahmed et al., 2000; Vartanian et al., 2011). However, circulating LPS is impermeable to the blood–brain barrier and does not gain access to the brain (Singh and Jiang, 2004; Banks and Robinson, 2010). Therefore, LPS is thought to induce ischemic tolerance by a systemic effect involving reprogramming of the immune system (Smith et al., 2002), which in turn protects the brain by suppressing postischemic proinflammatory gene expression, endothelial and microglial activation, as well as leukocyte infiltration (Bauer et al., 2000; Rosenzweig et al., 2004; Lin et al., 2009; Marsh et al., 2009a; Vartanian et al., 2011). However, the cell type(s) on which systemic LPS acts to induce the widespread changes in the immune system underlying the development of cerebral PC remains to be defined.
Monocytes are BM-derived cells characterized by surface expression of CD11b and CD115, which are present in large quantities in the spleen (Swirski et al., 2009). In the circulation, they consist of functionally diverse subsets (Geissmann et al., 2003): Ly6C+CCR2+CX3CR1low “inflammatory” monocytes, which contribute to the inflammatory process (Swirski et al., 2007; Tacke et al., 2007; Swirski et al., 2010), and Ly6C−CCR2−CX3CR1high “resident” monocytes, which patrol the vasculature and participate in the resolution of inflammation (Auffray et al., 2007; Nahrendorf et al., 2007). A third subset, monocytic myeloid-derived suppressor cells (Serafini et al., 2004; Huang et al., 2006; Bronte, 2009), is present in the BM and shares the same surface markers with inflammatory monocytes (Youn et al., 2008; Ostrand-Rosenberg, 2010; Forghani et al., 2012).
All monocytes subtypes have been implicated in ischemic injury in different organs, in which they can play either protective or destructive roles (Arnold et al., 2007; Nahrendorf et al., 2007, 2010), but their involvement in ischemic PC remains unclear. Considering that monocytes express TLR4 and are a prime target of LPS (Muzio et al., 2000; Biswas and Lopez-Collazo, 2009), it is conceivable that they participate in LPS-PC. Indeed, LPS administration induces BM egress of selected monocyte subtypes such as Ly6C+ monocytes, which have the potential of modulating systemic immune responses (Serbina et al., 2008; Shi et al., 2011). Here, we investigated the role of peripheral immune cells in the neuroprotection observed after LPS-PC in mice. We found that LPS treatment induces a Ly6Chi monocyte population of splenic monocytes endowed with a powerful neuroprotective capacity against experimental ischemic brain injury, which is mediated through suppression of meningeal inflammation. The findings highlight a previously unrecognized neuroprotective potential of a distinct monocyte subset and raise the prospect of novel cell-based stroke therapies based on monocyte-induced immunomodulation.
Materials and Methods
Mice
All procedures were approved by the institutional animal care and use committee of Weill Cornell Medicine and were performed in accordance with ARRIVE guidelines (Kilkenny et al., 2011). Experiments were performed in 7- to 8-week-old WT (JAX 000664), inducible nitric oxide synthase KO (iNOS−/−; MacMicking et al., 1995), IL-10−/− (JAX 002251, B6.129P2-Il10tm1Cgn/J), CCR2RFP/RFP (JAX 017586, B6.129(Cg)-Ccr2tm2.1Ifc/J), Cox2−/− (Morham et al., 1995), or GFP-transgenic (JAX 006567, C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ) male mice on a C57BL/6J background obtained from The Jackson Laboratory (JAX) and from in-house colonies (iNOS−/− and Cox2−/− mice).
Middle cerebral artery occlusion (MCAO)
Transient focal cerebral ischemia was induced using the intraluminal filament model of MCAO as described previously (Jackman et al., 2011). Under isoflurane anesthesia (maintenance 1.5–2%), the MCA was occluded for 35 min. Reperfusion was confirmed by measuring the cerebral blood flow (CBF) in the MCA territory by transcranial laser Doppler flowmetry (Periflux System 5010; Perimed). Only animals with CBF reduction of >85% during MCAO and CBF recovered by >80% after 10 min of reperfusion were included in the study (Jackman et al., 2011). Rectal temperature was monitored and kept constant (37.0 ± 0.5°C) during the surgical procedure and in the recovery period until the animals regained full consciousness. Topical lidocaine and bupivacaine (0.25%, 0.1 ml, transdermal) were used for preoperative analgesia and buprenorphine (0.5 mg/kg every 24 h, s.c.) as postoperative analgesia for 48 h.
Measurement of infarct volume and neurological deficit
As described in detail previously (Jackman et al., 2011), blinded assessment of infarct volume, corrected for swelling, was quantified 3 d after ischemia using Nissl stain on 12 30-μm-thick coronal brain sections using image analysis software (MCID; Imaging Research).
Sensorimotor deficit was assessed 1 d before and 3 d after MCAO using the tape test performed by an investigator blinded to the treatment groups (Benakis et al., 2016). Mice were tested at the start of the dark phase of the circadian day/night cycle. Briefly, an adhesive tape was placed on the dorsal aspect of both forepaws. Latency of tape contact with the mouth and tape removal was scored on videos recorded over 180 s. In the test performed 1 d before MCAO, mice received 3 trials with 10 min rest cycles in between and, on day 3 after MCAO, mice received 2 trials. Results were expressed as time average of tape contact or tape removal latency in each test for the contralesional and lesional side.
LPS in vivo PC
LPS was repurified from a commercial LPS preparation (Salmonella enterica serotype typhimurium; Sigma-Aldrich; L7261) according to a previously published protocol (Manthey and Vogel, 1994). Mice were injected with either purified LPS (0.5 mg/kg, i.p.) or saline (100 μl, i.p.) 24 h before MCAO.
Splenectomy
Total splenectomy was performed under aseptic conditions and isoflurane anesthesia. Meloxicam (2 mg/kg, s.c.) and buprenorphine (0.5 mg/kg, s.c.) were given for preemptive analgesia. A 1 cm parasagittal incision was performed through the skin and abdominal musculature and the spleen was exteriorized. Vascular pedicles were ligated using silk suture (6/0) and then the splenic artery and vein were transected between the ligation and the spleen, removing the latter. For sham operations, a laparotomy incision was made, but no splenic tissue was resected. Buprenorphine (0.5 mg/kg q24 h, s.c.) was given for 48 h postoperatively. Mice were allowed to recover for 14 d before undergoing MCAO.
Cell isolation
Mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and transcardially perfused with heparinized PBS. The spleen was dissected and the head was cut. Next, the skull was cleaned from skin and muscles. The upper portion of the skull was separated from the brain and the meninges (dura/arachnoid) were recovered from the interior of the skull under a dissection microscope. Meninges and spleen were placed on a premoistened 70 μm cell strainer and gently homogenized in PBS. Brain cell isolation was performed by mechanical method or enzymatic digestion with Liberase DH (Roche Diagnostics) when cell sorting was performed, as described previously (Garcia-Bonilla et al., 2014b; Benakis et al., 2016). Briefly, for enzymatic brain cell isolation, brain hemispheres were separated from the cerebellum and olfactory bulb and gently triturated in HEPES-HBSS buffer containing the following (in mm): 138 NaCl, 5 KCl, 0.4 Na2HPO4, 0.4 KH2PO4, 5 d-glucose, and 10 HEPES using a Gentle MACS dissociator (Miltenyi Biotec) following the manufacturer's instructions. The suspension was digested with 62.5 μg/ml Liberase and 50 U/ml DNase I at 37°C for 45 min in an orbital shaker at 100 rpm. For mechanical cell isolation, brain hemispheres were homogenized in RPMI 1640 medium (Mediatech) using a Dounce homogenizer. Brain cells isolated from enzymatic or mechanical procedures were washed and subjected to discontinuous 70/30% Percoll (GE Healthcare) density gradient centrifugation. Enriched-mononuclear cells were collected from the interphase. BM cells were flushed out from the femurs and tibias and filtered through a 40 μm cell strainer. Blood was drained from the submandibular vein into tubes containing sodium heparin and erythrocytes were lysed. Cell suspensions were subsequently stained for flow cytometric analysis.
Flow cytometric analysis
Cells were stained using rat monoclonal antibodies (Table 1) and analyzed on a MACSQuant Analyzer 10 (Miltenyi Biotec). Brain-infiltrating leukocytes were identified as CD45hi and further gated by CD11b and CD11c markers. CD45hiCD11b−CD11c+ were identified as dendritic cells (DCs). From the CD45hiCD11b+CD11c− gate, both monocytes/macrophages (MMøs) and neutrophils were identified as CD45hiCD11b+CD115+ and CD45hiCD11b+Ly6G+, respectively. MMøs were further separated as Ly6Chi or Ly6Clo. The same gating strategy was used to phenotype leukocyte in the meninges and peripheral organs. Appropriate isotype controls, “fluorescence minus one” staining, and staining of negative populations were used to establish sorting parameters. Absolute cell numbers and frequencies were recorded. Samples were acquired and analyzed by an investigator blinded to the treatment groups.
Table 1.
Antibodies used in the study
| Antigen | Label | Clone | Isotype | Supplier |
|---|---|---|---|---|
| Antibodies used for cytometric analysis and sorting | ||||
| CD16/CD32 | 93 | Rat IgG2b, λ | BioLegend | |
| CD45 | BV 510 | 30F-11 | Rat IgG2b | BioLegend |
| CD11b | APC/Cy7 | M1/70 | Rat IgG2b, κ | BioLegend |
| CD11c | PE/Cy7 | N418 | Armenian hamster IgG | BioLegend |
| Ly6C | FITC | HK1.4 | Rat IgG2a, κ | BioLegend |
| Ly6G | PerCP/Cy5.5 | 1A8 | Rat IgG2a, κ | BioLegend |
| CD115 | PE | AFS98 | Rat IgG2a, κ | BioLegend |
| CD4 | F488 | RM4-5 | Rat IgG2a, κ | BioLegend |
| TCR β | PerCP/Cy5.5 | H57-597 | Armenian hamster | BioLegend |
| CD8a | PE/Cy7 | 53-6.7 | Rat IgG2a, κ | BioLegend |
| TCR γδ | APC | GL3 | Armenian hamster | BioLegend |
| CD19 | APC/Cy7 | 6D5 | Rat IgG2a, κ | BioLegend |
| CCR2 | APC | 475301 | Rat IgG2b | R&D Systems |
| Antibodies used for monocyte purification by immunodepletion | ||||
| CD117 | Biotin | 2B8 | Rat IgG2b, κ | BioLegend |
| CD45R (B220) | Biotin | RA3-6B2 | Rat IgG2a, κ | BioLegend |
| CD49 | Biotin | HMa2 | Armenian hamster IgG | eBioscience |
| TER 119 | Biotin | TER-119 | Rat IgG2b, κ | BioLegend |
| CD5 | Biotin | 3 7.3 | Rat IgG2a, κ | BioLegend |
| Ly6G | Biotin | 1A8 | Rat IgG2a | BioLegend |
| Antibodies used for neutrophil purification by immunodepletion | ||||
| CD117 | Biotin | 2B8 | Rat IgG2b, κ | BioLegend |
| CD45R (B220) | Biotin | RA3-6B2 | Rat IgG2a, κ | BioLegend |
| CD49 | Biotin | HMa2 | Armenian hamster IgG | eBioscience |
| TER 119 | Biotin | TER-119 | Rat IgG2b, κ | BioLegend |
| CD5 | Biotin | 3 7.3 | Rat IgG2a, κ | BioLegend |
| CD115 | Biotin | AFS98 | Rat IgG2a | BioLegend |
| F4/80 | Biotin | BM8 | Rat IgG2a, κ | BioLegend |
Fluorescence activated cell sorting
Brain cells isolated by the enzymatic procedure were stained for CD45, CD115, Ly6G, and Ly6C as described above. Leukocytes isolated from brains of mice injected with LPS or saline that did not undergo MCAO were identified as CD45hi and further gated by CD115 and Ly6G and Ly6C markers. “Inflammatory” or “resident” MMøs, identified as CD45hiCD115+Ly6G−Ly6Chi or Ly6Clo, respectively, were sorted on a FACSVantage cytometer (BD Biosciences) and collected in TRIzol LS (Invitrogen) for transcriptomic profiling. Brains from three to four mice were pooled.
Monocyte and neutrophil isolation by immunodepletion from BM
BM was flushed out from femurs and tibias and cells were dispersed by aspiration through a 20 gauge needle. After erythrolysis, BM cells were resuspended in MACS buffer (PBS supplemented with 2% FBS, 2 mm EDTA; 100 μl/107 cells). Cells were incubated with a biotinylated antibody mixture (Table 1) and purified with anti-biotin microbeads according to the manufacturer's instructions (Miltenyi Biotec). Afterward, cells were washed, resuspended in sterile physiological saline (5 × 106 cells/ml), and administered by retroorbital injection (intraveous) in anesthetized mice. Cell preparations contained >90% Ly6Chi monocytes as determined by flow cytometry analysis. Neutrophils were also purified from mouse BM of LPS-preconditioned animals by immunomagnetic negative enrichment essentially as described above with a different biotinylated antibody mixture (Table 1). Cell preparations contained >90% neutrophils as determined by flow cytometry (anti-Ly6G-PerCP-Cy5.5, clone 1A8; BioLegend).
Generation of BM-derived macrophages (BMMs) and myeloid-derived suppressor cells (MDSCs)
Generation of BMMs, classically activated macrophages (CAMs), or alternatively activated macrophages (AAMs) was conducted as described previously (Weischenfeldt and Porse, 2008) with modifications. After erythrocyte lysis, isolated BM cells were resuspended (1 × 106 cells/ml) in BM medium (DMEM/F12, 10% FBS, 20% L929-conditioned medium, 100 U/ml penicillin, and 100 μg/ml streptomycin) and cultured in 5% CO2 at 37°C for 7 d. Fresh medium was added every 2–3 d. Polarization to CAM was induced by 100 U/ml IFN-γ (PeproTech) for 48 h and 10 ng/ml LPS for the last 24 h. AAMs were obtained by incubating BMMs with 10 ng/ml recombinant murine IL-4 (R&D Systems) for 24 h. Fully differentiated adherent macrophages were washed twice with warm DMEM/F12 and collected by detaching them with a stream of ice-cold DMEM/F12 containing 10% FBS. Cells were collected by centrifugation and resuspended in sterile saline (5 × 105 cells/100 μl). MDSCs were generated according to a previously published protocol (Highfill et al., 2010). Briefly, isolated BM cells were cultured in MDSC medium (DMEM, 10% FBS, 10 mm HEPES, 1.5 mm l-arginine, 1.5 mm l-asparagine, 1.5 mm l-glutamine, and 0.05 mm β-mercaptoethanol) in the presence of recombinant murine granulocyte macrophage colony-stimulating factor (250 U/ml; BioLegend) and granulocyte colony-stimulating factor (100 ng/ml; Peprotech) for 4 d. IL-13 (BioLegend) was added (80 ng/ml) on day 3. Cells were harvested at day 4 and MDSCs were isolated from each culture by immunomagnetic positive selection using anti-CD11b-PE antibody, anti-PE magnetic microbeads, and LS column separation (Miltenyi Biotec). The purity of isolated cell populations was found to be >90% by flow cytometry. Suppressor function was tested for inhibition of splenic CD4+ T-cell proliferation in the presence of anti-CD3 and anti-CD28 antibodies (see Fig. 4B).
Figure 4.
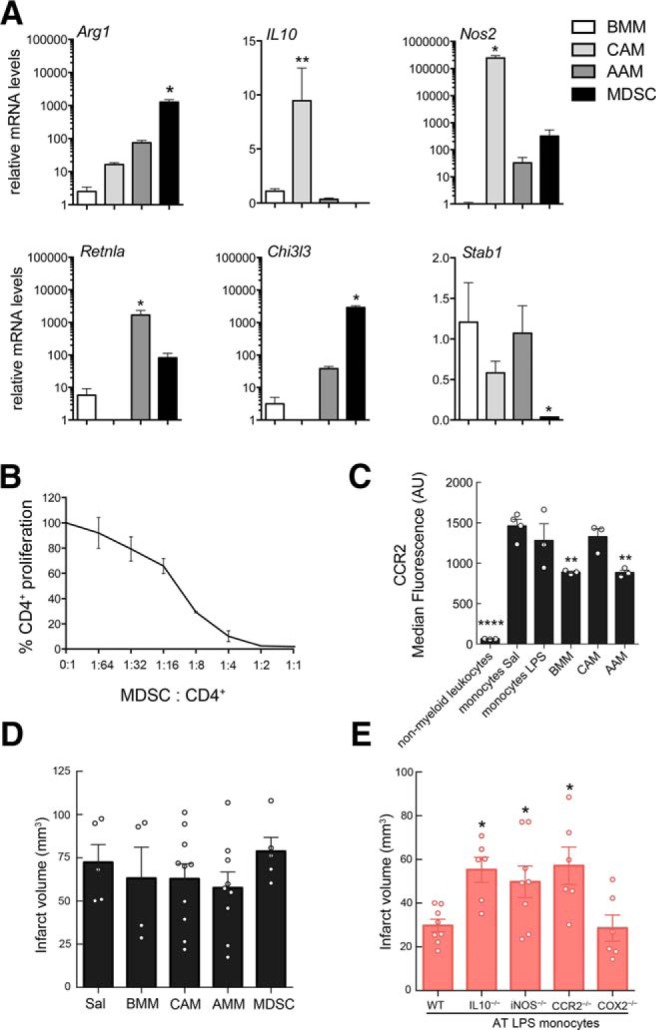
IL-10, iNOS, and CCR2 are required for the neuroprotection induced by LPS-primed monocytes. A, Quantification of Arg1, IL-10, iNOS (Nos2), Retnla, Chi3l3, and Stab1 gene expression in cultured BMMs, classical or alternative activated BMMs (CAMs and AAMs, respectively), and MDSCs by qRT-PCR. Statistical analysis: Kruskal–Wallis followed by Dunn's test versus BMMs; Arg 1 (H = 8.291, p = 0.0013), *p = 0.05; IL-10 (H = 10.97, p < 0.0001), **p = 0.01; Nos2 (H = 9.859, p = 0.003), *p = 0.05; Retnla (H = 9.929, p = 0.0012), *p = 0.05; Chi3l3 (H = 10.24, p = 0.006), *p = 0.05; Stab1 (H = 7.750, *p = 0.0286), *p = 0.05; n = 3–4/group. B, Increment cell numbers of splenic CD4+ cells were stimulated with anti-CD3 and anti-CD28 antibodies in the presence of MDSCs. Percentage of CD4+ T-cell proliferation was calculated to assess suppressor function (n = 2). x-axis values represent MDSC/splenic CD4+cell ratio. C, Quantification of the median fluorescence of CCR2 signal analyzed by flow cytometry of in nonmyeloid leukocytes, BM monocytes from saline (SAL)- or LPS-treated mice, cultured BMMs, and classical or alternative activated BMMs (CAMs and AAMs, respectively). Statistical analysis: one-way ANOVA F(5,13) = 26.13, p < 0.0001) followed by Holm–Sidak's post hoc test, **p < 0.01; ****p < 0.0001; n = 3–4/group. D, Infarct volumes of mice receiving BMM (+7 h post-MCAO), BMMs that underwent either classical (CAMs) or alternative (AAMs) activation, or BMMs that were polarized to MDSCs were not different from those of mice treated with saline (SAL) (n = 4–10 mice/group). Statistical analysis: Kruskal–Wallis (H = 2.833, p = 0.7257). E, Mice receiving monocytes (7 h after MCAO) isolated from IL-10−/−, iNOS−/−, or CCR2−/− LPS-preconditioned mice, but not from COX2−/− LPS-preconditioned mice, had larger infarcts compared with the mice receiving monocytes isolated from WT LPS-preconditioned mice (n = 6–8 mice/group). AT, Adoptive transfer. Statistical analysis: one-way ANOVA (F(3,32) = 4.175, p = 0.0040) followed by Holm–Sidak's post hoc test, *p < 0.05.
Transcriptomic profiling
Total RNA was extracted from sorted Ly6Chi brain MMøs for analysis of gene expression by Illumina Mouse WG-6 version 2.0 Expression BeadChips. Total RNA was amplified with TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (Epicenter). All procedures were conducted according to the manufacturer's instructions by the Genomics Shared Resource supported by Roswell Park Cancer Institute and National Cancer Institute Grant P30CA016056. Data were checked for quality with Illumina's GenomeStudio gene expression module version 1.9.0. Summary data were background subtracted and normalized by variance stabilization using the beadarray (version 2.20.2) (Dunning et al., 2007) and limma (version 3.26.8) (Ritchie et al., 2015) packages within the R statistical environment (version 3.2.3). Analysis of differential expression was conducted in limma for probe sets filtered by spot detection (p < 0.05) and difference in mean signal intensity between treatment groups (difference >4). Significantly changed genes were subjected to analysis of canonical pathways curated by DAVID Functional Annotation Bioinformatics Microarray Analysis (Sherman et al., 2007). Heat maps were generated in GenePattern (Reich et al., 2006) using genes selected by the differential gene expression analysis. Expression data have been deposited for public access in the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE84327.
qRT-PCR
Quantitative determination of gene expression was examined as described prevously (Garcia-Bonilla et al., 2015). Total RNA was extracted from ischemic hemispheres and meninges using TRIzol reagent. The RNeasy Plus Mini Kit (Qiagen) was used to extract RNA from BM cells. Next, cDNA was synthetized with iScript reverse transcription supermix for qRT-PCR (Bio-Rad). qRT-PCR was conducted with cDNA in duplicate reactions using the Maxima SYBR Green/ROX qPCR Master Mix (2×) (Thermo Scientific). The reactions were incubated at 50°C for 2 min and then at 95°C for 10 min. A PCR cycling protocol consisting of 15 s at 95°C and 1 min at 60°C for 45 cycles was used for quantification. Primer sequences (Invitrogen) are described in Table 2. Data were expressed as relative fold change over sham-operated control mice calculated by the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Table 2.
Primer sequences used for qRT-PCR
| Gene | Sequence |
|---|---|
| Arg1 | 5′-CATCAACACTCCCCTGACAAC-3′ |
| 5′-GTACACGATGTCTTTGGCAGA-3 | |
| Ccl2 | 5′-AGGTGTCCCAAAGAAGCTGTA-3′ |
| 5′-ATGTCTGGACCCATTCCTTCT-3′ | |
| Ccl5 | 5′-ATATGGCTCGGACACCACTC-3′ |
| 5′-GTGACAAACACGACTGCAAGA-3′ | |
| Chi3l3 | 5′-AGGCTGCTACTCACTTCCACA-3′ |
| 5′-TTTTCTCCAGTGTAGCCATCC-3′ | |
| Csf1 | 5′-TGAGTCTGTCTTCCACCTGCT-3′ |
| 5′-CCCACAGAAGAATCCAATGTC-3′ | |
| Csf2 | 5′-AGCTCTGAATCCAGCTTCTCA-3′ |
| 5′-TGGTCCCTTTAAGGCAGAAAT-3′ | |
| Csf3 | 5′-AGTGTTCCCAAACTGGGTTCT-3′ |
| 5′-TTAGGGACTTCGTTCCTGTGA-3′ | |
| Cxcl1 | 5′-GCGAAAAGAAGTGCAGAGAGA-3′ |
| 5′-AAACACAGCCTCCCACACAT-3′ | |
| Cxcl5 | 5′-TGAGAAGGCAATGCTGTCAT-3′ |
| 5′-CCAGGCTCAGACGTAAGAACA-3′ | |
| Stab | 5′-CTGAAAGCTTCGGCTACAAGA |
| 5′-TTCTTTCTATCAGGAGGCAAGG-3′ | |
| Cxcl10 | 5′-ATCACTCCCCTTTACCCAGTG-3′ |
| 5′-GGAGGAGTAGCAGCTGATGTG-3′ | |
| Cxcl12 | 5′-GTCTAAGCAGCGATGGGTTC-3′ |
| 5′-TAGGAAGCTGCCTTCTCCTG-3′ | |
| Hprt | 5′-AGTGTTGGATACAGGCCAGAC-3′ |
| 5′-CGTGATTCAAATCCCTGAAGT-3′ | |
| Il-10 | 5′-TGGAGCAGGTGAAGAGTGATT-3′ |
| 5′-CACACTGCAGGTGTTTTAGCTT-3′ | |
| Nos2 | 5′-TCACCACAAGGCCACATCGGATT-3′ |
| 5′-AGCTCCTCCAGAGGGGTAGGCT-3′ | |
| Retnla | 5′-TCCTGGAACCTTTCCTGAGAT-3′ |
| 5′-GATGCAGATGAGAAGGGAACA-3′ |
Histology
Histology of whole-mount meninges was performed as described previously (Louveau and Kipnis, 2015) with some modifications. Mice were anesthetized with sodium pentobarbital and perfused transcardially with ice-cold PBS followed by 4% paraformaldehyde (PFA) in PBS. Brains were extracted from the skull and the skullcap was postfixed in 4% PFA-PBS overnight. Next, the meninges (dura/arachnoid) were washed with PBS and dissected from the skull under a surgical microscope. Whole meninges were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) in PBS (PBST) for 30 min, blocked with 5% normal donkey serum (NDS) in 0.1% PBST for 1 h, and incubated with anti-collagen IV (1:400, rabbit polyclonal; #ab6586; Abcam, RRID:AB_305584) antibody for 2 h in 1%NDS-0.1% PBST. The meninges were washed with 0.1% PBST, incubated with Cy5-conjugated secondary antibody (1:200; Jackson ImmunoResearch Laboratories), and mounted on slices using ProLong Gold Antifade mountant with DAPI (Thermo Fisher Scientific). All staining procedures were performed at room temperature. Immunostaining was visualized by epifluorescent (Olympus IX83) or confocal (Leica TCS SP5) microscopy.
Dissected brains and spleens were postfixed in 4% PFA-PBS overnight at 4°C, immersed in 30% sucrose solution, and frozen. Coronal brain sections and transverse sections of the spleen were cut (16 μm thickness) using a cryostat and mounted on slides. For nuclear staining, spleen and brain sections were permeabilized with 0.5% PBST and then incubated with TO-PRO-3 iodide (Thermo Fisher Scientific) for 30 min at room temperature. For brain immunostaining, coronal sections were permeabilized with 0.5% PBST, blocked with 5% normal donkey serum (NDS), and incubated with antibodies against the endothelial cell marker glucose transporter 1 (GLUT-1; 1:200, rabbit polyclonal antibody; Calbiochem), the microglia/macrophage marker ionized calcium binding adaptor molecule 1 (Iba1; 1:500, rabbit polyclonal antibody; Wako Chemicals), and transforming growth factor beta (TGF-β) cytokine (1:200; catalog #MAB1835, RRID:AB_357931) in 1% NDS-0.1% PBST at 4°C overnight. After 3 10 min washes with 0.1% PBST, brain slices were incubated with a Tetramethylrhodamine (TRITC)- or a Cy5-conjugated secondary antibody (1:200; Jackson ImmunoResearch Laboratories). Slices were washed with 0.1% PBST, mounted with FluorSave Reagent (Millipore), and visualized by confocal microscopy.
Arginase and NOS activity
Arginase activity was assayed essentially as described previously (Corraliza et al., 1994). Briefly, 1 × 105 isolated BM monocytes from saline or LPS-treated mice (24 h) were lysed in 50 μl of 0.1% Triton X-100 containing pepstatin, aprotinin, and antipain and incubated on a shaker for 30 min at room temperature. The enzyme was activated after adding 50 μl of 10 mm MnCl2, 50 mm Tris-HCl, pH 7.5, at 55°C for 10 min. After addition of equal volume of 0.5 m arginine, pH 9.7, arginine hydrolysis was performed at 37°C for 60 min. Reactions were stopped by acidification and the urea formed was colorimetrically quantified at 540 nm after addition of α-isonitrosopropiophenone and heating at 100°C for 45 min. NOS activity was assessed by measuring nitrite production. Briefly, isolated BM monocytes were resuspended in RPMI 1640 supplemented with 10% FBS and penicillin/streptomycin at a density of 5 × 105 cells/ml and restimulated with 100 U/ml IFN-γ and 100 ng/ml LPS for 24 h at 37°C in a humidified atmosphere containing 5% CO2. Nitrite in supernatants was determined by the Griess reaction according to the manufacturer's suggestions (Promega, catalog #g2930).
IL-10 protein level determination
IL-10 production in isolated BM monocytes was measured by ELISA (Thermo Fisher Scientific, catalog #BMS614/2, RRID:AB_2575685). Briefly, isolated BM monocytes were incubated in RPMI 1640 supplemented with 10% FBS and penicillin/streptomycin and restimulated with 100 ng/ml LPS for 16 h at 37°C in a humidified atmosphere containing 5% CO2. Secreted IL-10 was assayed in cell culture supernatants.
Experimental design and statistical analysis
Adoptive transfer of in vivo-preconditioned monocytes
A first study was performed to test whether the transfer of isolated monocytes from LPS-PC mice induces neuroprotection in mice that undergo MCAO. To this end, monocytes were purified by immunodepletion from BM cells of 7- to 8-week-old WT mice treated with saline or LPS (0.5 mg/kg in 100 μl saline, i.p.) 24 h previously. A different researcher blinded to the surgical procedures injected the isolated monocytes (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.) into mice at different time points before or after MCAO. The mice were randomly allocated to receive either LPS or saline monocytes. The infarct volume was analyzed 72 h after MCAO to assess brain damage and compared with mice that received either saline or LPS (intraperitoneally) or that received adoptively transferred neutrophils (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.).
A second study was conducted to test whether iNOS, IL-10, CCR2, or Cox2 molecules are important in conferring the protective effect of the monocytes. In these experiments, monocytes were purified by immunodepletion from BM cells of 7- to 8-week-old WT, iNOS−/−, IL-10−/−, CCR2RFP/RFP, or Cox2−/− mice treated with saline or LPS (0.5 mg/kg in 100 μl saline, i.p.) 24 h previously. The monocytes were injected (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.) into mice 7 h after MCAO. Infarct volumes were analyzed 72 h after MCAO.
Adoptive transfer of BMMs and MDSCs
A third study was conducted to assess whether BMMs that underwent classical (CAM) or alternative (AAM) activation and MDSCs also have the capacity to provide neuroprotection to mice undergoing MCAO when adoptively transferred. Mice were randomly allocated to receive BMMs, CAMs, AAMs, or MDSCs (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.) 7 h after MCAO. An investigator blinded to surgical procedures conducted macrophage transfers. Infarct volumes were analyzed 72 h after MCAO.
Adoptive transfer of ex vivo-preconditioned monocytes
In a forth study, we tested the translational potential of transferring monocytes that were ex vivo treated with LPS into mice undergoing MCAO. In these experiments, mouse monocytes isolated from BM of 7- to 8-week-old naive mice as described above and human peripheral blood monocytes (79% CD14+CD16low cells) purchased from Lifeline Cell Technology were incubated with purified LPS (100 ng/ml) or vehicle (PBS) in RPMI with 10% FBS (2 × 106 cells/ml) at 37°C and 5% CO2 for 2 h. Next, the monocytes were washed, resuspended in sterile saline, and injected (5 × 105 cells/100 μl of saline, i.v.) into mice 7 h after MCAO. Mice were randomly allocated to LPS or PBS ex vivo monocyte groups. An investigator blinded to surgical procedures and monocyte treatment performed monocyte transfer experiments. Infarct volumes were analyzed 72 h after MCAO.
Role of the spleen in the protection afforded by preconditioned monocytes
A fifth study was conducted to determine the splenic contribution to the neuroprotective effect exerted by preconditioned monocytes. In a first substudy, we performed tracking experiments of labeled monocytes to investigate whether preconditioned monocytes have a splenic origin. In a second substudy, we tested whether the protection afforded by preconditioned monocytes is lost in mice without spleen.
Experiments of substudy 1.
To investigate whether monocytes mobilize from the spleen after LPS-PC, splenic monocytes were labeled with fluorescent latex beads. To this end, intraabdominal surgery was conducted to inject 50 μl (3.64 × 1010 particles/ml) of Fluoresbrite Yellow Green Microspheres (0.50 μm Ø; Polysciences) into the mouse spleen capsule 7 h after saline or LPS treatment. Mice were anesthetized under isoflurane (maintenance 1.5–2%) and shaved on the middle left side of the abdomen. A 1.0 cm parasagittal incision was made to enter in the peritoneal cavity and exteriorize the spleen to perform the fluorescent latex (Lx+) injection. Meloxicam (2 mg/kg, s.c.) and buprenorphine (0.5 mg/kg, s.c.) were given for preemptive analgesia. Splenic and mobilized fluorescent Lx+ monocytes to blood, brain, and meninges were quantified 24 h after LPS by flow cytometry. Moreover, GFP+-expressing monocytes were ex vivo treated with LPS and transferred (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.) into mice 7 h after MCAO. Mice were allowed to survive for 24, 48, and 72 h relative to MCA occlusion. Histological analysis of spleen, BM, lymph nodes, lungs, brain, and meninges was conducted to map the location of mobilized GFP+ MMøs.
Experiments of substudy 2.
After total splenectomy or sham surgery (sham group), mice were allowed to recover for 14 d and then randomly assigned to LPS or saline intraperitoneal injection before MCAO. Infarct volume was evaluated 72 h after MCAO. In separate groups, MCAO was induced in either splenectomized or sham mice 14 d after surgery. Ex vivo LPS-treated monocytes (5 × 105 cells in 100 μl of sterile saline/mouse, i.v.) were injected into stroke mice 7 h after reperfusion. Infarct volume was evaluated 72 h after MCAO.
Statistical analysis
Data were analyzed using GraphPad Prism version 7 software. Intergroup differences were assessed for parametric data (Shapiro–Wilk normality test) by Student's t test, one-way ANOVA, or two-way ANOVA followed by post hoc tests as appropriate. For nonparametric data, Mann–Whitney or Kruskal–Wallis test was used. Data are expressed as mean ± SEM and differences were considered statistically significant p < 0.05 at a 95% confidence level.
Results
LPS-PC increases Ly6Chi MMøs in brain and meninges
First, we sought to determine whether administration of neuroprotective doses of LPS (Tasaki et al., 1997; Marsh and Stenzel-Poore, 2008) leads to an increase in immune cells in the CNS. LPS administration induced the recruitment of MMøs to the brain compared with mice receiving saline (Fig. 1A,D). The increase was observed 24 h after LPS and was sustained for at least 72 h, a time window consistent with the protective effect afforded by LPS-PC (Rosenzweig et al., 2007). Although neutrophils (Ly6G+ cells) increased 3.7-fold in the blood 24 h after LPS (Fig. 1B), they were increased in the brain only at 24 h, an effect that did not reach statistical significance (Fig. 1D). To determine the localization of LPS-induced MMøs within the brain, we conducted histological analysis in animals after intravenous administration of Lx+. Histological analysis showed that Iba1+Lx+ MMøs displayed an amoeboid morphology and intermediate Iba1 expression. They were found in the parenchyma as well within cortical vessels making tight contact with the endothelium (Fig. 1C). These findings also indicate that LPS-PC leads to entry of circulating monocytes into the brain.
Figure 1.

Systemic LPS-PC induces selective recruitment of inflammatory monocytes to brain and meninges. A, Flow cytometry analysis of isolated brain cells separated P1, P2, P3, and P4 populations based on CD45 and Ly6C expression. P1 identified infiltrating brain leukocytes (CD45hi) 24 h after LPS injection (0.5 mg/kg, i.p.); P2 corresponded to microglia (CD45intLy6C−); P3 identified endothelial cells (CD45−Ly6C+); and P4 corresponded to CD45−Ly6C− cells. Further analysis of CD115 and Ly6G expression on infiltrating leukocytes (P1) identified recruited monocytes (CD45hiCD115+Ly6G−) and neutrophils (CD45hiCD115−Ly6G+). An increase of infiltrating MMøs was observed in LPS-treated mice compared with saline-injected mice. B, Heat map analysis of leukocyte numbers in peripheral blood (PB) or spleen (SP) from LPS-treated mice over saline-injected mice. Frequencies of leukocyte populations were calculated for each tissue at 24, 48, 72, or 96 h after treatment. Log2 fold change was calculated for LPS over saline for each endpoint. Statistical analysis: unpaired two-tailed Student's t test, *p < 0.05; #p < 0.01; §p < 0.001. C, Representative images of CD115+Lx+ cells in the brain. Top image shows an Lx+ monocyte (green), stained for TGF-β (red), in close association to GLUT1-positive (blue) endothelial cells. Bottom image shows an Lx+ (green) and Iba1-intermediate (red) monocyte cell next to Iba1-high microglia with stellate morphology. D, CD115+ cells quantification in the brain indicated a sustained increase of MMøs in LPS-treated mice during the first 72 h after LPS injection. A transient increase in the Ly6G+ cell number was observed at 24 h in LPS-injected mice, although statistical changes were not found in brain neutrophils between saline and LPS treatment over the first 72 h (n = 5–14 mice/group). Statistical analysis: two-way ANOVA (interaction, F(2,44) = 0.07020, p = 0.9323; time, F(2,44) = 0.4909, p = 0.6154; treatment, F(1,44) = 55.76, p < 0.0001) followed by Sidak's post hoc test, **p < 0.05; ****p < 0.000. E, Left, Quantification of the total cell number of brain CD115+Ly6Chi (dark color) and CD115+Ly6Clo (light color) in saline- or LPS-treated mice 24 h after treatment. The percentage of CD115+Ly6Chi of the total CD115+ is given in brackets. Right, Percentage of blood inflammatory monocytes (CD115+Ly6Chi) in saline- or LPS-treated mice 24 h after treatment (n = 12–13 mice/group). Increased Ly6Chi number of MMøs was observed in the brains of LPS-treated mice. Statistical analysis: two-way ANOVA (interaction, F(1,46) = 31.16, p < 0.0001; treatment, F(1,46) = 69.33, p < 0.0001; subpopulation, F(1,46) = 37.92, p < 0.0001) followed by Sidak's post hoc test, ***p < 0.0001; NS, no statistical significance. F, Total MMø (CD115+) and inflammatory MMø (CD115+Ly6Chi) number was increased in meninges of mice 24 h after either saline (n = 8) or LPS (n = 7) injection. Neutrophils (Ly6G+) did not change. Statistical analysis: unpaired two-tailed Student's t test (t = 16.93, df = 13, ****p < 0.0001) for total CD115+ cells; unpaired two-tailed Student's t test (t = 7.207 df = 1, ****p < 0.0001) for CD115+Ly6Chi cells.
We then investigated whether the brain-associated MMøs were of the “inflammatory” (Ly6Chi) or “resident” (Ly6Clo) phenotype. Ly6Chi MMøs were significantly higher in brains 24 h after LPS compared with saline (Fig. 1E). This was not correlated with an increase in the peripheral blood, in which the number of CD115+Ly6Chi cells after LPS was not different from saline controls (Fig. 1E). In addition to the brain, CD115+ Ly6Chi cells also increased in the meninges 24 h after LPS treatment (Fig. 1F), whereas no change in meningeal neutrophils was observed (Fig. 1F). Nonmyeloid immune cells in brain and meninges were not affected by LPS-PC (data not shown). Therefore, LPS administration leads to accumulation of predominantly CD115+Ly6Chi MMøs in the brain and meninges.
Adoptive transfer of monocytes from preconditioned donors protects naive mice from ischemic brain injury
Having established that LPS-PC leads to accumulation of CD115+Ly6Chi MMøs to brain and meninges, we investigated whether these cells contribute to the neuroprotection observed after LPS-PC. To this end, we isolated BM monocytes 24 h after LPS or saline administration and transferred them into naive mice undergoing MCAO (Fig. 2A). More than 90% of immunopurified BM monocytes were Ly6Chi (Fig. 2B). Adoptive transfer of monocytes (5 × 105 cells, i.v.) from LPS-treated mice 7 h, but not 24 h, after MCAO resulted in a reduction in infarct volume similar to that of mice receiving LPS 24 h before MCAO (Fig. 2C,D). Adoptive transfer of monocytes 24 h before MCAO resulted in partial protection that was not statistically significant (Fig. 2D). Monocytes isolated from mice 24 h after saline administration did not elicit neuroprotection when adoptively transferred to naive mice 7 h after MCAO (Fig. 2E). Similarly, adoptive transfer of neutrophils isolated from the BM of LPS-treated mice did not confer neuroprotection after adoptive transfer into naive hosts 7 h after MCAO (Fig. 2E). These findings indicate that Ly6Chi monocytes isolated from LPS-treated mice and transferred into naive mice reproduce in full the protective effect of LPS-PC.
Figure 2.

Adoptive transfer of LPS-primed monocytes isolated from preconditioned donors decreases infarct volume in stroke recipient mice. A, Experimental design for adoptive transfer of monocytes in stroke mice. Mice were injected with either LPS or saline and monocytes were isolated from BM by immunodepletion 24 h later. Saline or LPS monocytes (5 × 105 monocytes/100 μl) were injected intravenously into stroke mice 24 h before transient MCAO or 7 or 24 h after MCAO. Infarct volume was determined by cresyl violet staining 72 h after MCAO. B, Isolated BM cells were stained for CD11b, CD115, Ly6G, CD117, and Ly6C markers before (start) and after (purified) immunodepletion. Stained cells were assayed by flow cytometry. Monocytes were isolated with high purity as evidenced by the expression of CD11b, CD115, and Ly6C markers and lack of expression of the neutrophil marker Ly6G and the hematopoietic stem cell marker CD117. C, Representative images of brain infarcts in saline-injected mice (SAL), LPS-preconditioned mice (LPS), or mice that received monocytes isolated from LPS-preconditioned mice 24 h before MCAO (−24 h) or 7 or 24 h after MCAO (+7 h and +24 h, respectively). Mice that received monocytes from saline-injected mice are indicated as SAL +7 h and mice that received neutrophils isolated from LPS-injected mice are indicated as PMN +7 h. The infarct (pale areas) is outlined in brain coronal sections stained with cresyl violet 72 h after MCAO. AT, Adoptive transfer. D, Infarct volume was significant smaller in either LPS-preconditioned mice (LPS i.p.) or in mice receiving adoptively transferred LPS-monocytes at +7 h (n = 9–12). Statistical analysis: Kruskal–Wallis (H = 15.65, p = 0.0035) followed by Dunn's post hoc test. *p < 0.05; ***p < 0.001. E, Adoptive transfer of monocytes isolated from saline-injected mice or neutrophils (PMN) from LPS-preconditioned mice at +7 h after MCAO did not reduce infarct volumes (n = 8–11). Statistical analysis: one-way ANOVA F(3,32) = 7.747, p = 0.0006, followed by Holm–Sidak's post hoc test, *p < 0.05.
Transcriptomic profiling of protective monocytes
Next, we investigated whether LPS-induced brain-associated monocytes exhibit a gene expression profile different from those of saline-treated animals. Transcriptomic profiling of brain MMøs identified 270 genes that were upregulated (>4-fold) in MMøs isolated from the brains of LPS-treated animals compared with MMøs from saline controls, whereas 414 were downregulated (Fig. 3A). Upregulated genes were associated with immune and inflammatory responses, regulation of GTPase activity and cholesterol homeostasis, whereas downregulated genes included members of cell adhesion, cell proliferation, positive transcriptional regulation, and angiogenesis pathways (Fig. 3B). Genes associated with alternative macrophage polarization were highly upregulated, including IL-10 and arginase 1 (Fig. 3C). IL-10 and Arginase 1 (Arg1) mRNA, as assessed by RT-PCR, were also increased in monocytes isolated from the BM of LPS-treated mice (Fig. 3D), in which a corresponding increase in IL-10 protein expression and arginase enzymatic activity was also observed (Fig. 3E). Moreover, iNOS (Nos2) mRNA, a gene involved in LPS-PC (Kawano et al., 2007; Kunz et al., 2007), was also upregulated in BM monocytes isolated from LPS-treated mice (Fig. 3D) and was associated with increased NO production (Fig. 3E). Other genes linked to AAMs, such as YM1 (Chi3l1), Fizz1/RELMα (Retnla), and stabilin 1 (Stab1), were also upregulated (Fig. 3F). Therefore, the “protective” monocytes induced by LPS administration express selected genes typical of AAMs.
Figure 3.

Transcriptomic analysis of brain-associated monocytes from LPS-preconditioned mice. A, Log2 fold-change versus p-value plot of microarray data analysis of gene expression in isolated brain MMøs from LPS-preconditioned mice over saline-treated mice (n = 2–3, n = 8–12), highlighting downregulated genes (blue, <4-fold) and upregulated genes (red, >4-fold). B, Gene Ontology (GO) analysis of gene expression revealed upregulation of genes associated with immune and inflammatory response, regulation of GTPase activity, and cholesterol homeostasis and downregulation of genes that are members of cell adhesion, cell proliferation, positive transcriptional regulation, and angiogenesis pathways. C, Heat map showing the top 20 genes upregulated and the top 20 genes downregulated in LPS versus saline (SAL)-treated mice. D, Quantification of Arg1, IL-10, and iNOS gene (Nos2) expression by qRT-PCR in mouse BM monocytes. Monocytes were isolated 24 h after LPS or SAL injection. Data are expressed as relative n-fold changes over SAL group. Arg1, IL-10, and Nos2 mRNA were significantly upregulated in LPS-primed monocytes. Statistical analysis: Arg1, Mann–Whitney test (U = 0, ****p < 0.0001); IL-10, Mann–Whitney test (U = 0, ****p < 0.0001); Nos2, unpaired two-tailed Student's t test (t = 2.895, df = 13, *p = 0.0125). E, Quantification of arginase activity by determination of urea, IL-10 protein level by ELISA assay, and iNOS activity by Griess reaction showing increased activities or protein level in BM monocytes of LPS-preconditioned mice compared with BM monocytes of saline-treated mice. Statistical analysis: urea, Mann–Whitney test (U = 0, **p < 0.0012); IL-10, unpaired two-tailed Student's t test (t = 6.749, df = 10, ****p < 0.0001); NO, unpaired two-tailed Student's t test (t = 4.83, df = 8, **p = 0.0013), respectively. F, Quantification of genes associated with alternatively activated macrophage polarization showing upregulated mRNA levels in BM monocytes isolated 24 h after LPS treatment over saline treatment. Statistical analysis: Retnla, unpaired two-tailed Student's t test (t = 4.371, df = 9, **p = 0.0018); Chi3l3, unpaired two-tailed Student's t test (t = 13.71, df = 9, ****p < 0.0001), Stab1, unpaired two-tailed Student's t test (t = 5.426, df = 9, ***p = 0.0004).
Unlike monocytes, macrophages and MDSCs are not neuroprotective
Given that AAMs have been observed in the brain after ischemia and could be beneficial due to their anti-inflammatory activity (Miró-Mur et al., 2016), we compared the neuroprotective ability of LPS-induced monocytes with that of BMMs that underwent classical (CAM) or alternative (AAM) activation. In addition, we assessed the protective capacity of MDSCs, a myeloid cell population with strong anti-inflammatory activity upregulated in the periphery after stroke (Serafini et al., 2004; Huang et al., 2006; Bronte, 2009; Liesz et al., 2015). To this end, we generated CAMs, AAMs, and MDSCs from BMMs in vitro by exposing BMMs to IFN-γ/LPS, IL-4, or IL-4/IL-13, respectively (Weischenfeldt and Porse, 2008; Highfill et al., 2010). Successful polarization was confirmed by monitoring the mRNA expression of genes associated with the respective subpopulations including Arg1, IL-10, Nos2, Retnla, Chi3l3, and Stab1 (Fig. 4A). MDSCs were tested for their ability to suppress CD4+ T-cell proliferation (Fig. 4B). All in vitro-generated macrophage populations expressed CCR2, a chemokine receptor needed for macrophage entry into the brain and association with meninges (Fig. 4C). Cells (5 × 105) were then adoptively transferred to mice 7 h after MCAO and infarct volumes were determined 72 h later. We found that none of the in vitro-differentiated BMM populations reduced infarct volume after adoptive transfer (Fig. 4D). Therefore, unlike LPS-induced protective monocytes, CAMs, AAMs, and MDSCs are unable to ameliorate ischemic brain injury.
iNOS, IL-10, and CCR2 are required for the beneficial effect of monocytes
Because Nos2 and IL-10 gene expression was upregulated in LPS-induced monocytes, we investigated whether iNOS or IL-10 was required for their neuroprotective effects. To this end, BM monocytes isolated from iNOS−/− or IL-10−/− LPS-treated mice were adoptively transferred (5 × 105 cells) to WT mice 7 h after MCAO and infarct volume was determined 72 h later. Neither iNOS−/− nor IL-10−/− monocytes induced neuroprotection compared with LPS-induced WT monocytes (Fig. 4E). Because CCR2 is required for the recruitment of “inflammatory” monocytes to sites of ischemic inflammation (Swirski et al., 2009; Garcia-Bonilla et al., 2016), we adoptively transferred monocytes from CCR2−/− mice treated with LPS (Fig. 4E). We found that CCR2−/− monocytes failed to induce neuroprotection, suggesting that monocyte migration to the brain and/or meninges is required for the protection. In contrast, LPS-induced monocytes isolated from mice deficient for the inflammatory enzyme cyclooxygenase-2 (COX2) were able to protect from ischemic brain injury, attesting to the specificity of iNOS, IL-10, and CCR2 in the protective effect.
Trafficking of protective monocytes to spleen, meninges, and brain
Next, we investigated the potential source of the protective MMøs in mice undergoing LPS-PC. Because we observed a significant depletion of splenic monocytes 24 h after LPS-PC, whereas blood monocytes were not changed (Fig. 1B), we tested whether monocytes are mobilized from the spleen and recruited to the brain and meninges. The spleen is a monocyte reservoir that can be mobilized after ischemia in other organs (Swirski et al., 2009) and splenic monocytes have been implicated in stroke pathology (Seifert et al., 2012; Dotson et al., 2015; Kim et al., 2014). To specifically label phagocytes in the spleen, Lx+ beads (0.5 μm diameter) were injected into the spleen of mice 7 h after they were treated with either saline or LPS. Eighteen hours later, histological examination of the spleen revealed that Lx+ beads distributed throughout the subcapsular sinus and the red pulp (Fig. 5A). Flow cytometric analysis showed efficient uptake of Lx+ beads by splenic CD115+Ly6G− MMøs in LPS- and saline-treated animals (Fig. 5B,C). However, Lx+ monocytes were found in blood and MMøs in bthe rain and meninges in LPS-treated animals, but not saline controls, indicating that LPS-PC mobilizes splenic monocytes that are then recruited to brain and meninges (Fig. 5B,C).
Figure 5.

LPS induces trafficking of splenic monocytes to the brain and meninges. A, Representative image of the localization of Lx+ in the spleen 24 h after intrasplenic injection (1.8 × 109 particles). The beads (green) accumulated in the MZ and subcapsular red pulp. Fluorescent nuclear staining with the TO-PRO-3 (red) was used to reveal the structural anatomy of the spleen (transverse section). B, C, Representative flow cytometry histograms (B) and quantification (C) of MMøs (CD115+ cells) showing the percentage of MMøs containing Lx+ in the spleen, blood, brain, and meninges 24 h after either LPS-PC or saline injection in mice (n = 3–4/group). Total number of Lx+CD115+ cells in the brain and meninges are given in brackets. Lx+ was injected into the spleen 7 h after saline or LPS treatment. Statistical analysis: unpaired two-tailed Student's t test, blood (t = 2.77, df = 6, *p = 0.0321), brain (t = 7.81, df = 6, ***p = 0.0002) and meninges (t = 5.22, df = 6, **p = 0.0019). D, Representative image of the spleen (left) and meninges (right) from mice that underwent 24 h of MCAO and received GFP+ monocytes (5 × 105 cells, i.v.) isolated from LPS-preconditioned mice at 7 h after MCAO. Transverse section of the spleen was stained with DAPI (red) for nuclear staining. Adoptively transferred monocytes accumulated in the red pulp of the spleen. In the meninges, monocytes were associated with meningeal blood vessel revealed by collagen IV (red) immunohistochemistry. E, Quantification of adoptively transferred monocytes (AT) in the meninges of WT mice 24 h after MCAO. Mice received either GFP+CCR2+/+ monocytes (CCR2+/GFP) or RFP+CCR2−/− monocytes (CCR2RFP/RFP; 5 × 105 cells, i.v.) isolated from GFP-WT or CCR2RFP/RFP LPS-preconditioned mice, respectively. GFP or RFP fluorescence was used to track transferred BM monocytes (n = 4 mice/group). The genetic deletion of CCR2 resulted in a decrease of meninge-associated monocytes. Statistical analysis: unpaired two-tailed Student's t test, blood (t = 2.96, df = 5, *p = 0.0315). F, Infarct volume analysis in either sham-operated (Sham) or splenectomized (Splenectomy) mice that received saline or LPS treatment (i.p.) 24 h before MCAO. Sham-operated mice treated with LPS showed reduction of infarct volume, whereas no statistical significances were observed between splenectomized mice treated with saline (SAL) or LPS (n = 6–10 mice/group). Statistical analysis: one-way ANOVA (F(3,29) = 5.608, p = 0.0037) followed by Bonferroni's post hoc test, **p < 0.05. G, Infarct volume analysis in either sham-operated (Sham) or splenectomized (Splenectomy) mice that received ex vivo LPS-primed monocytes 7 h after MCAO (LPS exmono). Sham-operated mice had significant smaller infarcts than splenectomized mice (n = 9–11 mice/group). Statistical analysis: Mann–Whitney test (U = 0.03 *p < 0.05).
To gain an insight into the trafficking of adoptively transferred protective monocytes, we isolated BM monocytes from mice ubiquitously expressing GFP treated with LPS 24 h earlier and transferred them to WT mice 7 h after MCAO. Histological analysis of spleen, brain, and meninges was performed 24 h after MCAO. In contrast to monocytes in mice treated with LPS (Fig. 5B,C), adoptively transferred GFP+ monocytes were found in the spleen and meninges, but not the brain parenchyma (Fig. 5D). The transfer to the meninges required CCR2 because CCR2−/− monocytes did not efficiently migrate to the meninges (Fig. 5E); therefore, the spleen and meninges are major sites of monocyte/MMø localization both in LPS-preconditioned mice and in mice adoptively transferred with monocytes from LPS-treated mice undergoing MCAO.
Role of the spleen in the neuroprotection afforded by LPS-PC monocytes
In view of the above results supporting a splenic source of preconditioned monocytes, we next investigated whether the spleen is required in the ischemic neuroprotection triggered by monocytes. To this end, mice underwent surgery to remove their spleens and, 14 d after recovery, splenectomized mice were treated with LPS or saline intraperitoneally 24 h before MCAO induction. Analysis of their infarct volumes 72 h later showed similar large infarcts in both LPS- and saline-treated splenectomized mice, indicating that LPS-PC did not induce neuroprotection after splenectomy (Fig. 5F). Moreover, ex vivo LPS-PC monocytes were adoptively transferred into either splenectomized mice or sham-operated mice undergoing 7 h of MCAO (Fig. 5G). Adoptive transfer of LPS monocytes decreased the infarct volume in sham-operated mice, but not in splenectomized mice. These data reveal that a significant loss in monocytic protection is observed in mice without spleens.
LPS-PC results in increased meningeal and decreased brain immune cell accumulation after stroke
We then investigated whether the presence of PC-induced MMøs in brain and meninges has an impact on the postischemic infiltration of inflammatory cells in brain. Mice underwent LPS or saline treatment 24 h before stroke and the cell populations in brain and meninges were assessed 48 h after MCAO, a time point when peripheral immune cell infiltration is maximal (Garcia-Bonilla et al., 2014b; Benakis et al., 2016). Although MMøs were increased in the meninges of LPS-PC mice compared with saline treated controls (Fig. 6A), brain MMø were unchanged (Fig. 6B). In contrast, the number of neutrophils was not different in the meninges between groups, but brain-infiltrating neutrophils were reduced in LPS-PC mice (Fig. 6A,B). CD4+ T cells, CD8+ T cells, γδT cells, B cells, and NK cells trended lower without reaching statistical significance (data not shown). Together, these data indicate that PC-induced MMøs in the meninges are associated with reduced neutrophil trafficking into the brain after a stroke.
Figure 6.

LPS-PC increases meningeal monocyte/macrophages and decreases brain neutrophil recruitment after MCAO. Flow cytometry analysis of immune cell infiltration in the meninges (A) and the brain (B) of saline or LPS-preconditioned mice 48 h after MCAO (n = 8 mice/group). Flow cytometry plots depict gating strategy. Infiltrating leukocytes (CD45hi) were phenotyped as MMøs (MMø) by high expression of CD11b and CD115 and low expression of CD11c and Ly6G (CD115+ cells). Inflammatory MMø were subclassified by high Ly6C expression (CD115+Ly6Chi). Neutrophils (PMN) were identified by high expression of CD11b and Ly6G and low expression of CD11c and CD115 (Ly6G+ cells). A, Graph plots show increased inflammatory MMø (CD115+ and CD115+Ly6Chi cells) in the meninges of LPS-preconditioned mice following MCAO. Statistical analysis: CD115+, unpaired two-tailed Student's t test, blood (t = 2.381, df = 13, *p = 0.0332); CD115+Ly6Chi, unpaired two-tailed Student's t test, blood (t = 2.186, df = 13, *p = 0.0477); B. Graph plots show decreased brain neutrophils (PMN) in LPS-preconditioned mice following MCAO. Statistical analysis: Ly6G+, unpaired two-tailed Student's t test, blood (t = 2.502, df = 13, *p = 0.0265).
Ex vivo treatment with LPS induces a neuroprotective phenotype in mouse and human monocytes
Although our results are consistent with a direct effect of LPS on monocytes leading to a protective phenotype, indirect activation by downstream effectors induced by LPS such as proinflammatory cytokines, lipid mediators, or DAMPs cannot be excluded. To address whether LPS acts directly on monocytes, we exposed monocytes isolated from naive animals to LPS (100 ng/ml, 2 h) or vehicle (PBS) ex vivo before injecting them (5 × 105 cell, i.v.) into mice 7 h after MCAO. Mice receiving LPS-treated monocytes showed smaller infarcts (−55 ± 10%; p < 0.05) than mice receiving PBS treated monocytes (Fig. 7A), a protective effect comparable to that observed in LPS-PC mice (Fig. 2D). Importantly, the protection was lost when LPS-stimulated monocytes were lysed (Fig. 7A). The reduction in brain injury was accompanied by a better neurological outcome as assessed by the tape test 72 h after occlusion (Fig. 7B). As in monocytes isolated from LPS-treated mice, monocytes exposed in vitro to LPS for 2 h showed increased IL-10 and Nos2 expression (Fig. 7C). To test whether the protective activity could also be elicited by human monocytes, we exposed purified human peripheral blood monocytes to saline or LPS for 2 h and transferred them into mice that underwent stroke as described above. Transfer of human monocytes that were exposed to LPS, but not saline, significantly reduced infarct volumes after MCAO (Fig. 7D). The degree of reduction was comparable to that observed in mice receiving murine LPS-treated monocytes compared with mice receiving either human or mouse PBS monocytes (−45 ± 10% vs −55 ± 10%, respectively; p > 0.05).
Figure 7.

Adoptive transfer of ex vivo LPS-primed mouse and human monocytes exerts protection in recipient mice after MCAO. A, Infarct volume analysis in mice that adoptively received ex vivo PBS-treated monocytes (PBS exmono), LPS-treated (LPS; 100 ng/ml, 2 h) monocytes (LPS exmonno), or lysed LPS-treated monocytes 7 h after MCAO (LPS lysed exmono). Mice receiving ex vivo LPS-primed monocytes showed reduction of infarct volume (n = 7–8 mice/group). Statistical analysis: one-way ANOVA F(2,19) = 4.933, p = 0.0188), followed by Bonferroni's post hoc test, *p < 0.05. B, Assessment of sensorimotor function by tape test performance revealed that mice receiving ex vivo LPS-primed monocytes had reduced latency to contact the tape on the impaired paw (contralesional side) than mice receiving PBS-treated monocytes at 3 d after MCAO (n = 13 mice/group). Statistical analysis: two-way ANOVA (interaction, F(1,49) = 7.828, p = 0.0073; treatment, F(1,49) = 5.915, p = 0.0187; time, F(1,49) = 18.4, p < 0.0001) followed by Sidak's post hoc test, **p < 0.01. C, Quantification of IL-10 and Nos2 (iNOS) expression by qRT-PCR of ex vivo PBS- or LPS-treated monocytes showed upregulation of both genes 2 h after LPS treatment (n = 4/group). Statistical analysis: unpaired two-tailed Student's t test, IL-10 (t = 15.19, df = 6, ****p < 0.0001) and Nos2 (t = 16, df = 5, ****p < 0.0001). D, Infarct volume analysis in stroke mice receiving ex vivo PBS- or LPS-treated human monocytes 7 h after MCAO. LPS-primed human monocytes significantly reduced infarct volumes after MCAO compared with mice receiving PBS-primed human monocytes (n = 9–10 mice/group). Statistical analysis: unpaired two-tailed Student's t test (t = 2.267, df = 17, *p = 0.0367). E, Histology of the meninges after adoptive transfer of GFP+ monocytes ex vivo treated with LPS. Monocytes transferred 7 h after MCAO were recruited to the meninges, where they associated with vessels (extravascular and intravascular localization) identified by collagen IV expression. The graph shows the number of adoptively transferred monocytes (AT) that accumulated in the meninges at 48 h MCAO of mice that received either PBS or LPS ex vivo-treated GFP+ monocytes. No difference in the number of accumulated monocytes was found between groups (n = 4 mice/group). Statistical analysis: unpaired two-tailed Student's t test (t = 0.5148, df = 6, p = 0.6251). F, Gene expression of inflammatory molecules in the meninges of mice receiving ex vivo PBS- or LPS-primed monocytes 48 h after MCAO. Decreased Cxcl5, Csf2, Csf3, and Cxcl10 gene upregulation was observed in the meninges of mice receiving LPS-primed monocytes (n = 10–11 mice/group). Statistical analysis: unpaired two-tailed Student's t test, Cxcl5 (t = 3.6298, df = 7, *p = 0.0084); Csf2 (t = 4.4886, df = 7, *p = 0.0028); Csf3 (t = 2.6324, df = 7, *p = 0.0337), and Cxcl10 (t = 2.7547, df = 7, *p = 0.0283).
We also sought to determine whether adoptively transferred monocytes exposed to LPS ex vivo were recruited to the brain and meninges. BM monocytes from GFP transgenic mice were exposed to vehicle (PBS) or LPS and transferred into mice 7 h after MCAO. We found that both PBS- and LPS-primed monocytes were present in the meninges, but not in the brain, after stroke (Fig. 7E). Transferred monocytes were mainly seen intravascularly or perivascularly in vessels in or near the superior sagittal sinus. These findings suggest that the presence of MMøs in the meninges is not sufficient to confer neuroprotection and priming of the monocytes by LPS is also needed.
LPS-primed monocytes suppress inflammatory cytokines in the meninges
To address whether adoptive transfer of protective monocytes would alter the inflammatory response in meninges after stroke, we conducted mRNA analysis of inflammatory molecules involved in leukocyte chemotaxis (Ccl2, Ccl5, Cxcl1, Cxcl3, Cxcl5, Cxcl10, and Cxcl12) and survival/activation (Csf1, Csf2, and Csf3) in the meninges of mice receiving LPS-primed monocytes 48 h after MCAO. All inflammatory molecules were upregulated in the meninges except for Ccl5 (Fig. 7F). The neutrophil regulatory factors Cxcl5, Csf2, and Csf3 and the lymphocyte regulatory factor Cxcl10 were significantly decreased in mice receiving LPS-primed monocytes. These data indicate that LPS-primed monocytes in the meninges suppress the postischemic expression of inflammatory cytokines involved in leukocyte trafficking to the brain.
Discussion
We have found that LPS-PC leads to accumulation of “inflammatory” MMøs in the brains and meninges of preconditioned animals and that LPS-PC monocytes are sufficient to induce neuroprotection after adoptive transfer. Although previous studies have focused on the effects of LPS-PC on gene regulation within the brain (Stevens et al., 2011), here, we investigated whether LPS-PC induces a peripheral immune response that contributes to cerebral ischemic tolerance and modifies stroke outcome. We found that monocytes are recruited to brain and meninges after LPS-PC. This was specific for monocytes because neutrophils did not accumulate in the brain or meninges after LPS-PC even though their frequency was strongly increased in the peripheral blood. Monocytes and/or MMøs were observed at intravascular and perivascular sites, raising the possibility that they may signal to vascular and parenchymal cells, leading to an altered immune response after cerebral ischemia. We did not address whether this response was specific to brain and meninges, but findings that MMøs are increased in the kidney after LPS-PC (Hato et al., 2018) could indicate that similar immune cell recruitment is observed in other organs.
Importantly, we observed that monocytes isolated from the BM of mice that underwent LPS-PC were able to confer robust neuroprotection when adoptively transferred to naive mice 7 h after MCAO, indicating that monocytes are sufficient to induce ischemic tolerance. Adoptive transfer of monocytes isolated from saline-treated animals did not alter stroke outcome, suggesting that LPS-induced gene expression is required to establish the protective phenotype. In contrast, adoptively transferred neutrophils isolated from the LPS-preconditioned animals or in vitro differentiated CAMs, AAMs, and MDSCs were not able to confer neuroprotection. Therefore, the capacity to confer protection from ischemic brain injury is restricted to a selected monocyte subpopulation.
Our data also show that the protective phenotype of adoptively transferred monocytes is dependent on the expression of CCR2, IL-10, and iNOS. CCR2 is the major chemokine receptor involved in monocyte infiltration after brain ischemia (Gliem et al., 2012; Garcia-Bonilla et al., 2016). Here, we show that CCR2 is also required for meningeal recruitment of monocytes. Interestingly, several studies have reported increased injury after MCAO in CCR2−/− mice, which lack circulating inflammatory monocytes (Gliem et al., 2012; Chu et al., 2015; Garcia-Bonilla et al., 2016; Wattananit et al., 2016), an observation consistent with a neuroprotective activity of monocytes recruited to the ischemic lesion. Furthermore, hypoxic and ischemic PC are also dependent on CCL2/CCR2 signaling (Rehni and Singh, 2012; Stowe et al., 2012), indicating that recruitment of protective monocytes might be a global mechanism underlying brain PC. IL-10 has been proposed as a necessary component of ischemic tolerance in the myocardium (Cai et al., 2012) and is upregulated in the plasma, but not the brain, after LPS-PC (Vartanian et al., 2011). These results are consistent with a role for IL-10 production in circulating immune cells, possibly monocytes, as observed in our study. Previous studies have identified iNOS-derived NO as an essential mediator of ischemic and LPS-PC (Kawano et al., 2007; Kunz et al., 2007). Our results support these previous findings and support the hypothesis that monocyte-derived NO contributes to ischemic tolerance in LPS-PC.
Using labeling of splenic monocyte/macrophages by subcapsular Lx+ injection, we also found that LPS treatment mobilizes monocytes from the spleen into circulation and that splenic monocytes became associated with brain and meninges. The contribution of splenic monocytes to stroke pathology has been a matter of debate. Although several studies have shown a role for the spleen, specifically splenic monocytes, in promoting ischemic brain injury (Ajmo et al., 2008; Ostrowski et al., 2012; Seifert et al., 2012; Dotson et al., 2015), others failed to do so (Kim et al., 2014; Zierath et al., 2017). We did not specifically address the deleterious effects of splenic monocytes in brain ischemia, but our studies in splenectomized mice support the conclusion that splenic monocytes, when exposed to a systemic PC stimulus such as LPS, can be protective against ischemic brain injury. In addition, the spleen was required to unleash the neuroprotective capacity of adoptively transferred LPS-preconditioned monocytes. Several potential mechanisms could account for this finding. First, monocytes may need to home to the spleen to become licensed to exhibit their neuroprotective phenotype. Similar licensing has been observed in other models. For example, it has been shown that interaction with splenic stromal cells is required to elicit immunosuppressor functions in DCs (Zhang et al., 2004; Xu et al., 2012). Alternatively, adoptively transferred monocytes might instruct other immune cells in the spleen to change their inflammatory phenotype. It has been reported that adoptively transferred Ly6Chi monocytes can transform into marginal zone (MZ) macrophages (A-Gonzalez et al., 2013). The macrophages in the MZ and subcapsular red pulp are strategically placed to interact with splenic immune cells and bloodborne mediators produced by a systemic PC stimulus or after stroke. It is therefore possible that splenic MMøs, depending on their activation state, instruct other splenic immune cells poised to participate in the inflammatory response after stroke to exhibit a proinflammatory or anti-inflammatory phenotype.
Our study also identifies the meninges as a key immunoregulatory compartment involved in PC and ischemic brain injury. Whereas protective MMøs were found in both brain and meninges after LPS-PC, they were only found in the meninges after adoptive transfer. These findings emphasize a potential role for the meninges in mediating the neuroprotection afforded by LPS-primed monocytes. We have previously shown that meningeal γδT cells orchestrate immune cell trafficking after ischemic brain injury by promoting neutrophil infiltration into the affected brain parenchyma (Benakis et al., 2016). Here, we demonstrate that meningeal MMøs are capable of dampening the postischemic inflammatory response by suppressing the production of neutrophil-activating and chemotactic factors such as Csf2, Csf3, and Cxcl5. We have shown previously that Csf3 is an endothelial-derived cytokine responsible for neutrophil activation after stroke (Garcia-Bonilla et al., 2015). Downregulating Csf3 and related cytokines in the meninges may suppress the inflammatory response in neutrophils, thereby limiting their ability to invade the brain parenchyma. Therefore, we propose a reduced infiltration of damping neutrophils as the mechanism by which monocytes protect against ischemic injury.
Another major finding of this study is that monocytes can be primed in vitro to express a neuroprotective phenotype. Therefore, exposure of monocytes to LPS for 2 h was sufficient to induce their protective potential. Together with the favorable therapeutic window, the quick induction of the neuroprotective phenotype should allow for the development of clinical protocols for stroke therapy. BM mononuclear cells (BMMCs) are increasingly used as a cell therapy for a variety of brain injuries, including stroke, both in preclinical and clinical settings (Rosado-de-Castro et al., 2016). Although the identity of the immune cells within the heterogeneous BMMC pool responsible for the protective effects has not been established, one study showed that BMMCs depleted of myeloid cells lost their protective potential after transient MCAO in rats, whereas depletion of the lymphocyte and erythrocyte lineage had no effect (Yang et al., 2016). Similarly, in rats treated with human umbilical cord blood after permanent MCAO, monocytes have been singled out as the protective cell population and peripheral blood CD14+ monocytes were sufficient to improve long-term outcome (Womble et al., 2014). The finding that human monocytes can be reprogrammed in vitro to be neuroprotective is of translational and clinical relevance. Although we did not address the mechanism by which human PC monocytes exert their protective effects, it is likely that their effector mechanisms are also dependent upon IL-10 and NO. Importantly, iNOS and IL-10 are upregulated in LPS-treated human monocytes and human IL-10 shows cross-species activity on mouse cells (Tan et al., 1993).
In summary, our study identifies a distinct population of splenic monocytes as key effector cells in the profound protective effect afforded by LPS-PC. After exposure to LPS either in vitro or in vivo, these cells are able to modulate meningeal and parenchymal immune responses after MCAO, limiting ischemic injury and improving functional outcome with a clinically relevant therapeutic window. Furthermore, our findings demonstrate that in vitro priming of monocytes with TLR4 agonists triggers protective potential against ischemic brain injury and raise the possibility that similar approaches could be used to enhance the effectiveness of monocyte-based cell therapies in ischemic stroke patients.
Footnotes
This work was supported by the National Institutes of Health (Grant NS34179 to C.I. and Grant NS081179 to J.A.), the American Heart Association (Grant 16SDG30970036 to L.G), and the Feil Family Foundation.
The authors declare no competing financial interests.
References
- A-Gonzalez N, et al. (2013) The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nat Immunol 14:831–839. 10.1038/ni.2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH, He YY, Nassief A, Xu J, Xu XM, Hsu CY, Faraci FM (2000) Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 31:193–199. 10.1161/01.STR.31.1.193 [DOI] [PubMed] [Google Scholar]
- Ajmo CT Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR (2008) The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res 86:2227–2234. 10.1002/jnr.21661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069. 10.1084/jem.20070075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F (2007) Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317:666–670. 10.1126/science.1142883 [DOI] [PubMed] [Google Scholar]
- Banks WA, Robinson SM (2010) Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behav Immun 24:102–109. 10.1016/j.bbi.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer P, Welbourne T, Shigematsu T, Russell J, Granger DN (2000) Endothelial expression of selectins during endotoxin preconditioning. Am J Physiol Regul Integr Comp Physiol 279:R2015–R2021. 10.1152/ajpregu.2000.279.6.R2015 [DOI] [PubMed] [Google Scholar]
- Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, Iadecola C, Anrather J (2016) Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med 22:516–523. 10.1038/nm.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 30:475–487. 10.1016/j.it.2009.07.009 [DOI] [PubMed] [Google Scholar]
- Bronte V. (2009) Myeloid-derived suppressor cells in inflammation: uncovering cell subsets with enhanced immunosuppressive functions. Eur J Immunol 39:2670–2672. 10.1002/eji.200939892 [DOI] [PubMed] [Google Scholar]
- Cai ZP, Parajuli N, Zheng X, Becker L (2012) Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol 107:277. 10.1007/s00395-012-0277-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HX, Broughton BR, Kim HA, Lee S, Drummond GR, Sobey CG (2015) Evidence that Ly6Chi monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke 46:1929–1937. 10.1161/STROKEAHA.115.009426 [DOI] [PubMed] [Google Scholar]
- Corraliza IM, Campo ML, Soler G, Modolell M (1994) Determination of arginase activity in macrophages: a micromethod. J Immunol Methods 174:231–235. 10.1016/0022-1759(94)90027-2 [DOI] [PubMed] [Google Scholar]
- Dotson AL, Wang J, Saugstad J, Murphy SJ, Offner H (2015) Splenectomy reduces infarct volume and neuroinflammation in male but not female mice in experimental stroke. J Neuroimmunol 278:289–298. 10.1016/j.jneuroim.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning MJ, Smith ML, Ritchie ME, Tavaré S (2007) beadarray: r classes and methods for illumina bead-based data. Bioinformatics 23:2183–2184. 10.1093/bioinformatics/btm311 [DOI] [PubMed] [Google Scholar]
- Forghani P, Khorramizadeh MR, Waller EK (2012) Natural suppressor cells; past, present and future. Front Biosci (Elite Ed) 4:1237–1245. [DOI] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Benakis C, Moore J, Iadecola C, Anrather J (2014a) Immune mechanisms in cerebral ischemic tolerance. Front Neurosci 8:44. 10.3389/fnins.2014.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Moore JM, Racchumi G, Zhou P, Butler JM, Iadecola C, Anrather J (2014b) Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J Immunol 193:2531–2537. 10.4049/jimmunol.1400918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Racchumi G, Murphy M, Anrather J, Iadecola C (2015) Endothelial CD36 contributes to postischemic brain injury by promoting neutrophil activation via CSF3. J Neurosci 35:14783–14793. 10.1523/JNEUROSCI.2980-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J, Brea D, Iadecola C, Anrather J (2016) Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J Neuroinflammation 13:285. 10.1186/s12974-016-0750-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. 10.1016/S1074-7613(03)00174-2 [DOI] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S (2012) Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71:743–752. 10.1002/ana.23529 [DOI] [PubMed] [Google Scholar]
- Hato T, Zollman A, Plotkin Z, El-Achkar TM, Maier BF, Pay SL, Dube S, Cabral P, Yoshimoto M, McClintick J, Dagher PC (2018) Endotoxin preconditioning reprograms S1 tubules and macrophages to protect the kidney. J Am Soc Nephrol 29:104–117. 10.1681/ASN.2017060624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR (2010) Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is upregulated by interleukin-13. Blood 116:5738–5747. 10.1182/blood-2010-06-287839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH (2006) Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66:1123–1131. 10.1158/0008-5472.CAN-05-1299 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368. 10.1038/nn.2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman K, Kunz A, Iadecola C (2011) Modeling focal cerebral ischemia in vivo. Methods Mol Biol 793:195–209. 10.1007/978-1-61779-328-8_13 [DOI] [PubMed] [Google Scholar]
- Kawano T, Kunz A, Abe T, Girouard H, Anrather J, Zhou P, Iadecola C (2007) iNOS-derived NO and nox2-derived superoxide confer tolerance to excitotoxic brain injury through peroxynitrite. J Cereb Blood Flow Metab 27:1453–1462. 10.1038/sj.jcbfm.9600449 [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; National Centre for the Replacement, Refinement and Reduction of Amimals in Research (2011) Animal research: reporting in vivo experiments–the ARRIVE guidelines. J Cereb Blood Flow Metab 31:991–993. 10.1038/jcbfm.2010.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Yang J, D Beltran CD, Cho S (2014) Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 34:1411–1419. 10.1038/jcbfm.2014.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T. (2002) Ischemic tolerance. J Cereb Blood Flow Metab 22:1283–1296. 10.1097/01.WCB.0000040942.89393.88 [DOI] [PubMed] [Google Scholar]
- Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C (2007) Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 27:7083–7093. 10.1523/JNEUROSCI.1645-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, Yang H, Na SY, Akhisaroglu M, Fleming T, Eigenbrod T, Nawroth PP, Tracey KJ, Veltkamp R (2015) DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci 35:583–598. 10.1523/JNEUROSCI.2439-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY, Huang CC, Chang KF (2009) Lipopolysaccharide preconditioning reduces neuroinflammation against hypoxic ischemia and provides long-term outcome of neuroprotection in neonatal rat. Pediatr Res 66:254–259. 10.1203/PDR.0b013e3181b0d336 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Louveau A, Kipnis J (2015) Dissection and immunostaining of mouse whole-mount meninges. Protocol Exchange. Nature Publishing Group. [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N (1995) Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81:641–650. 10.1016/0092-8674(95)90085-3 [DOI] [PubMed] [Google Scholar]
- Manthey CL, Vogel SN (1994) Elimination of trace endotoxin protein from rough chemotype LPS. Innate Immunity 1:84–91. [Google Scholar]
- Marsh BJ, Stenzel-Poore MP (2008) Toll-like receptors: novel pharmacological targets for the treatment of neurological diseases. Curr Opin Pharmacol 8:8–13. 10.1016/j.coph.2007.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP (2009a) Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci 29:9839–9849. 10.1523/JNEUROSCI.2496-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Williams-Karnesky RL, Stenzel-Poore MP (2009b) Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158:1007–1020. 10.1016/j.neuroscience.2008.07.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró-Mur F, Pérez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, Planas AM (2016) Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun 53:18–33. 10.1016/j.bbi.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O (1995) Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83:473–482. 10.1016/0092-8674(95)90125-6 [DOI] [PubMed] [Google Scholar]
- Muzio M, Bosisio D, Polentarutti N, D'amico G, Stoppacciaro A, Mancinelli R, van't Veer C, Penton-Rol G, Ruco LP, Allavena P, Mantovani A (2000) Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol 164:5998–6004. 10.4049/jimmunol.164.11.5998 [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204:3037–3047. 10.1084/jem.20070885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Pittet MJ, Swirski FK (2010) Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121:2437–2445. 10.1161/CIRCULATIONAHA.109.916346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S. (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59:1593–1600. 10.1007/s00262-010-0855-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski RP, Schulte RW, Nie Y, Ling T, Lee T, Manaenko A, Gridley DS, Zhang JH (2012) Acute splenic irradiation reduces brain injury in the rat focal ischemic stroke model. Transl Stroke Res 3:473–481. 10.1007/s12975-012-0206-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradillo JM, Fernández-López D, García-Yébenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I (2009) Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem 109:287–294. 10.1111/j.1471-4159.2009.05972.x [DOI] [PubMed] [Google Scholar]
- Rehni AK, Singh TG (2012) Involvement of CCR-2 chemokine receptor activation in ischemic preconditioning and postconditioning of brain in mice. Cytokine 60:83–89. 10.1016/j.cyto.2012.05.009 [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38:500–501. 10.1038/ng0506-500 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47–e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado-de-Castro PH, de Carvalho FG, de Freitas GR, Mendez-Otero R, Pimentel-Coelho PM (2016) Review of preclinical and clinical studies of bone marrow-derived cell therapies for intracerebral hemorrhage. Stem Cells Int 2016:4617983–18. 10.1155/2016/4617983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP (2004) Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 35:2576–2581. 10.1161/01.STR.0000143450.04438.ae [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, Meller R, Simon RP, Stenzel-Poore MP (2007) Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab 27:1663–1674. 10.1038/sj.jcbfm.9600464 [DOI] [PubMed] [Google Scholar]
- Seifert HA, Leonardo CC, Hall AA, Rowe DD, Collier LA, Benkovic SA, Willing AE, Pennypacker KR (2012) The spleen contributes to stroke induced neurodegeneration through interferon gamma signaling. Metab Brain Dis 27:131–141. 10.1007/s11011-012-9283-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V (2004) Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother 53:64–72. 10.1007/s00262-003-0443-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG (2008) Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol 26:421–452. 10.1146/annurev.immunol.26.021607.090326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman BT, Huang da W, Tan Q, Guo Y, Bour S, Liu D, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007) DAVID knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics 8:426. 10.1186/1471-2105-8-426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, Leiner I, Li MO, Frenette PS, Pamer EG (2011) Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 34:590–601. 10.1016/j.immuni.2011.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Jiang Y (2004) How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology 201:197–207. 10.1016/j.tox.2004.04.015 [DOI] [PubMed] [Google Scholar]
- Smith RM, Lecour S, Sack MN (2002) Innate immunity and cardiac preconditioning: a putative intrinsic cardioprotective program. Cardiovasc Res 55:474–482. 10.1016/S0008-6363(02)00288-2 [DOI] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP (2011) Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 31:8456–8463. 10.1523/JNEUROSCI.0821-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowe AM, Wacker BK, Cravens PD, Perfater JL, Li MK, Hu R, Freie AB, Stüve O, Gidday JM (2012) CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J Neuroinflammation 9:33. 10.1186/1742-2094-9-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117:195–205. 10.1172/JCI29950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ (2009) Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325:612–616. 10.1126/science.1175202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Wildgruber M, Ueno T, Figueiredo JL, Panizzi P, Iwamoto Y, Zhang E, Stone JR, Rodriguez E, Chen JW, Pittet MJ, Weissleder R, Nahrendorf M (2010) Myeloperoxidase-rich ly-6C+ myeloid cells infiltrate allografts and contribute to an imaging signature of organ rejection in mice. J Clin Invest 120:2627–2634. 10.1172/JCI42304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194. 10.1172/JCI28549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JC, Indelicato SR, Narula SK, Zavodny PJ, Chou CC (1993) Characterization of interleukin-10 receptors on human and mouse cells. J Biol Chem 268:21053–21059. [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM (1997) Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 748:267–270. 10.1016/S0006-8993(96)01383-2 [DOI] [PubMed] [Google Scholar]
- Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP (2011) LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation 8:140. 10.1186/1742-2094-8-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Birch SE, He R, Tawadros P, Szaszi K, Kapus A, Rotstein OD (2010) Remote ischemic preconditioning by hindlimb occlusion prevents liver ischemic/reperfusion injury. Ann Surg 251:292–299. 10.1097/SLA.0b013e3181bfda8c [DOI] [PubMed] [Google Scholar]
- Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O, Schwartz M, Kokaia Z (2016) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36:4182–4195. 10.1523/JNEUROSCI.4317-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Porse B (2008) Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008:pdb.prot5080. 10.1101/pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- Womble TA, Green S, Shahaduzzaman M, Grieco J, Sanberg PR, Pennypacker KR, Willing AE (2014) Monocytes are essential for the neuroprotective effect of human cord blood cells following middle cerebral artery occlusion in rat. Mol Cell Neurosci 59:76–84. 10.1016/j.mcn.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yi H, Guo Z, Qian C, Xia S, Yao Y, Cao X (2012) Splenic stroma-educated regulatory dendritic cells induce apoptosis of activated CD4 T cells via fas ligand-enhanced IFN-γ and nitric oxide. J Immunol 188:1168–1177. 10.4049/jimmunol.1101696 [DOI] [PubMed] [Google Scholar]
- Yang B, Parsha K, Schaar K, Xi X, Aronowski J, Savitz SI (2016) Various cell populations within the mononuclear fraction of bone marrow contribute to the beneficial effects of autologous bone marrow cell therapy in a rodent stroke model. Transl Stroke Res 7:322–330. 10.1007/s12975-016-0462-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181:5791–5802. 10.4049/jimmunol.181.8.5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Tang H, Guo Z, An H, Zhu X, Song W, Guo J, Huang X, Chen T, Wang J, Cao X (2004) Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol 5:1124–1133. 10.1038/ni1130 [DOI] [PubMed] [Google Scholar]
- Zierath D, Shen A, Stults A, Olmstead T, Becker KJ (2017) Splenectomy does not improve long-term outcome after stroke. Stroke 48:497–500. 10.1161/STROKEAHA.116.016037 [DOI] [PMC free article] [PubMed] [Google Scholar]