

Abstract
Mucopolysaccharidosis IIIB (MPS IIIB) is an autosomal recessive lysosomal storage disorder. In comparison to Hurler syndrome (MPS I) and Hunter syndrome (MPS II), characteristic facial and physical features tend to be milder and progression of neurological symptoms may initially be slower. Obvious neurological and behavioural symptoms may not appear until age 2–6 years, but once they begin, progression is relentless, leading to death by the early 20s. Although there is currently no known cure for MPS IIIB, enzyme replacement clinical trials are showing hope for delay in the progression of symptoms. Early diagnosis is therefore necessary before neurological symptoms have progressed. In our case, MPS IIIB was diagnosed at an early age because recurrent wheezing and otitis media in conjunction with hepatomegaly were recognised as more than trivial findings. A thorough examination and a definitive proactive decision to perform a liver biopsy resulted in early diagnosis of a rare disease.
Keywords: liver disease, paediatrics, congenital disorders, asthma, genetics
Background
Mucopolysaccharidosis III (MPS III or Sanfilippo syndrome) is an autosomal recessive lysosomal storage disorder that was first reported in 1963 in connection with excessive urinary excretion of glycosaminoglycans (GAG).1 Based on the specific enzyme deficiency, MPS III comprises four different biochemical subtypes (A, B, C and D).2 The four cannot be differentiated clinically, however. The worldwide prevalence rate of MPS ranges from 0.28 to 4.1 per 100 000 people. The frequency of subtypes is greatest in type A, followed by type B and type C. Type D was discovered in 1980 and appears to be the least common, though there are few estimates of its prevalence. In Okinawa prefecture, type B is the most common.3 MPS III type B (MPS IIIB) is caused by a deficiency of α-N-acetylglucosaminidase, which plays a role in the catabolism of heparan sulfate. This enzyme deficiency leads to accumulation of heparan sulfate in lysosomes,4 causing severe central nervous system degeneration.5 Subtle dysmorphisms may be present, but skeletal and visceral manifestations are uncommon. There are three phases of the natural history of MPS. The first neurological symptoms are mild and appear between the ages of 1 and 3 years. In the second phase, at approximately 3–6 years, behaviour disorders, intellectual deterioration and sleep disorders are followed by aggressiveness and hyperactivity. The third stage usually begins in the early teenage years. Psychomotor skills decline and the ability to speak is lost. The disease progresses relentlessly and leads to a vegetative state.5 This disproportionate involvement of the Central Nervous System (CNS) is unique to MPS III. The prognosis is poor with death occurring in most cases by the late teens or early 20s. There are currently no known cures; however, clinical trials to replace deficient enzymes are beginning and gene therapy for MPS III is launching.6 Delays in diagnosis of MPS III are common due to the subtle physical characteristics and the slow progression of the neurological disease.7 It is important to diagnose this disease at the earliest stages in order to maximise the effect of potential new treatments such as enzyme replacement therapies. Early diagnosis can also help direct genetic consultation for parents who wish to know more about their reproductive options, including prenatal screening and diagnosis.
Case presentation
A 2 and 6/12-year-old Japanese boy presented to our hospital secondary to recurrent episodes of wheezing and otitis media. He was the product of an uncomplicated pregnancy, delivered by normal vaginal delivery at 39 weeks of gestation. Birth weight was 3055 g and 5 min Apgar score was 10. He was the first child born to non-consanguineous healthy parents. His physical growth was normal and his developmental quotient (DQ) of 84.5 was at the borderline of the mild delay/normal range (DQ 71–84=mild/moderate delay, ≥85=normal). He walked and uttered single words at age 1.6 years and spoke in sentences at age 2.5 years. Frequent episodes of wheeze and otitis media started at age 8 months. On his fourth hospitalisation for an asthma-like attack, he was noted to have hepatomegaly and was referred to a paediatric gastroenterologist.
Investigations
The physical examination revealed relatively coarse facial features, including a bulging forehead, flat nasal bridge and relatively large lips (figure 1), valgus elbows (excess carrying angle), extensive Mongolian spots (figure 2) and hepatomegaly (the liver edge could be felt 8 cm below the right costal margin). Ophthalmic disease and congenital heart disease were not recognised. Thoracic X-ray showed wedge-shaped vertebral bodies (figure 3). Brain MRI showed relatively large lateral ventricles (figure 4). Abdominal ultrasonography showed enlarged liver size with hyperechogenic hepatic parenchyma. Liver biopsy specimens showed substantial colloidal iron staining of the hepatocytes and Kupffer cells (figure 5). Lucent lysosomes with reticulo-particulate content were also seen on liver biopsy (×7000, electron micrograph) (figure 6). In addition, granular histiocytes (Buhot cells) were seen in the bone marrow (figure 7). Serum biochemical and haematological investigations were within normal limits. Urine uronic acid was elevated at 181 mg/g creatinine (normal range 29.7±13.3), and thin-layer chromatographic analysis of urinary GAG demonstrated accumulation of heparan sulfate (42%). The α-N-acetylglucosaminidase level for leucocytes was <0.6 nmol/mg protein/17 hours (normal range 11.6–28.2). Together with our genetic specialist, we proposed molecular genetic testing to the parents, but they declined this testing for their child.
Figure 1.
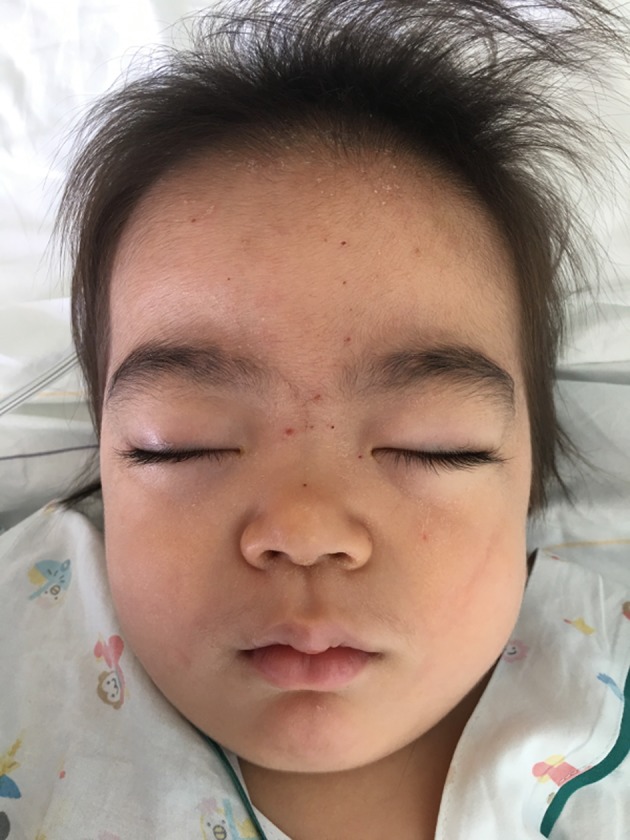
Characteristic facial appearance, showing coarse face and hirsutism.
Figure 2.
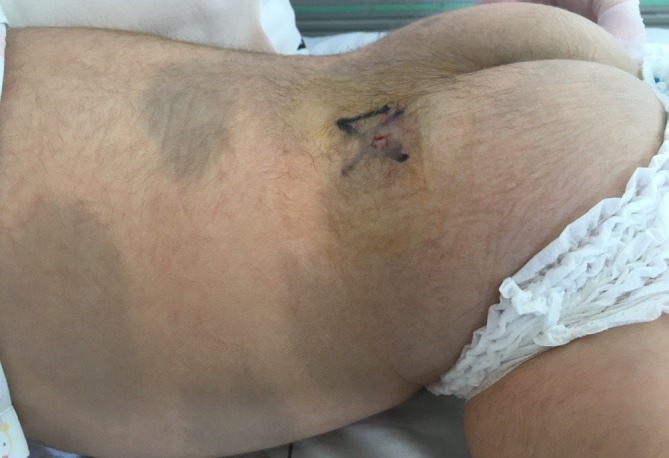
Extensive Mongolian spots on his back.
Figure 3.
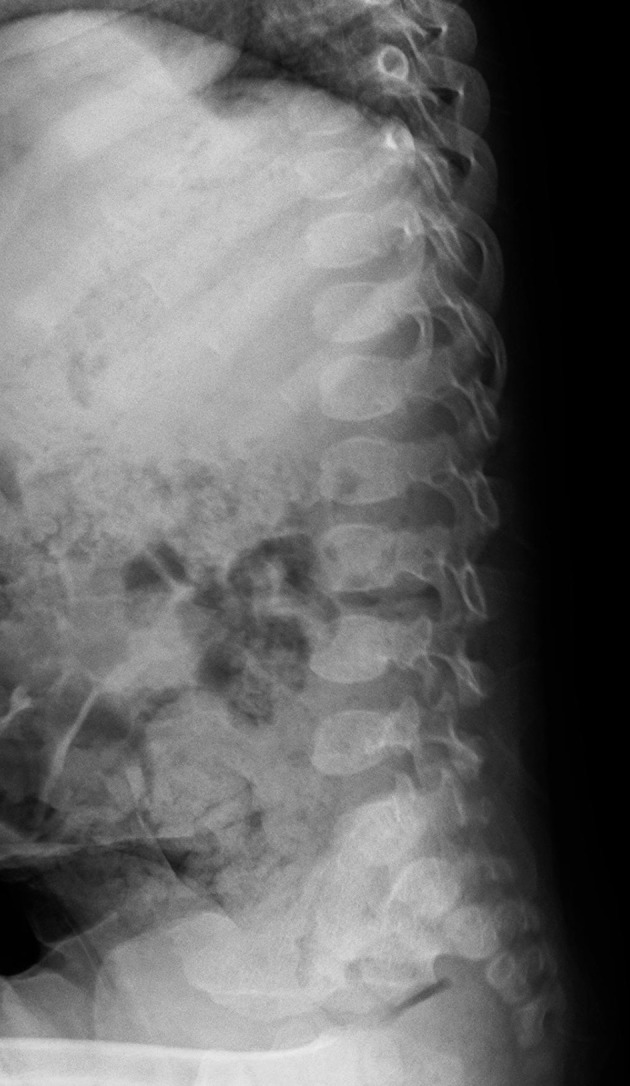
Lumber spine, lateral view, at 2 years of age, showing rounding of the vertebral bodies.
Figure 4.
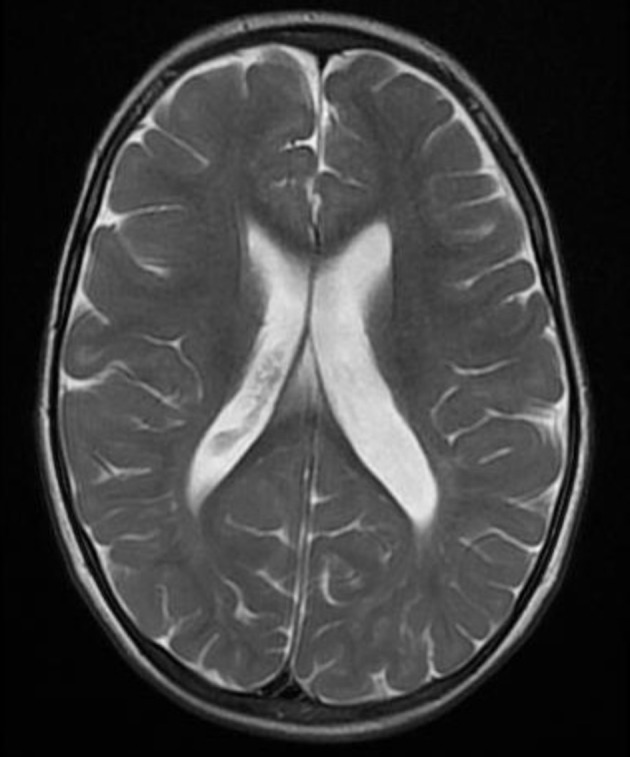
T2-weighted MRI shows enlarged lateral ventricles.
Figure 5.
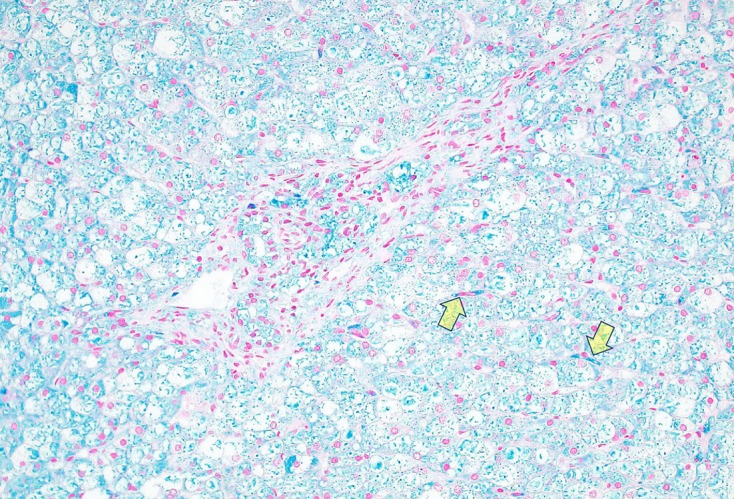
Stained Kupffer cells (arrowheads) and acid mucopolysaccharide in hepatocytes stained blue with colloidal iron stain (×200, colloidal iron staining).
Figure 6.
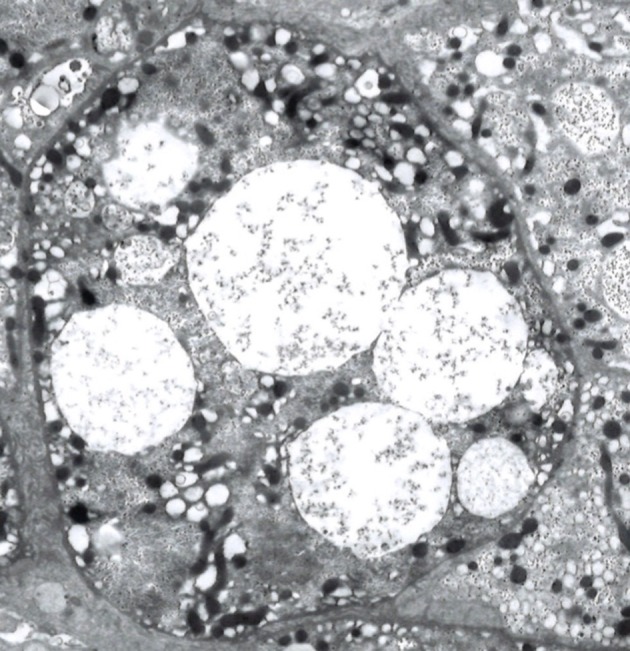
Clear or lucent lysosomes with reticulo-particulate contents in a hepatocyte (×3000, electron micrograph).
Figure 7.

A granular histiocyte (Buhot cell) present in the bone marrow (×1000, HE staining).
Outcome and follow-up
During the hospitalisation, the patient underwent polysomnography and was diagnosed with adenoidal enlargement and sleep apnoea syndrome. He was scheduled to undergo adenoidectomy with a further plan for follow-up of the comorbidities (recurrent otitis media and wheezing) and for ongoing assessment of his growth and development.
Discussion
Delays in diagnosis of MPS III are common, due in large part to the mild physical features when the children are young. In this case, the combination of recurrent wheezing episodes, recurrent otitis media and hepatomegaly were recognised as findings suggestive of MPS III. Liver biopsy played a major role in the definitive diagnosis.
Recurrent wheezing symptoms are common in patients with mucopolysaccharidoses.8 Despite this, lysosomal storage diseases are not listed in textbooks as part of the differential diagnosis for wheezing.9 Therefore, these patients are often initially diagnosed with just bronchial asthma and/or chronic otitis media. The airway problems are secondary to cranial and spinal abnormalities (eg, flattened nasal bridge, short neck, high epiglottis, mandibular abnormalities and abnormal cervical vertebrae) and GAG deposition in the mouth, nose and throat.8 The management of recurrent wheezing episodes in MPS IIIB is challenging. Inhaled steroids may be effective in decreasing airway inflammation and patients may respond better to anticholinergic bronchodilator agents than to beta-agonists.10
Our patient had hepatomegaly. Liver enlargement has a variety of causes, such as inflammation, infiltration of cells, malignant tumours, increased size of the vascular space, increased size of the biliary space, idiopathic aetiologies and storage diseases.11 Only about half of patients with MPS III display hepatomegaly.12 In our patient, liver transaminases and inflammatory markers (white blood cell count and C-reactive protein (CRP)) were not elevated. Ultrasonography showed hyperechogenic hepatic parenchyma, which can be seen with metabolic diseases (some storage diseases) or fatty liver (obesity, malnutrition, hyperalimentation or corticosteroids).7 Our patient underwent liver biopsy and bone marrow biopsy which, as anticipated, revealed accumulation of GAG. This leads to the diagnosis of MPS. Clear or lucent lysosomes with reticulo-particulate content are of limited diagnostic value, but are helpful findings to rule in MPS.13 To differentiate from other lysosomal storage diseases, we performed a bone marrow biopsy. Gaucher cells in Gaucher’s disease and foamy cells in Niemann-Pick disease are well known as pathological findings in the bone marrow.14 In our case, Buhot cells were also seen in the bone marrow (figure 7), indicating an excess of heparan sulfate.15 MPS IIIB was ultimately diagnosed by the extremely low level of α-N-acetylglucosaminidase.
Early facial and physical abnormalities in MPS III tend to be subtle and progression of neurological symptoms is initially slow. From the time of early school age, however, the neurological and behavioural symptoms progress aggressively, leading to death by the early twenties. There is currently no definitive treatment for MPS III. Intrathecal enzyme replacement treatments (ERT) for MPS I, MPS II and MPS III had initially been felt to show promise, but these treatments have not been effective and their use has been discontinued.16 Intracranial ERT for MPS IIIB is currently being studied, but it is unclear at this point if it will be effective. Gene therapy clinical trials for MPS III using different administration methods (intravenous, intracranial and intraventricular) are currently underway with results not yet published.17 For these treatments to be effective, early diagnosis is necessary before irreversible neurological symptoms have progressed. Because this disease cannot be identified early by physical examination or general blood tests, neonatal screening has been proposed. This is still in the pilot study stage in Japan. In our patient, we were able to diagnose MPS IIIB at an early age by proactively performing a liver biopsy and bone marrow biopsy. Generally, abnormal X-ray findings such as those in figure 3 can lead to early diagnosis of MPS. However, it may be difficult for some radiologists to recognise the abnormalities, especially those who are not specialists for rare conditions like lysosomal storage diseases. Therefore, in order to diagnose rare diseases, it is essential not to overlook physical findings that may seem trivial and to conduct definitive examinations and procedures in situations where they are called for.
Learning points.
Frequent episodes of wheeze and otitis media, when coupled with hepatomegaly and developmental delay, can be initial indicators of lysosomal storage diseases.
If there is unexplained hepatomegaly, liver biopsy and bone marrow biopsy should be considered to find lysosomal storage diseases.
If we consider a rare disease, it is necessary not to overlook trivial abnormal physical findings and to definitively conduct diagnostic examinations and procedures if indicated.
Footnotes
Contributors: TY conceptualised and designed the study, collected data, carried out the initial analyses, drafted the initial manuscript and reviewed and revised the manuscript. TLH conceptualised and designed the study, supervised data collection and critically reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Parental/guardian consent obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Sanfilippo SJ, Podosin R, Langer L, et al. Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J Pediatr 1963;63:837–8. 10.1016/S0022-3476(63)80279-6 [DOI] [Google Scholar]
- 2.Valstar MJ, Ruijter GJ, van Diggelen OP, et al. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008;31:240–52. 10.1007/s10545-008-0838-5 [DOI] [PubMed] [Google Scholar]
- 3.Tanaka A, Kimura M, Lan HT, et al. Molecular analysis of the alpha-N-acetylglucosaminidase gene in seven Japanese patients from six unrelated families with mucopolysaccharidosis IIIB (Sanfilippo type B), including two novel mutations. J Hum Genet 2002;47:484–7. 10.1007/s100380200070 [DOI] [PubMed] [Google Scholar]
- 4.Chinen Y, Tohma T, Izumikawa Y, et al. Sanfilippo type B syndrome: five patients with an R565P homozygous mutation in the alpha-N-acetylglucosaminidase gene from the Okinawa islands in Japan. J Hum Genet 2005;50:357–9. 10.1007/s10038-005-0258-4 [DOI] [PubMed] [Google Scholar]
- 5.Chuang SC, Hwu WL, Wu CC, et al. Diagnosis of mucopolysaccharidosis type IIIB. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 1996;37:320–3. [PubMed] [Google Scholar]
- 6.BioMarin Presents Interim Data of Phase 1/2 Study of BMN 250 for Treatment of Sanfilippo B Syndrome (MPS IIIB) at 13th International Congress of Inborn Errors of Metabolism (ICIEM). 2017. http://investors.biomarin.com/2017-09-06-BioMarin-Presents-Interim-Data-of-Phase-1-2-Study-of-BMN-250-for-Treatment-of-Sanfilippo-B-Syndrome-MPS-IIIB-at-13th-International-Congress-of-Inborn-Errors-of-Metabolism-ICIEM-2017 (accessed 28 Dec 2017).
- 7.Spranger J, et al. Mucopolysaccharidoses : Kliegman RM, Stanton BF, Schor NF, Nelson textbook of Pediatrics. 19th edn Philadelphia: Saunders, 2011:512–4. [Google Scholar]
- 8.Berger KI, Fagondes SC, Giugliani R, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis 2013;36:201–10. 10.1007/s10545-012-9555-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Federico MJ. Wheeze : Bajaj L, Hambidge SJ, Kerby G, Berman’s pediatric decision making. 5th edn Philadelphia: Mosby, 2011:410–2. [Google Scholar]
- 10.Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev 2011;12:133–8. 10.1016/j.prrv.2010.10.005 [DOI] [PubMed] [Google Scholar]
- 11.Boamah LM, Balistreri WF. Manifestations of liver disease : Kliegman RM, Stanton BF, Schor NF, et alNelson textbook of Pediatrics. 19th edn Philadelphia: Saunders, 2011:1374–5. [Google Scholar]
- 12.Wijburg FA, Węgrzyn G, Burton BK, et al. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr 2013;102:462–70. 10.1111/apa.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghadially FN. Lysosomes in mucopolysaccharidoses In: Ghadially FN, ed Ultrastructural pathology of the cell and matrix, fourth edn. Oxford;Butterworth-Heinemann, 1997:748–9. [Google Scholar]
- 14.Burrow TA, Grabowski GA. Lysosomal storage disease : Sokol RJ, Balistreri WF, Liver disease in children. fourth edn Cambridge: Cambridge University press, 2014:546–66. [Google Scholar]
- 15.Garcia-Riego A, Prats JM, Zarranz JJ. Sanfilippo disease: Buhot cells in the cerebrospinal fluid. Acta Cytol 1978;22:282–4. [PubMed] [Google Scholar]
- 16.Shire announces top-line results for phase ii/iii clinical trial in children with hunter syndrome and cognitive impairment. https://globenewswire.com/news-release/2017/12/19/1266050/0/en/Shire-Announces-Top-Line-Results-for-Phase-II-III-Clinical-Trial-in-Children-with-Hunter-Syndrome-and-Cognitive-Impairment.html (accessed 30 Apr 2018).
- 17.Abeona therapeutics reports top-line data from phase 1/2 gene therapy trial in MPS IIIA. https://globenewswire.com/news-release/2018/02/08/1336416/0/en/Abeona-Therapeutics-Reports-Top-Line-Data-from-Phase-1-2-Gene-Therapy-Trial-in-MPS-IIIA.html (accessed 30 Apr 2018).