

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer death in the United States yet data are scant regarding host factors influencing pancreatic carcinogenesis. Increasing evidence support the role of the host microbiota in carcinogenesis but its role in PDAC is not well established. Herein, we report that antibiotic-mediated microbial depletion of KrasG12D/PTENlox/+ mice showed a decreased proportion of poorly differentiated tumors compared to microbiota-intact KrasG12D/PTENlox/+ mice. Subsequent 16S rRNA PCR showed that ~50% of KrasG12D/PTENlox/+ mice with PDAC harbored intrapancreatic bacteria. To determine if a similar observation in humans correlates with presence of PDAC, benign and malignant human pancreatic surgical specimens demonstrated a microbiota by 16S bacterial sequencing and culture confirmation. However, the microbial composition did not differentiate PDAC from non-PDAC tissue. Furthermore, murine pancreas did not naturally acquire a pancreatic microbiota, as germ-free mice transferred to specific pathogen-free housing failed to acquire intrapancreatic bacteria over time, which was not augmented by a murine model of colitis. Finally, antibiotic-mediated microbial depletion of Nod-SCID mice, compared to microbiota-intact, showed increased time to PDAC xenograft formation, smaller tumors, and attenuated growth. Interestingly, both xenograft cohorts were devoid of intratumoral bacteria by 16S rRNA PCR, suggesting that intrapancreatic/intratumoral microbiota is not the sole driver of PDAC acceleration. Xenografts from microbiota-intact mice demonstrated innate immune suppression by immunohistochemistry and differential regulation of oncogenic pathways as determined by RNA sequencing. Our work supports a long-distance role of the intestinal microbiota on PDAC progression and opens new research avenues regarding pancreatic carcinogenesis.
The human pancreas possesses a microbiota that does not discriminate adenocarcinoma from normal tissue but the intestinal microbiota does accelerate pancreatic carcinogenesis through a long-distance mechanism. Targeting of intestinal microbes may prove beneficial in curtailing pancreatic cancer development and progression.
Introduction
Despite progress determining the temporal relationship of genetic mutations in pancreatic carcinogenesis (1–3), evidence of distinct histopathologies in pancreatic ductal adenocarcinoma (PDAC) (4,5), and potential immune mediators (6,7), little is known regarding modifiable host factors that potentiate this deadly disease, which is now the third most common cause of cancer death in the United States (8). While risk factors for the development of PDAC have been associated with various pro-inflammatory states, including smoking (9), pancreatitis (10) and obesity (11), as well as individual genetic predisposition (12,13), the majority of cases are sporadic without an identifiable risk factor. Therefore, aggressive and paradigm-shifting approaches are needed in order to identify factors of pancreatic carcinogenesis that will potentially allow for early detection and risk modification.
The host microbiota comprises trillions of cells that make up the commensal, symbiotic and pathobiont milieu of microorganisms that share the body space of every individual. Disruptions in microbial–host networks have been associated with numerous human pathological conditions, including cancer (14–16). Previous observations suggest that patients with PDAC may have a distinct oral and fecal microbiome compared to those without pancreatic cancer (17–19). Specifically, recent reports demonstrate an association with the presence of Porphyromonas gingivalis and risk of developing PDAC (17,18). Furthermore, a concentration dependent risk reduction in PDAC development was noted in patients with increased abundance of the phylum Fusobacteria and its genus Leptotrichia (18). A protective effect was likewise seen in patients with higher serum antibody titers to commensal oral bacteria (odds ratio 0.55, 95% confidence interval 0.36–0.83) (17). Finally, gut microbiota may allow disease discrimination with decreased microbial phyla diversity, increased Bacteriodetes, and decreased Firmicutes and Proteobacteria in PDAC cases compared to healthy controls (19). However, the functional impact of these microbial differences on pancreatic cancer is currently unknown and represents only association with a disease state.
In this study, we investigated the impact of host microbiota on pancreatic carcinogenesis and sought to determine whether it is mediated by a local or distant effect. Herein we report the presence of a microbiota in the human pancreas, without a clear ‘disease-associated profile’ between cancer and non-PDAC tissue. Importantly, experiments utilizing PDAC xenograft and a genetically engineered mouse model (GEMM) of PDAC support the concept of a long-distance effect of intestinal bacteria on pancreatic carcinogenesis, which may be mediated by innate immune suppression and dysregulation of oncogenic pathways within the tumor.
Materials and methods
Human tissue acquisition
All aspects of human data and tissue acquisition and use were approved by the Institutional Review Board (IRB) at the University of Florida (Protocol # IRB201600873). Written informed consent was obtained from patients who were scheduled to undergo pancreatectomy for benign and malignant surgical diseases to allocate a portion of resected tissue for research purposes. An inner portion of pancreatic tissue from the surgical specimen was sterilely collected upon surgical resection, aliquoted into sterile cryogenic tubes to be snap frozen in liquid nitrogen, and subsequently stored at −80°C until used. Pathology from tissue used for culture and sequencing was confirmed by separate analysis of tissue sections. Tissue denoted as ‘normal’ represented histologically normal tissue acquired as part of determining surgical margin status during pancreatectomy or resected for non-malignant purposes (e.g. benign pancreatic cyst). Tissue denoted as ‘cancer’ represented histologically proven PDAC.
Tissue processing and histologic analysis
Cell line tumor xenografts or pancreas from KrasG12D/+/PTENlox/+/Pdx1-Cre mice (hereby referred to as simply ‘KrasG12D/PTENlox/+’) were sterilely harvested by clipping the mouse fur and sterilizing the flank or abdomen with a solution of 2% w/v chlorhexidine gluconate in 70% v/v isopropyl alcohol prior to resection. A representative portion of each xenograft and KrasG12D/PTENlox/+ mouse pancreas was formalin-fixed and paraffin embedded. Slides were cut at 5 μm thickness and stained with haematoxylin and eosin using routine protocols for pathologic analysis and tumor grade scoring (for pancreas only). Pancreas sections were scored by a blinded animal pathologist for pancreatic intraepithelial neoplasia (PanIN) and carcinoma grade. A total of 50 pancreatic lobules were evaluated per pancreas and scored based on the most malignant lesion present (i.e. if a lobule had PanIN 2 and PDAC it was scored as PDAC). Swiss-roll colon samples from germ-free (GF) Il10−/− experiments (see ‘Microbial Culture and Manipulation’ below) were formalin-fixed and paraffin embedded as described, and haematoxylin and eosin staining for colitis was performed. Degree of colitis was scored in a blinded fashion, as previously described (20).
Immunohistochemistry was performed on unstained xenograft slides for the immune cell marker, CD45. Tissue sections were de-paraffinized in xylene and dehydrated by a graduated ethanol series. For antigen retrieval, slides were boiled in 0.01 M sodium citrate buffer (pH 6) for 20 min. Non-specific binding was blocked by normal goat serum (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. Endogenous peroxidase activity was inhibited with 3% (v/v) hydrogen peroxide in Tris-buffered saline. Primary antibody rabbit anti-CD45 (Abcam, Cambridge, MA) was diluted (1:200) in blocking buffer (Vector Laboratories), applied to specimens, and incubated overnight at 4°C. Tissue sections then were incubated with goat anti-rabbit secondary antibody (Vector Laboratories) for 20 min at room temperature. The ABC reagent from Vectastain ABC kit (Vector Laboratories) was used to incubate slides for 30 min and subsequently the signal was detected by DAB reaction. Slides were counterstained, dehydrated with xylene, ethanol and mounted with permount. Scoring was performed in a blinded fashion for 10 random high power fields (at 40×). Photomicrographs were taken at 5× and 40× magnification with a Leica DM5500 B microscope system (Leica Microsystems Inc., Buffalo Grove, IL) running LAS X acquisition software (ver 3.3.3).
Bacterial DNA extraction and 16S rRNA sequencing
Bacterial whole genome DNA from collected stool or tissue samples was extracted using the DNeasy PowerLyzer PowerSoil Kit (Qiagen) according to manufacturer specifications. Human pancreatic tissue DNA was extracted from ~50 mg of pancreas specimen based on guidelines set by the Human Microbiome Project (protocol #07-001) utilizing the PowerMag Microbiome RNA/DNA isolation kit. The V1–V3 region hypervariable region of the 16S rRNA gene was amplified using primer pair 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′). Both the forward and the reverse primers contained universal Illumina paired-end adapter sequences, as well as unique individual 4–6 nucleotide barcodes between PCR primer sequence and the Illumina adapter sequence to allow multiplex sequencing. PCR products were visualized on an agarose gel, before samples were purified using the Agencourt AMPure XP kit (Beckman Coulter, Brea, CA) and quantified by qPCR with the KAPA Library Quantification Kit (KK4824, KAPA Biosystems). Equimolar amount of samples were then pooled and sequenced with an Illumina MiSeq (Illumina, Inc; San Diego, CA) paired end, 300 bases long, and produced 16 797 256 reads for each end. See Supplementary Methods, available at Carcinogenesis Online, for description of 16S rRNA sequencing analysis.
Tissue Culture and Maintenance
The human pancreatic cancer cell lines BxPC3 (American Type Culture Collection, ATCC, Manassas, VA) and L3.6pl were used for all in vivo xenograft animal studies in order to utilize a primary and metastatic cell line, respectively, harboring both the wild type (BxPC3) and mutant (L3.6pl) isoform of the Kras oncogene. The cell line, L3.6pl, was originally derived from liver metastases, which developed in nude mice orthotopically injected with the COLO-357 human pancreatic cancer cell line as previously described (21). Authentication of cell lines was confirmed by ATCC utilizing their STR profiling cell authentication service. Cell lines were grown in RPMI (BxPC3) or Dulbecco’s Modified Eagle Medium (L3.6pl) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were maintained in a humidified incubator at 37°C and 5% CO2 until ready for use in subcutaneous xenograft studies.
Generation and utilization of the genetically engineered mouse model of pancreatic cancer
The Institutional Animal Care and Use Committee (IACUC) at the University of Florida approved all aspects of animal care and use for all described animal experiments and national guidelines for the care and use of animals were followed. Breeding of KrasG12D/PTENlox/+ mice was performed by study personnel and animals housed in specific pathogen-free (SPF) conditions in a single dedicated room throughout the experiment. Depletion of the intestinal microbiota was with an antibiotic (Abx) cocktail created by reconstitution of streptomycin (2 g/l), gentamicin (0.5 g/l), bacitracin (1 g/l) and ciprofloxacin (0.125 g/l) in autoclaved water and provided to the animals ad libitum. Stool was routinely collected in a negative pressure hood and snap frozen for routine aerobic and anaerobic culture to confirm depletion of the intestinal microbiota (Supplementary Figure 1A, available at Carcinogenesis Online). See Supplementary Methods, available at Carcinogenesis Online, and Data for a description of the generation and utilization of the KrasG12D/+/PTENlox/+/Pdx1-Cre mice used in this study.
Animal pancreatic cancer xenograft studies
All aspects of animal care and use for these experiments were approved by the IACUC at the University of Florida (UF) and national guidelines for the care and use of animals were followed. After environmental acclimation for 1 week in SPF housing at UF, the intestinal microbiota of 4–6 week-old female NOD.CB17-Prkdcscid/J (‘Nod-SCID’) mice (The Jackson Laboratory, Bar Harbor, ME) was depleted with the same cocktail of broad spectrum antibiotics as described above. Sex-specific mice were chosen secondary to data that exist that show the murine gut microbiota can be influenced by sex hormones (22). As such, we chose for our xenograft studies to be performed in female mice as the cell lines used in the study were both originally derived from female patients. Additionally, previous analysis of the Human Microbiome Project and other studies have demonstrated that sex is not the main driver of microbial composition and habitat location (23,24). Depletion of the intestinal microbiota was confirmed prior to utilization of mice in xenograft implantation studies (Supplementary Figure 1B, available at Carcinogenesis Online). See Supplementary Methods, available at Carcinogenesis Online, for a description of the generation of PDAC heterotopic xenografts.
Intestinal inflammation, permeability assay and bacterial translocation
For barrier function experiments, 10–12-week-old GF Il10−/− (129SvEv) mice were transferred from gnotobiotic isolators to SPF housing and gavaged once with 1 × 108 cfu Campylobacter jejuni (strain 81-176) as described previously (25). Twelve days post-infection, these mice were euthanized per animal care guidelines. Four hours prior to euthanasia, 0.6 mg/g FITC-dextran (Sigma) was administered by oral gavage. Blood was collected via a terminal heart bleed into EDTA vacutainer tubes and allowed to clot at room temperature. Tubes were centrifuged at 3000 rpm (10 min at 4°C) and serum collected and stored at −80°C. The concentration of serum FITC was subsequently measured by a Synergy HTX microplate reader (Biotek, Winooski, VT) at an excitation/emission wavelength of 485/528nm. Additionally, colon and pancreas were harvested via sterile technique, as described, in a negative pressure hood. Swiss-roll of colon was formalin-fixed and paraffin embedded for histological analysis as described above. Longitudinal portions of pancreas (to encompass the head, body and tail) were snap frozen and subsequently used for bacterial culture, while the remaining portions were snap frozen for 16S bacterial PCR analysis as described. Each mouse pancreas was weighed and homogenized under anaerobic conditions using a motorized pestle in sterile PBS. Routine bacterial cultures were subsequently performed under both aerobic and anaerobic conditions.
For culture of fecal samples, fresh or frozen portions of collected mouse feces was weighed, thawed in anaerobic conditions and homogenized using a motorized pestle in sterile PBS. Samples were then plated in duplicate on brain heart infusion (BHI) or Brucella (BRU) agars (Anaerobe Systems, Morgan Hill, CA). Half of the plates were incubated for up to 72 h in anaerobic conditions, and the other half were incubated aerobically.
Statistical analysis
All non-sequence related statistical analysis (i.e. tumor growth characteristics, etc) was performed using GraphPad Prism 6 (La Jolla, CA). The Shapiro–Wilk test was utilized to determine normality within continuous datasets and compared with Student’s t-test or Mann–Whitney, where appropriate. Categorical variables were compared with Pearson’s chi-squared or Fisher’s exact test, where appropriate. A two-sided significance level of 0.05 and a 95% confidence interval were used for all statistical analyses. Methods for analysis of bacterial DNA extraction, 16S rRNA gene sequence, and RNA sequence analysis are described in Supplementary Methods, available at Carcinogenesis Online. All sequencing reads have been deposited in NCBI BioProject ID: PRJNA427203.
Results
The intestinal microbiota accelerates pancreatic carcinogenesis in the KrasG12D/PTEN−/+ mouse model of pancreatic cancer
Oral and intestinal biotas have previously been associated with pancreatic cancer but no direct relationship has been determined (17–19). In order to bridge this knowledge gap, we first sought to define the role of intestinal microbiota in pancreatic carcinogenesis and whether it can impact its progression from a distant site using the KrasG12D/PTENlox/+ mouse model of pancreatic cancer. This mouse model of PDAC develops adenocarcinoma based on the presence of a constitutively activated Kras oncogene (G12D transition mutation) with partial loss of PTEN tumor suppression within the murine pancreas (26). After weaning, the intestinal microbiota of 1 month old mice (n = 7) was depleted with an Abx cocktail and confirmed by routine stool culture (Supplementary Figure 1A, available at Carcinogenesis Online). These mice and the microbiota-intact control cohort (n = 7) were sacrificed at 3 months of age, pancreata sterilely harvested, and pathologically scored as described (Figure 1A and B). Compared to microbiota-depleted mice, the presence of microbiota resulted in a statistically significant difference in the pathologic grade between groups. Specifically, the cohort of mice with depleted microbiota had more PanIN lesions total (147 lesions) compared to microbiota-intact mice (86 lesions) and there was an apparent shift to PDAC in the microbiota-intact mice (223 total malignant lobules scored) compared to those that lacked a microbiota (161 malignant lobules; Figure 1C; P < 0.0001). Comparison of the malignant lobules between cohorts further revealed a higher proportion of grade 3 (i.e. poorly differentiated) PDAC in the microbiota-intact cohort compared to microbiota-depleted (89.7 versus 34.8%, respectively; Figure 1D; P < 0.0001). A similar decreased rate of carcinogenic progression was observed in a second independent cohort, that demonstrated the mean incidence of normal, PanIN and adenocarcinoma in microbiota-intact mice (n = 3) of 25, 41 and 33%, respectively compared to 22 79 and 0% in microbiota-depleted KrasG12D/PTENlox/+ mice (n = 4) at 3 months of age (P < 0.0001). We next assayed presence of bacteria in pancreata of KrasG12D/PTENlox/+ mice. PCR analysis using universal 16S rRNA bacterial primers of the harvested pancreas demonstrated a 2 log increase in bacterial PCR copy number in a representative set of control samples compared to the Abx-treated cohort (Figure 1E; P = 0.004). Confirmatory microbial culture performed in anaerobic and aerobic conditions demonstrated the presence of bacteria in the pancreata of control KrasG12D/PTENlox/+ mice [253887 and 8786 colony forming units (cfu)/gram of pancreatic tissue, respectively].
Figure 1.

Microbiota accelerates progression to high-grade pancreatic adenocarcinoma in the KrasG12D/PTENlox/+ mouse model. Mixed gender KrasG12D/PTENlox/+ mice were weaned at 1 month of age and the microbiota depleted with broad spectrum antibiotics for an additional 2 months [Microbiota (-)]. Mice were euthanized at 3 months of age and 50 lobules from each harvested pancreas pathologically examined for cancer grade (A, microbiota intact, representative image) and PanIN (B, microbiota depleted, representative image). Images presented at 5× magnification with inset at 40x magnification. The presence of microbiota accelerated the progression of lobules from PanIN to PDAC in the mice with microbiota present (C). Additionally, for KrasG12D/PTENlox/+ mice with cancer, there was a significant difference in PDAC grades between cohorts with grade 3 (poorly differentiated) cancer in the mice with an intact microbiota compared to microbiota depleted (D). Confirmation of microbiota depletion in antibiotic treated mice was made by bacterial PCR using universal primers (E). Dotted line in (E) indicates lower limit of detection for PCR detection, respectively (*P ≤ 0.05; #P < 0.001).
The human pancreas contains a microbiota
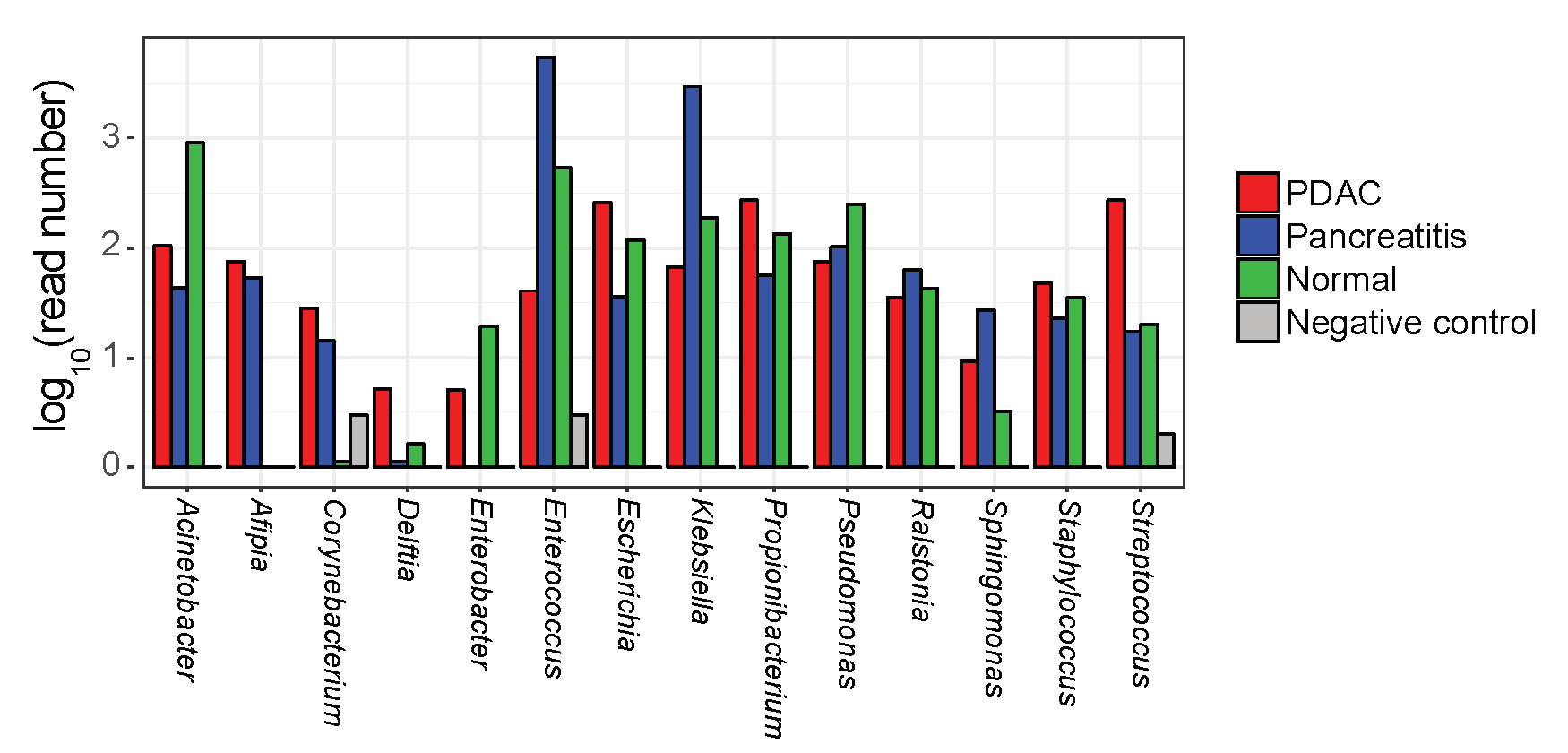
To extend upon our observation of bacteria in the pancreas of KrasG12D/PTENlox/+ mice, we performed 16S rRNA gene sequencing on bacterial genomic DNA isolated from sterile, surgically resected human pancreas samples pathologically confirmed as normal (n = 7), pancreatitis (n = 4) or PDAC (n = 16). Clinicopathologic factors for these patients are detailed in Table 1. Only samples with more than 500 reads were included for further analysis and presented, which accounts for differences in specimen numbers (see Supplementary Methods, available at Carcinogenesis Online). Genomic analysis showed that these pancreata, including non-PDAC specimens, harbor a microbiota (Figure 2A–C) that was not from external contamination (see Supplementary Methods and Figure 2, available at Carcinogenesis Online). However, based on principle coordinate analysis, there was no clustering in microbial populations at the genus level between healthy pancreas, pancreatitis, and PDAC specimens. No association between PDAC pathological stage and beta diversity or bacterial taxa (using linear discriminant analysis effect size [LEfSe]) was detected (P > 0.05, data not shown). Although there appeared to be microbial discrimination between normal and pancreatic cancer (Figure 2A), this was not significant after False Discovery Rate (FDR) correction (FDR = 0.06) for testing the first five principle coordinate analysis axes. Additionally, there was no difference in genus richness or diversity by Chao1 and Shannon indices, respectively (Figure 2D and E, Supplementary Tables 1 and 2). While several distinct genera such as Acinetobacter (P = 0.03), Afipia (P = 0.07), Enterobacter (P = 0.07) and Pseudomonas (P = 0.06) were observed (Figure 2F), no statistical link between specific genera (nor families, orders, classes or phyla) and disease state (normal versus PDAC) was identified after FDR correction (Supplementary Tables 3–7, available at Carcinogenesis Online). To confirm that bacteria colonized human pancreata, new lysates from independent malignant pancreatic specimens were cultured under aerobic and anaerobic conditions which confirmed the presence of culturable bacteria from the human pancreas with a mean of ~1 × 105 (aerobic) and ~1 × 105 (anaerobic) cfu/g of tissue after 48 h of culture. Subsequent 16S rRNA gene sequencing of these bacterial colonies and cross-reference taxonomy confirmed the presence of genera originally detected by the 16S sequencing of PDAC tissue (Supplemental Table 8, available at Carcinogenesis Online). Notably, the genera Corynebacterium, Enterobacter, Escherichia, Propionibacterium, Staphylococcus and Streptococcus were common between the sequencing sets and represented 35% of the total detected genera. Similar analysis using QIIME closed-reference OTUs generated at 100% similarity showed 23% of the OTUs are common between the two sets. In summary, the human pancreas contains a microbiota whose community composition failed to discriminate between normal and disease states.
Table 1.
Clinicopathologic factors for the patients from which 16S rRNA gene sequencing was performed on pancreas samplesa
| Total samples (n = 27) |
|
|---|---|
| Age (years) | |
| Mean (range) | 66.3 (40–84) |
| Median | 68.5 |
| SD | 10.8 |
| Sex, n (%) | |
| Male | 10 (45.4) |
| Female | 12 (54.6) |
| Pathology (n) | |
| Normal | 7 |
| Pancreatitis | 4 |
| PDAC | 16 |
| T stageb, n (%) | |
| 1 | 0 |
| 2 | 3 (18.8) |
| 3 | 13 (81.2) |
| 4 | 0 |
| N stageb, n (%) | |
| 0 | 2 (12.5) |
| 1 | 14 (87.5) |
| Overall survival (months)b | |
| Median | 11 |
| Range | 0–47 |
aOnly patients with pancreatic samples with >500 reads on 16S rRNA gene sequencing are included.
bStaging and survival data represent only patients with PDAC.
Figure 2.

The human pancreas harbors a microbiota that does not discriminate between disease states. 16S bacterial DNA sequencing from surgically resected human pancreatic specimens demonstrates no clustering or differences in bacterial populations at the genus level by principle coordinate analysis based on pathologic state of the pancreatic tissue (A–C). There was no difference in species richness on Chao1 index (D) or in species diversity on Shannon index (E). Sequencing did, however, demonstrate differences in several bacterial genera between disease states but these were not significant after FDR correction (F). Common genera between 16S sequencing of PDAC tissue and confirmatory 16S sequencing of cultured bacteria from additional independent PDAC tissue samples (Supplementary Table 8, available at Carcinogenesis Online) are underlined in (F).
Acquisition of pancreatic microbiota is not a physiologic process
To determine the temporal relationship of pancreatic microbiota acquisition, a cohort of GF 129SvEv mice were transferred to SPF conditions and orally gavaged with 1 × 105 SPF microbiota and housed for 1, 2, 4 and 8 weeks. After which, each pancreas was harvested and bacterial colonization determined by 16S rRNA PCR amplification. Whereas bacteria were detected by universal bacterial PCR in the stools of conventionally derived mice (Figure 3A), no evidence of colonization was observed in the pancreata of these mice (Figure 3B). Given that defective intestinal permeability is associated with bacterial translocation to peripheral organs (27), we next used Il10−/− mice, a model with defective intestinal permeability (28) and infected them orally with Campylobacter jejuni (strain 81–176) to accelerate this defective permeability (29), in order to determine the ability of intestinal microbes to translocate to the pancreas. The C. jejuni infected Il10−/− mice developed severe colitis (score: 3.66) compared to non-infected control Il10−/− mice (score: 0.288, P < 0.001; Figure 3C and D). In addition, C. jejuni infected Il10−/− mice showed increased intestinal permeability as measured by serum FITC-dextran assay compared to control (median serum concentration 1283 versus 167.7 ng/ml, respectively; P = 0.003; Figure 3E). Interestingly, 16S rRNA PCR analysis and culture assay failed to detect evidence of bacterial colonization in both groups (data not shown). These data suggest that acquisition of pancreatic bacteria is not a physiologic process, even under conditions of intestinal inflammation.
Figure 3.

Pancreatic microbes are not acquired from the intestine in a temporal relationship despite altered intestinal barrier function. PCR of stool (A) and pancreas (B) using universal bacterial primers demonstrated the presence of bacteria in stool of conventionally derived mice (germ-free 129SvEv mice that were transferred from gnotobiotic isolators to SPF housing conditions after oral gavage with 1 × 105 SPF bacteria) but failure of the pancreas to acquire microbes after 1, 2, 4 and 8 weeks of intestinal colonization. Electrophoresis blots have been cropped at the top and bottom for the sake of space limitations. Compared to non-gavaged controls (C), the colons of GF IL10−/− mice transferred to SPF housing and gavaged with Campylobacter jejuni (D) had increased colitis on haematoxylin and eosin (colitis score: 0.288 versus 3.66, respectively; P < 0.001). Serum FITC-dextran measurement demonstrated increased serum FITC-dextran in C. jejuni gavaged Il10−/− mice compared to non-gavaged controls, confirming increased intestinal permeability (E; **P ≤ 0.01).
Non-local effects are important in microbiota-mediated pancreatic carcinogenesis
Pancreatic tumor progression may hypothetically be influenced by both the presence of bacteria in the microenvironment of the pancreas and by intestinal microbiota exerting a long-distance effect. To dissect these contributions, we used a subcutaneous xenograft model of PDAC to circumvent any potential intrapancreatic bacterial role in pancreatic carcinogenesis and instead focus on the influence of extrapancreatic bacteria. The intestinal microbiota was depleted in Nod-SCID mice with a broad-spectrum oral Abx cocktail and confirmed by routine stool culture (Supplementary Figure 1B, available at Carcinogenesis Online). Subsequently, the pancreatic cancer cell lines BxPC3 and L3.6pl were heterotopically injected into the flank of these mice. The in vitro proliferation of these cells was independent of exposure to the Abx cocktail as there was no statistical difference in proliferation rates, as measured by reduction of Alamar Blue (Supplementary Methods and Supplementary Figure 3, available at Carcinogenesis Online). The presence of intestinal microbiota resulted in accelerated pancreatic carcinogenesis with an increased rate of successful xenograft engraftment in the BxPC3 cell line compared to the microbiota-depleted cohort (100 versus 67%, respectively; P = 0.05; Figure 4A). No difference in engraftment rates were seen with L3.6pl xenografts between the microbiota-intact and microbiota-depleted cohorts (71.4 versus 88.9%, respectively; P = 0.3; Figure 4A). Comparing the xenografts that successfully grew, an intact microbiota resulted in a statistically significant decreased mean latency time for detection [(BxPC3: 28.1 versus 50.8 days, respectively; P < 0.0001); L3.6pl: 21.2 versus 42.9 days; P < 0.0001, respectively; Figure 4B], increased tumor volume over time (BxPC3 P = 0.01–0.05; L3.6pl P = 0.01–0.03; Figure 4C), and increased rates of growth (BxPC3 P = 0.01–0.05; L3.6pl P = 0.04; Figure 4D). To confirm that xenografts were devoid of bacteria, which may explain these phenotypic differences, we performed bacterial culture and 16S rRNA PCR analysis on murine tumor samples. While the presence of bacteria was detected in harvested KrasG12D/PTEN−/+ pancreata by culture and PCR (Figure 1E), which was sensitive to Abx treatment (Supplementary Figure 1A, available at Carcinogenesis Online), there was no signal detected in xenografts (Figure 4E). This suggests that the acceleration of PDAC development in these models was secondary to the influence of the intestinal microbiota.
Figure 4.

The intestinal microbiota accelerates pancreatic carcinogenesis in a murine xenograft model. Xenografts were established in 4–6-week-old Nod-SCID mice after the depletion of the intestinal microbiota with broad spectrum antibiotics. After confirmation of microbiota depletion by routine culture methods, the human pancreatic cancer cell lines, BxPC3 and L3.6pl, were heterotopically injected into the flank of the mice. Presence of the intestinal microbiota resulted in improved engraftment efficiency (A), accelerated detection of xenograft growth (B), larger tumors (C) and increased xenograft growth rate (D). Time points of statistical significance for BxPC3 and L3.6pl are located below and above significant values, respectively (*P ≤ 0.05; #P < 0.001). Pancreatic cancer xenografts lack bacterial colonization regardless of antibiotic treatment as isolated bacterial genomic DNA from harvested pancreatic cancer xenografts using a universal 16S bacterial primer failed to detect bacteria (E). Predicted 580bp PCR product from the universal 16S bacterial primer set is indicated (M, molecular weight marker). Electrophoresis blot (E) has been cropped at the top and bottom for the sake of space limitations and controls used from the same PCR reaction but unable to be ran on the same gel are cropped into the right panel of (E).
In summary, these data demonstrate that intestinal microbes have the capacity to influence pancreatic carcinogenesis in an extra-intestinal, long-distance manner that is independent of a pancreatic microbiota or the pancreatic stromal microenvironment.
The intestinal microbiota influences human and murine transcriptomic changes in pancreatic cancer xenografts
To determine the intratumoral transcriptomic changes that occur in response to the microbiota, Illumina HiSeq 3000 RNA sequencing (RNAseq) of sterilely harvested BxPC3 xenografts was performed. In order to interrogate both the human tumoral component (from the BxPC3 cells) and the murine component (from tumor infiltrating murine cells), the trimmed and filtered reads were aligned to independent human and murine reference sequences (see Supplementary Methods, available at Carcinogenesis Online). This demonstrated numerous differentially expressed genes in both the human and mouse subsets, known to be involved in carcinogenesis, depending on the state of the intestinal microbiota Supplementary Figure 4, available at Carcinogenesis Online). For example, the presence of the intestinal microbiota resulted in the upregulation of human pro-carcinogenic genes such as TNC (6.9 log2 fold, P = 0.008), PLXNA4 (3.4 log2 fold, P = 0.008), and CXCL10 (2.5 log2 fold, P = 0.008). Conversely, microbiota depletion resulted in the upregulation of many pathways associated with patient survival and tumor suppression such as DAPK2 (2.35 log2 fold, P = 0.008), KLF9 (2.32 log2 fold, P = 0.008) and LUM (log2 fold 1.8, P = 0.008). Differentially expressed murine genes in the presence of the microbiota included the upregulation of pro-carcinogenic genes involved in progression and immune surveillance such as ST14 (2.35 log2 fold, P = 0.04), CCR2 (1.8 log2 fold, P = 0.004), and FABP4 (1.6 log2 fold, P = 0.04). Conversely, microbiota depletion resulted in the upregulation of many anti-cancer pathways such as WIF1 (3 log2 fold, P = 0.004), SCARA5 (2.6 log2 fold, P = 0.004) and CDKN1C (1.8 log2 fold, P = 0.03). While it may be argued that the administration of antibiotics may alter the tumoral transcriptome, we compared our reads to a previously published sequencing database of antibiotic induced genes (30) and determined that the differentially expressed genes within our datasets are not directly inducible by antibiotics (data not shown).
Innate immune cells infiltrate pancreatic cancer xenografts in microbiota depleted mice
Given the similar phenotypes between our immunocompetent KrasG12D/PTENlox/+ murine model and the immunodeficient Nod-SCID xenograft model, as well as our RNAseq data and prior literature implicating immune cells in tumor surveillance (31), if immune cells played a role in the observed phenotype we hypothesized that it would be via the innate system. We therefore sought to evaluate the presence of innate immune cell infiltration in the PDAC xenografts. Using immunohistochemistry for CD45, our data demonstrate a statistically significant increase in CD45 positive cells within the PDAC xenografts of microbiota depleted mice (P = 0.009; Figure 5). This suggests that the observed phenotype is associated with microbiota-mediated innate immune system suppression.
Figure 5.

Intact intestinal microbiota associates with decreased CD45 immune cell infiltration in pancreatic cancer xenografts. Immunohistochemistry for CD45 was performed on PDAC xenografts from mice with an intact (A, representative image) or antibiotic depleted (B, rep resentative image) intestinal microbiota. Images presented at 5× magnification with inset at 40× magnification. CD45 positive cells (denoted by arrows) were scored in 10 random high-powered fields (40× ) for each xenograft. There was a statistically significant decrease (C, P = 0.009) in the CD45+ cell infiltration in xenografts derived from microbiota-intact mice (*P ≤ 0.05), suggesting innate immune cell suppression.
Discussion
Pancreatic ductal adenocarcinoma is a deadly disease with a death rate that has remained stable for several decades (8). Paradigm shifting research is necessary to accelerate the prevention, detection and treatment of this aggressive malignancy. In this study, we identified an acceleration of the natural progression of PanIN to PDAC in the KrasG12D/PTENlox/+ murine model of PDAC; there was a greater incidence of cancer formation, particularly poorly differentiated cancers, when a microbiota was present. Our observation of bacteria within the pancreata of microbiota-intact KrasG12D/PTENlox/+ mice was extended to human samples in which it was noted that the human pancreas possesses a microbiota but it does not appear to differentiate between normal and pancreatic cancer after FDR correction. Utilizing germ-free mice, this pancreatic microbiota does not appear to form under normal physiologic conditions, even in the presence of defective intestinal membrane function. Finally, we identified intestinal biota as an important mediator of pancreatic cancer progression since increased PDAC xenograft tumorigenicity was observed in microbiota-intact mice compared to microbiota-depleted mice, which may be secondary to regulation of intratumoral cancer pathways or innate immune suppression by the microbiota.
While other non-intestinal tissue sites such as the breast (32), respiratory tract (33), and skin (34) have been shown to possess a microbiota that associate with tissue-specific malignancies, the pancreatic microbiota was not correlated with carcinogenesis in our limited patient dataset in contrast to a recent report (35). Nevertheless, microbiota analysis using a larger cohort will be needed to make a definitive conclusion regarding the relationship between bacteria and carcinogenesis in the pancreas. Despite the lack of correlation with PDAC in our dataset, the presence of a pancreatic microbiota in PDAC patients aligned well with two recent reports showing presence of bacteria in patients with pancreatic cancer (35,36). Interestingly, the report by Geller and colleagues showed low level bacteria in normal pancreatic specimens harvested from organ donors as measured by qPCR 16S PCR. This contrasts with our data where a pancreatic microbiota was detected by bacterial sequencing in all tested specimens (normal, pancreatitis and PDAC) but similar to Pushalkar and colleagues who identified bacteria within their PDAC and normal specimens (35). However, while we did not note discrimination between sample sets based on the state of the pancreas, the report by Pushalkar identified microbial differences between normal and PDAC samples but did not incorporate FDR correction and no information was given regarding the source of their normal pancreatic specimens. The reason for the discrepancies between studies is unclear but may be related to FDR correction, technique (qPCR versus 16S rRNA sequencing), or tissue source, given that our use of non-PDAC patient samples, versus tissue from healthy organ donors, may nevertheless possess a ‘field defect’ that allows entry of particular microbes. Nevertheless, strong similarity in the human PDAC microbiota was observed between our study and the studies by Geller and Pushalkar (35,36). Namely, bacterial genera Acinetobacter, Enterobacter, Pseudomonas, Delftia, Enterococcus, Streptococcus, Corynebacterium, Propionibacterium, Klebsiella, Sphingomonas and Staphylococcus were observed in our study and Geller with both studies identifying Klebsiella as disproportionally overrepresented in cancer versus normal specimens. In regards to the recent report by Pushalkar, their most abundant phyla in human PDAC samples were Proteobacteria (45%), Bacteroidetes (31%) and Firmicutes (22%), which was similar to our observation of Proteobacteria (45%) and Firmicutes (33%) while Bacteroidetes only had 3% abundance in our PDAC samples. The concordance between studies for the phyla Proteobacteria and Firmicutes presents a potential future area of investigation.
The impact of bacteria within the human pancreas and how they become established in this organ, is currently unknown. As the pancreas has direct access to the upper gastrointestinal tract via the pancreatic duct, it would be reasonable to surmise that colonization occurs as a result of bacterial reflux from the duodenum or biliary tree, as implicated in the recent study by Pushalkar et al. (35). However, the argument that the oral microbiota is the responsible flora for a potential role in PDAC development fails to take into account various studies that have shown differences in the stool microbiota between normal and malignant pancreas and the potential implication of this (19,35,37). While Pushalkar implicates the oral microbiota, its presence in the pancreas was limited to the need for a high concentration oral gavage of Enterococcus faecalis and Escherichia coli, species both native to the colon not oral cavity, and detected soon after gavage with a decrease over the ensuing 72 h. Additionally, based on our germ-free data, it appears that colonization of the pancreas via either the upper or lower gastrointestinal tract may not be the ‘default setting’ of this organ as bacteria were not detected in the pancreata of ex-GF mice transferred to SPF housing, nor the pancreas of Il10−/− mice with compromised intestinal permeability. However, bacteria were detected in the pancreas of KrasG12D/PTENlox/+ mice, a phenomenon also reported by Pushalkar et al. (35) using a similar GEMM of PDAC. It may be hypothesized that pancreatic pathology (either pancreatitis, PanIN formation or PDAC), in conjunction with increased bacterial translocation (oral, intestinal), represent key conditions for intra-pancreatic bacterial colonization. Additionally, a long-distance connection between pancreas-derived antimicrobial peptide and intestinal microbiota composition and function has been previously reported (38). One could speculate that a bilateral communication takes place between the pancreas and intestine that influence microbial seeding. At present, no studies demonstrate changes in the oral microbiota in function to PDAC development, and the field is limited to potential bacterial markers. Further studies will therefore be needed to decipher the mechanisms responsible for pancreas bacterial colonization and whether it originates from an oral or intestinal source.
Despite our documentation of a pancreatic microbiota, our study clearly identifies a long-distance modulation of pancreatic cancer progression by intestinal biota in both a GEMM and a subcutaneous xenograft model of pancreatic cancer. The lack of intra-tumor bacteria in the xenograft models, yet dramatic effect of antibiotic-mediated microbial depletion on tumor growth, clearly identifies intestinal biota as a key modulator of carcinogenesis. This may be related to the ability of the microbiota to alter pro- and anti-carcinogenic pathways with upregulation of known oncogenic genes such as tenascin C (39), plexin A4 (40) and CXCL10 (41) in the microbiota-intact mice versus the upregulation of tumor suppressor genes such as DAPK2 (42), lumican (43) and Wif1 (44) in the microbiota-depleted mice as demonstrated by RNAseq. Additionally, the decreased CD45 intratumoral immune cell infiltration in the microbiota-depleted Nod-SCID mice suggests that the microbiota suppresses innate immune cell infiltration as in other cancers (31) and in agreement with the study by Pushalkar et al. (35). However, a recent report suggests that the microbiota-mediated tumor response is secondary to adaptive immune system regulation (45). These studies underscore the critical need to interrogate these immune pathways and their interaction with the microbiota and pancreatic carcinogenesis. Differences in mutational burden between BxPC3 (Kras wildtype) versus L3.6pl (Kras mutant) may explain differences in engraftment rates between the cell lines regardless of intestinal microbiota status. Furthermore, recent data (35) corroborate our findings in that the effect of the microbiota appears to be independent of Kras mutational status. However, given that our xenograft and genetically engineered mice were not models of cancer initiation, as both possessed requisite genetic mutations known to cause PDAC, the acceleration of pancreatic carcinogenesis that we identified in the presence of microbiota, and lack of differentiation in human specimens based on 16S sequencing, suggests that either elements of the intestinal microbiota, or their metabolic products, accelerate pancreatic carcinogenesis in a remote fashion. For example, the intestinal microbiota has been shown to remotely impact other processes including atherosclerosis development and bone marrow immune function (46–48). Furthermore, the precedence of the intestinal microbiota influencing cancer development in a remote fashion has already been established (49,50) and such an interaction between host microbiota and cancer acceleration is further supported by our data. Whether the host microbiota can induce genetic aberrations that result in PDAC initiation is an area yet to be explored. Given that our model presented a system of intact versus depleted intestinal microbiota, with a distinct phenotype between the two, further studies must aim to decipher the direct causal species responsible for the observed phenotypes through more specific microbial manipulations.
Our study begins to elucidate a direct role for the microbiota in pancreatic cancer progression, beyond simple associations of microbial and disease presence. We report that normal and malignant pancreatic tissue harbor a microbiota but while this microbiota was not able to differentiate between disease states, the presence of intestinal microbes accelerated PDAC progression utilizing both a transgenic and xenograft mouse model of pancreatic cancer. This suggests that the host microbiota can have an impact on pancreatic carcinogenesis from a distant (i.e. non-pancreatic) site and not intrapancreatic as illustrated in a recent study (35). Furthermore, in contrast to this recent study, our observations were maintained in both a GEMM and a heterotopic mouse model of PDAC (as opposed to a orthotopic pancreatic model). Additionally, the lack of bacteria within the PDAC xenografts, even in microbiota-intact mice, supports a long-distance effect of the microbiota on pancreatic carcinogenesis. Intrapancreatic bacteria may therefore be more relevant to therapeutic response than cancer progression (36). These data open additional areas of investigation into the mechanisms of pancreatic carcinogenesis. Identification and confirmation of specific microbes and their potential metabolites that not only accelerate PDAC development, but also facilitate their translocation to the pancreas will open new avenues into the field of pancreatic cancer research.
Funding
This research was supported by the American Cancer Society - Norma and Rich DiMarco Mentored Research Scholar Grant (MRSG-17-228-01-TBG; RT), University of Florida, Department of Surgery (RT), and University of Florida Health Cancer Center Pilot Project Grant (RT, RZG, and CJ), Gatorade Trust through funds distributed by the University of Florida, Department of Medicine (MZK and CJ), and the University of Florida Health Cancer Center (RZG). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of Interest Statement: None declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abbreviations
- GEMM
genetically engineered mouse model
- PDAC
pancreatic ductal adenocarcinoma
- PanIN
pancreatic intraepithelial neoplasia
- SPF
specific pathogen-free
References
- 1. Hruban R.H., et al. (2000)Progression model for pancreatic cancer. Clin. Cancer Res., 6, 2969–2972. [PubMed] [Google Scholar]
- 2. Iacobuzio-Donahue C.A., et al. (2012)Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin. Cancer Res., 18, 4257–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey P., et al. ; Pancreatic Cancer Genome Initiative. (2016)Genomic analyses identify molecular subtypes of pancreatic cancer. Nature, 531, 47–52. [DOI] [PubMed] [Google Scholar]
- 4. Brat D.J., et al. (1998)Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol., 22, 163–169. [DOI] [PubMed] [Google Scholar]
- 5. Hruban R.H., et al. (2001)Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol., 25, 579–586. [DOI] [PubMed] [Google Scholar]
- 6. Zheng L., et al. (2013)Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology, 144, 1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson B.A. 3rd, et al. (2017)Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res., 23, 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. American Cancer Society. (2017). Cancer Facts & Figures 2017. American Cancer Society, Atlanta. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and- figures/2017/cancer-facts-and-figures-2017.pdf [Google Scholar]
- 9. Bosetti C., et al. (2012)Cigarette smoking and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol., 23, 1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duell E.J., et al. (2012)Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann. Oncol., 23, 2964–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aune D., et al. (2012)Body mass index, abdominal fatness and pancreatic cancer risk: a systematic review and non-linear dose-response meta-analysis of prospective studies. Ann. Oncol., 23, 843–852. [DOI] [PubMed] [Google Scholar]
- 12. Giardiello F.M., et al. (2000)Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology, 119, 1447–1453. [DOI] [PubMed] [Google Scholar]
- 13. Rebours V., et al. (2008)Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am. J. Gastroenterol., 103, 111–119. [DOI] [PubMed] [Google Scholar]
- 14. Huttenhower C., et al. (2014)Inflammatory bowel disease as a model for translating the microbiome. Immunity, 40, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iida N., et al. (2013)Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science, 342, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kostic A.D., et al. (2014)The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology, 146, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Michaud D.S., et al. (2013)Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut, 62, 1764–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fan X., et al. (2018)Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut, 67, 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ren Z., et al. (2017)Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget, 8, 95176–95191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rath H.C., et al. (1996)Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest., 98, 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruns C.J., et al. (1999)In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia, 1, 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Org E., et al. (2016)Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes, 7, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Human Microbiome Project, C. (2012)Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kovacs A., et al. (2011)Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb. Ecol., 61, 423–428. [DOI] [PubMed] [Google Scholar]
- 25. Lippert E., et al. (2009)Gnotobiotic IL-10; NF-kappaB mice develop rapid and severe colitis following Campylobacter jejuni infection. PLoS One, 4, e7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill R., et al. (2010)PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res., 70, 7114–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bischoff S.C., et al. (2014)Intestinal permeability–a new target for disease prevention and therapy. BMC Gastroenterol., 14, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madsen K.L., et al. (1999)Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis., 5, 262–270. [DOI] [PubMed] [Google Scholar]
- 29. Sun X., et al. (2012)Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology, 142, 86–95.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morgun A., et al. (2015)Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut., 64, 1732–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corrales L., et al. (2017)Innate immune signaling and regulation in cancer immunotherapy. Cell Res., 27, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hieken T.J., et al. (2016)The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep., 6, 30751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu G., et al. (2016)Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol., 17, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grice E.A., et al. (2011)The skin microbiome. Nat. Rev. Microbiol., 9, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pushalkar S., et al. (2018)The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov., 8, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geller L.T., et al. (2017)Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science, 357, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panebianco C., et al. (2017)Engineered resistant-starch (ERS) diet shapes colon microbiota profile in parallel with the retardation of tumor growth in in vitro and in vivo pancreatic cancer models. Nutrients, 9, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahuja M., et al. (2017)Orai1-mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab., 25, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cai J., et al. (2017)Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget, 8, 74406–74422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Müller M.W., et al. (2007)Association of axon guidance factor semaphorin 3A with poor outcome in pancreatic cancer. Int. J. Cancer, 121, 2421–2433. [DOI] [PubMed] [Google Scholar]
- 41. Delitto D., et al. (2015)Downstream mediators of the intratumoral interferon response suppress antitumor immunity, induce gemcitabine resistance and associate with poor survival in human pancreatic cancer. Cancer Immunol. Immunother., 64, 1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dansranjavin T., et al. (2006)E-cadherin and DAP kinase in pancreatic adenocarcinoma and corresponding lymph node metastases. Oncol. Rep., 15, 1125–1131. [PubMed] [Google Scholar]
- 43. Li X., et al. (2014)Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin. Cancer Res., 20, 6529–6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taniguchi H., et al. (2005)Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene, 24, 7946–7952. [DOI] [PubMed] [Google Scholar]
- 45. Sethi V., et al. (2018)Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology, Apr 7, pii:S0016-5085(18)30411-6. doi:10.1053/j.gastro.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karlsson F.H., et al. (2012)Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun., 3, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tang W.H., et al. (2014)The contributory role of gut microbiota in cardiovascular disease. J. Clin. Invest., 124, 4204–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berer K., et al. (2011)Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature, 479, 538–541. [DOI] [PubMed] [Google Scholar]
- 49. Rao V.P., et al. (2006)Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res., 66, 7395–7400. [DOI] [PubMed] [Google Scholar]
- 50. Lakritz J.R., et al. (2015)Gut bacteria require neutrophils to promote mammary tumorigenesis. Oncotarget, 6, 9387–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.