

Abstract
Trimethoprim (TMP) has been widely used since the 1960s, both alone and in combination with sulfamethoxazole. Unfortunately, information regarding the role that cytochrome P450 enzymes (P450s) play in the formation of TMP primary metabolites is scarce. Hence, we undertook in vitro studies to identify and more fully characterize the P450s that catalyze formation of six TMP primary metabolites: TMP 1-N-oxide (1-NO-TMP) and 3-N-oxide (3-NO-TMP), 3′- and 4′-desmethyl-TMP, a benzylic alcohol (Cα-OH-TMP), and an N-acetyl cysteine (NAC) adduct of TMP (Cα-NAC-TMP). Formation kinetics for each TMP metabolite in human liver microsomes (HLMs) were consistent with single-enzyme Michaelis-Menten kinetics, and Km values were markedly above (≥10-fold) the therapeutic concentrations of TMP (50 µM). The combined results from correlation studies between rates of metabolite formation and marker P450 activities in a panel of HLMs along with inhibition studies utilizing selective P450 inhibitors incubated with pooled HLMs suggested that 1-NO-TMP, Cα-NAC-TMP, and Cα-OH-TMP were predominantly formed by CYP3A4. In contrast, 3-NO-TMP was formed predominantly by CYP1A2 in HLMs and inhibited by α-naphthoflavone. 4′-Desmethyl-TMP, which is believed to be a reactive TMP metabolite precursor, was formed by several P450s, including CYP3A4, correlated with multiple P450 activities, but was inhibited primarily by ketoconazole (up to 50%), suggesting that CYP3A4 makes a major contribution to TMP 4′-demethylation. TMP 3′-demethylation was catalyzed by multiple P450s, including CYP2C9, correlated with CYP2C9 activity, and was inhibited by sulfaphenazole (up to 40%). Overall, CYP2C9 and CYP3A4 appear to be the most significant contributors to TMP primary metabolism.
Introduction
Trimethoprim-sulfamethoxazole (TMP-SMX) is a frequently prescribed antimicrobial for the treatment of urinary tract, skin, and soft tissue infections and the prevention of opportunistic infections in the immuno-compromised host (Hersh et al., 2008; Mofenson et al., 2009; Copp et al., 2011). This drug combination is also one of the most common causes of unpredictable, undesirable, and potentially severe idiosyncratic adverse drug reactions (IADRs) with symptoms that include rash, fever, lymphadenopathy, cytopenias, liver injury, or severe skin detachment, such as Stevens-Johnson syndrome or toxic epidermal necrolysis (Roujeau et al., 1995; Clarkson and Choonara, 2002). Historically, SMX has been suspected as the inciting agent in these IADRs, largely based on its ability to form a nitroso intermediate capable of covalent modification of proteins, which results in subsequent immunogenicity and antigenicity (Manchanda et al., 2002; Naisbitt et al., 2002). However, administration of TMP alone has also been associated with severe and even fatal IADRs (Nwokolo et al., 1988; Hawkins et al., 1993; Mortimer et al., 2005). Recently, we identified N-acetyl cysteine (NAC) adducts of TMP metabolites in the urine of children taking TMP-SMX, confirming bioactivation of TMP to reactive metabolite species in vivo, thus raising the possibility that TMP may also contribute to IADRs with TMP-SMX use (van Haandel et al., 2014). Incubations containing human liver microsomes (HLMs), NAC, and a NADPH-generating system formed all of the TMP-NAC adducts observed in urine, suggesting the possibility that cytochrome P450 enzymes (P450s) may be responsible for TMP bioactivation.
TMP is largely excreted unchanged in the urine. Several stable human urinary metabolites have previously been identified: the N-oxides, TMP 1-N-oxide (1-NO-TMP) and TMP 3-N-oxide (3-NO-TMP), which are generated by oxidation of nitrogen atoms in the pyrimidine ring; benzylic alcohol (Cα-OH-TMP), formed via oxidation of the methylene bridge; and 3′- and 4′-desmethyl-TMP, resulting from oxidative O-demethylation (Fig. 1) (Brooks et al., 1973; Sigel et al., 1973). Further in vitro data generated by Damsten et al. (2008) suggest that multiple O-demethylation reactions may occur, resulting in up to three free hydroxyl groups on the benzylic ring.
Fig. 1.
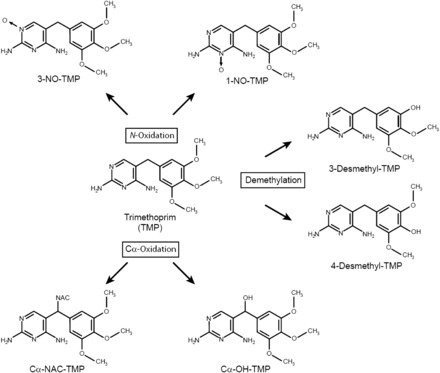
Metabolic scheme for the conversion of TMP to its primary oxidative metabolites.
In addition to the formation of these stable metabolites, evidence of TMP bioactivation has been demonstrated in vitro through the trapping of bioactivated TMP metabolites with small molecular weight thiols, such as NAC. TMP reactive metabolites are believed to form through both direct oxidation of TMP to an iminoquinone methide and via sequential, secondary oxidation of primary, demethylated TMP metabolites to form a paraquinone methide (Lai et al., 1999; Damsten et al., 2008). Consistent with the NAC-TMP adducts observed in vitro, we detected six NAC-TMP conjugates in the urine of children taking TMP-SMX (van Haandel et al., 2014).
In vitro results have demonstrated that TMP bioactivation is catalyzed by P450s, although the specific enzymes involved in generating both the stable and reactive metabolites have not been well defined. Damsten et al. (2008) described the formation of several NAC adducts and stable metabolites from TMP utilizing HLMs, rat liver microsomes, heterologously expressed P450 enzymes, and P450 BM3 mutants. Based only on data from incubations containing heterologously expressed human P450s 1A2, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4, Damsten et al. (2008) suggested that CYP1A2 and CYP3A4 were capable of forming the iminoquinone methide, and CYP2D6 was primarily responsible for catalyzing both the demethylation and subsequent secondary oxidation step leading to the paraquinone methide species, whereas CYP3A4 catalyzed multiple TMP demethylation reactions leading to quinone methide intermediates. Formation of reactive intermediates, such as the iminoquinone methide and the paraquinone methide species, is believed to be the initiating step in IADRs. Unfortunately, additional studies designed to more fully characterize the quantitative importance of individual P450s, such as correlation analyses with marker P450 activities in a panel of HLMs and inhibition studies with selective P450 isoform inhibitors, were not performed. We sought to fill this knowledge gap by more completely characterizing the role of P450s in the formation of TMP primary stable and reactive metabolites at therapeutically relevant concentrations of TMP (5–50 µM, based on plasma Cmax). Defining the metabolic pathways leading to the bioactivation of TMP is a critical first step to better understand factors that may contribute to variability in reactive metabolite formation and, potentially, the risk of developing IADRs.
Materials and Methods
Chemicals.
Liquid chromatography (LC)–mass spectrometry (MS) Optima grade acetonitrile, methanol, and water were purchased from Fisher Scientific (Fair Lawn, NJ). EDTA, formic acid (LC-MS grade), glucose-6-phosphate dehydrogenase, magnesium chloride, N-acetyl-l-cysteine (NAC), NADP, potassium phosphate dibasic, potassium phosphate monobasic, TMP, danazol, 4-methylpyrazole, montelukast, α-naphthoflavone, quinidine, sulfaphenazole, tranylcypromine, and Triton X-100 were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO). Benzylnirvanol and thiotepa were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). Ketoconazole was obtained from Research Biochemicals International (Natick, MA). All reagents were reagent grade quality or higher. 1-NO-TMP, 3-NO-TMP, 4′-desmethyl-TMP, Cα-OH-TMP, and Cα-NAC-TMP were custom synthesized by Artis-Chem Co. Ltd. (Shanghai, China). Purity of the standards was determined to be ≥98% by H NMR, LC-MS, and LCUV (liquid chromatography ultraviolet) at 214 nm.
Human Liver Microsomes and cDNA-Expressed Enzymes.
A reaction phenotyping kit (version 8; containing HLMs prepared from 16 individuals: 4 females, 12 males), pooled HLMs, and EasyCYP Bactosomes (manufactured by Cypex Ltd., Scotland, UK) were purchased from XenoTech, LLC (Lenexa, KS). The EasyCYP Bactosomes included the following cDNA-expressed human P450 enzymes: P450s 1A1, 1A2, 1B1, 2A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, 3A7, and 4A11. Each of the human P450 enzymes were coexpressed with P450 reductase, and CYP2B6, CYP2C8, CYP2C9, CYP2E1, CYP3A5, and CYP3A7 were coexpressed with cytochrome b5. Low reductase–containing microsomes were used where available. Microsomes were stored at −80°C and placed on ice prior to use.
In Vitro Incubation Conditions.
Standard in vitro enzyme incubations (200 µl) contained HLMs (20 µg of microsomal protein) or baculovirus cDNA–expressed P450 enzymes (4 pmol), potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), NAC (5 mM), and TMP (0–500 µM) dissolved in water. Reactions were initiated by the addition of a NADPH-generating system consisting of NADP (1 mM), glucose 6-phosphate (1 U/ml), and glucose-6-phosphate dehydrogenase (5 mM). Following the addition of the NADPH-generating system, reactions were incubated in a shaking water bath at 37 ± 1°C for 0–30 minutes and terminated by the addition of 200 µl of ice-cold methanol. Protein was precipitated by centrifugation at 10,000g for 10 minutes, and an aliquot of the supernatant was analyzed by high-performance liquid chromatography/MS via direct injection.
Preliminary experiments with TMP (0.5, 0.75, 1, 2.5, 5, 7.5, 10, 25, 50, 75, 100, 250, 300, 400, and 500 µM) and pooled HLMs (0.1, 0.5, and 1.0 mg of protein/ml) suggested that the rates of TMP metabolite formation were directly proportional to protein concentration (1.0 mg/ml) and time (60 minutes). Under these conditions, TMP biotransformation was less than 15%. Experiments utilizing HLMs that were warmed to 45°C for 5 minutes, placed on ice to cool, and then incubated with TMP (5 and 50 µM), NAC, and the NADPH-generating system (as described previously) were conducted to determine if flavin-containing monooxygenases (FMOs) could catalyze TMP primary metabolite formation. Similarly, HLMs treated with Triton X-100 (1% final concentration) were added to incubations containing TMP (5 and 50 µM), NAC, and the NADPH-generating system as described to determine the contribution of total P450 to TMP primary metabolite formation. Control incubations contained HLMs that were not subjected to heat nor treated with Triton X-100, TMP (5 and 50 µM), NAC, or the NADPH-generating system, as described earlier. Correlation experiments with HLMs were performed in triplicate with two replicate samples per condition. Kinetic experiments and experiments performed with cDNA-expressed P450s were conducted in duplicate.
Chemical-Inhibition Experiments.
Conversion of TMP (50 µM) to the primary metabolites of interest (1-NO-TMP, 3-NO-TMP, 3′-desmethyl-TMP, 4′-desmethyl-TMP, Cα-OH-TMP, and Cα-NAC-TMP) by pooled HLMs was determined in the presence or absence of selective, competitive P450 inhibitors at four concentrations encompassing the denoted ranges, including α-naphthoflavone (CYP1A2, 0.15–5 µM), tranylcypromine (CYP2A6, 0.2–6 µM), thiotepa (CYP2B6, 5–150 µM), montelukast (CYP2C8, 0.15–5 µM), sulfaphenazole (CYP2C9, 0.3–10 µM), benzylnirvanol (CYP2C19, 0.3–10 µM), quinidine (CYP2D6, 0.15–5 µM), 4-methylpyrazole (CYP2E1, 2–60 µM), danazol (CYP2J2, 0.02–0.6 µM), and ketoconazole (CYP3A4, 0.03–1 µM). These ranges of inhibitor concentrations were based on reported Ki (inhibition constant) values, with the highest concentration of inhibitor approximately 30 times Ki (range: Ki, 3 Ki, 10 Ki, and 30 Ki). In theory, the highest concentration of inhibitor used in these studies should virtually abolish (>90%) product formation by the inhibited enzyme. Inhibition experiments were conducted in triplicate with replicate incubations.
UPLC (Ultra-Performance Liquid Chromatography) Tandem MS Analysis.
Sample analyses were performed by UPLC tandem MS, as described by van Haandel et al. (2014). Optimization of the analysis protocol included the injection of 3µl of sample to minimize band broadening and the custom synthesis of reference compounds. The availability of reference standards allowed us to construct calibration curves for 1-NO-TMP, 3-NO-TMP, 4′-desmethyl-TMP, Cα-OH-TMP, and Cα-NAC-TMP. 3′-Desmethyl-TMP was quantitated from the 4′-desmethyl-TMP standard because of its structural similarity and due to the absence of available reference material. Calibration curves were constructed in MeOH-quenched incubations containing HLMs and 1, 3, 10, 30, 100, 300, or 400 µM concentrations of standard reference compounds.
Data Analysis.
Correlation coefficients (r) between the rates of 1-NO-TMP, 3-NO-TMP, 3′-desmethyl-TMP, 4′-desmethyl-TMP, Cα-OH-TMP, and Cα-NAC-TMP formation with the marker activities of P450 enzymes in the HLM reaction phenotype kit were determined using least-squares regression analysis. Significance was determined by Pearson's regression analysis using a two-tailed t test with α = 0.05 (JMP 10). Effects of the selective P450 inhibitors were determined by comparing the rate of individual metabolite formation as the percent of control by inhibitor concentration (Microsoft Excel 2007; Microsoft Corporation, Redmond, WA). Rate calculations with the heterologously expressed enzymes were corrected for metabolite formation by microsomes containing vector only. Data derived from kinetic incubation studies using HLMs and selected cDNA-expressed enzymes (CYP2C9 and 3A4) were analyzed by nonlinear regression to estimate Km (Michaelis constant) and Vmax values. These data were visually inspected via Eadie-Hofstee and Lineweaver-Burk plots and subsequently fit to a one-enzyme Michaelis-Menten model (GraphFit 5; Erithacus Software Ltd., Surrey, UK).
Results
Biotransformation of TMP by HLMs.
In the presence of oxygen, NAC, and an NADPH-generating system, HLMs converted TMP (5 and 50 µM) to six primary metabolites resulting from oxidation of TMP: 1-NO-TMP, 3-NO-TMP, 3′-desmethyl-TMP, 4′-desmethyl-TMP, Cα-OH-TMP, and Cα-NAC-TMP (Fig. 1). The identities of the metabolites formed were consistent with our previous in vitro and in vivo observations (van Haandel et al., 2014). 3′- And 4′-desmethyl-TMP were the most abundant metabolites formed and accounted for approximately 65 and 25% of total metabolite formation, respectively, regardless of substrate concentration examined. Of the remaining metabolites, only 1-NO-TMP had an abundance approaching 5%; each of the other minor metabolites constituted less than 2% of the total metabolite formation (Fig. 2).
Fig. 2.
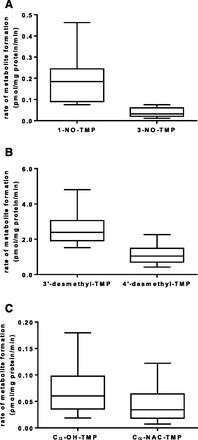
Variability in the formation rates of trimethoprim primary metabolites. TMP (5 µM) was incubated with HLMs prepared from individual donors (n = 16) as described under Materials and Methods. Microsomes from each donor were incubated in triplicate. Box and whisker plots were constructed for the rate of formation for each metabolite. The box extends from the 25th to the 75th percentile with the line in the middle at the median. The whiskers extend to the minimum and maximum values, respectively. (A) TMP N-oxidation (1- and 3-NO-TMP). (B) TMP demethylation (3′- and 4′-desmethyl-TMP). (C) TMP Cα-oxidation (Cα-OH-TMP and Cα-NAC-TMP).
To determine if FMOs might contribute to the formation of these metabolites, HLMs heat treated at 45°C for 5 minutes (causing heat inactivation of FMOs) were evaluated for their ability to convert TMP (5 and 50 µM) to primary metabolites. There was no difference in metabolite formation between heat-treated and control HLMs (results not shown). In contrast, incubations containing 1% Triton X-100 (which inhibits P450 activity), HLMs, and TMP (5 and 50 µM) demonstrated that Triton X-100 virtually eliminated TMP metabolite formation relative to control incubations (results not shown).
The kinetics of formation for the six primary TMP metabolites were estimated in pooled adult HLMs and in microsomes containing cDNA-expressed CYP2C9 and CYP3A4 enzymes (Table 1). CYP2C9 and CYP3A4 were selected because both enzymes were shown to either significantly contribute to direct bioactivation of TMP (Cα-NAC-TMP) or result in the abundant formation of primary metabolites (3′- and 4′-desmethyl-TMP) that could potentially undergo further bioactivation to potential reactive species (experimental results supporting these findings are described later). The aqueous solubility of TMP limited final incubation concentrations to 500 µM or less. Direct plots of substrate concentration versus rate of TMP metabolite formation indicated that substrate saturation did not occur for the formation of any of the TMP primary metabolites (i.e., the plateau phase was not achieved). Although this precluded an accurate estimation of Km and Vmax parameters, Eadie-Hofstee plots were constructed for the available data. Although there was a slight indication of substrate activation in the formation of Cα-OH-TMP, visual inspection of the Eadie-Hofstee plots for each of the six primary metabolites suggested that they could be fit to a single-enzyme Michaelis-Menten equation. Data were then fit to a single-enzyme model using the software Grafit 5 (Erithacus Software Limited, United Kingdom) to estimate the kinetic parameters Km and Vmax, which are shown in Table 1. Of note, all of the determined Km values for the six primary metabolites were above 450 µM and as high as 2 mM, markedly higher than therapeutic concentrations of TMP observed in vivo.
TABLE 1.
Estimated kinetic parameters for the formation of TMP primary metabolites in human liver microsomes and cDNA-expressed human P450 enzymes
Data are presented as the value ± S.E.
| Km | Vmax a,b | Clint c,d | |
|---|---|---|---|
| Section I.1 μM | |||
| Pooled HLMs | |||
| 1-NO-TMP | 1050 ± 120 | 69 ± 6 | 0.07 |
| 3-NO-TMP | 690 ± 76 | 5 ± 0.3 | 0.007 |
| 3′-desmethyl-TMP | 647 ± 55 | 453 ± 25 | 0.70 |
| 4′-desmethyl-TMP | 482 ± 42 | 146 ± 7 | 0.30 |
| Cα-OH-TMP | 2180 ± 400 | 108 ± 17 | 0.05 |
| Cα-NAC-TMP | 1310 ± 160 | 18 ± 2 | 0.01 |
| CYP2C9 | |||
| 1-NO-TMP | 531 ± 264 | 23.3 ± 6.87 | 0.044 |
| 3-NO-TMP | 428 ± 253 | 5.97 ± 1.95 | 0.014 |
| 3′-desmethyl-TMP | 1330 ± 400 | 2030 ± 470 | 1.53 |
| 4′-desmethyl-TMP | I.D. | I.D. | I.D. |
| Cα-OH-TMP | I.D. | I.D. | I.D. |
| Cα-NAC-TMP | I.D. | I.D. | I.D. |
| CYP3A4 | |||
| 1-NO-TMP | 971 ± 188 | 1320 ± 190 | 1.36 |
| 3-NO-TMP | N.D. | N.D. | N.D. |
| 3′-desmethyl-TMP | 1470 ± 510 | 2060 ± 571 | 1.40 |
| 4′-desmethyl-TMP | 1040 ± 231 | 1430 ± 240 | 1.38 |
| Cα-OH-TMP | 754 ± 122 | 1020 ± 110 | 1.35 |
| Cα-NAC-TMP | 784 ± 143 | 137 ± 18 | 0.17 |
CLint, intrinsic clearance; I.D., insufficient data to determine kinetic parameters; N.D., metabolite not detected.
Values are pmol/mg protein/minute for pooled HLMs.
Values are pmol/nmol P450/minute for heterologously expressed enzymes.
Values are µl/mg protein/minute for pooled HLMs.
Values are µl/nmol P450/minute for heterologously expressed enzymes.
TMP N-Oxidation (1- and 3-NO-TMP).
Incubations with microsomes containing one of 17 cDNA-expressed human P450 enzymes were conducted at two TMP concentrations spanning the therapeutic range (5 and 50 µM) to evaluate the ability of these enzymes to catalyze formation of TMP N-oxides. The selected TMP concentrations were based on observed in vivo TMP Cmax measurements (Hoppu and Arjomaa, 1984; Rylance et al., 1985). Several heterologously expressed P450 enzymes, including CYP1A1, CYP1B1, CYP2C19, CYP3A4, CYP3A5, and CYP3A7, were capable of catalyzing 1-NO-TMP formation (Fig. 3A, 5 µM data shown), whereas CYP1A1 and CYP1B1 were the heterologously expressed enzymes that catalyzed the highest rates of 3-NO-TMP formation.
Fig. 3.
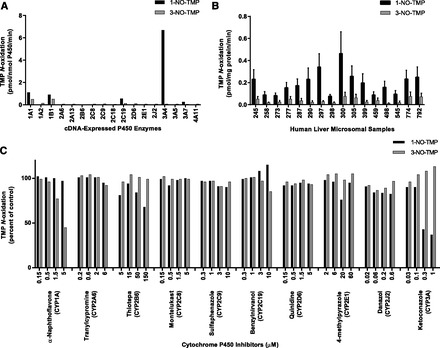
N-oxidation of TMP to primary metabolites. (A) 1- and 3-NO-TMP formation by human cDNA-expressed P450 enzymes. TMP (5 µM) was incubated with cDNA-expressed P450 enzymes as described under Materials and Methods. Rates shown are corrected for rates from microsomes containing vectors alone. Each bar represents the mean of the duplicate incubations. (B) Sample-to-sample variation in the rates of TMP 1- and 3-N-oxidation by HLMs at a substrate concentration of 5 µM. Each bar represents the mean of the triplicate experiments ± S.D. (C) Effects of various P450 isoform–selective inhibitors on the formation of 1- and 3-NO-TMP by pooled HLMs at a substrate concentration of 50 µM, as described under Materials and Methods. Each bar represents the mean of the triplicate incubations. Uninhibited mean rates of 1- and 3-NO-TMP formation were 8.2 and 1.6 pmol/mg protein/min, respectively.
A panel of HLMs prepared from 16 individual donors and characterized for P450 and FMO activities using marker substrate reactions was examined for the ability to catalyze the 1- and 3-N-oxidation of TMP. At a substrate concentration of 5 µM, each of the microsomal preparations catalyzed the formation of both N-oxides, and the rate of TMP N-oxide formation varied >6-fold; 1-N-oxide formation (rates ± S.D.) ranged from 0.08 ± 0.02 to 0.46 ± 0.20 pmol/mg protein/min, whereas 3-N-oxide formation ranged from 0.01 ± 0.005 to 0.08 ± 0.04 pmol/mg protein/min (Figs. 2A and Fig. 3B). Rates of TMP 1- and 3-N-oxide formation for each of the microsomal samples increased proportionally (approximately) with a 10-fold increase in substrate concentration (50 µM; results not shown). The sample-to-sample variation in the rates of 1-NO-TMP formation correlated significantly (P < 0.01) with CYP2A6, CYP2C8, CYP2C9, and CYP3A4 activities, whereas 3-NO-TMP formation correlated significantly with CYP1A2, CYP2C8, and CYP2C9 activities, but not with any other P450 or FMO activities (Table 2).
TABLE 2.
Correlation analysis (r) of the relationship between the rates of TMP primary metabolite formation with the sample-to-sample variation in cytochrome P450 activities in human liver microsomes
| TMP Concentration | 1-NO-TMP | 3-NO-TMP | 3'-Desmethyl-TMP | 4′-Desmethyl-TMP | Cα-OH-TMP | Cα-NAC-TMP |
|---|---|---|---|---|---|---|
| CYP1A2 | ||||||
| 5 μM | 0.444 | 0.710** | 0.578* | 0.437 | 0.247 | 0.232 |
| 50 μM | 0.434 | 0.785** | 0.555* | 0.409 | 0.235 | 0.221 |
| CYP2A6 | ||||||
| 5 μM | 0.633** | 0.435 | 0.554* | 0.566* | 0.506* | 0.493 |
| 50 μM | 0.587** | 0.391 | 0.512* | 0.558* | 0.488 | 0.488 |
| CYP2B6 | ||||||
| 5 μM | 0.499* | 0.145 | 0.055 | 0.352 | 0.521* | 0.510* |
| 50 μM | 0.404 | 0.077 | 0.000 | 0.316 | 0.459 | 0.457 |
| CYP2C8 | ||||||
| 5 μM | 0.734** | 0.884** | 0.750** | 0.930** | 0.302 | 0.292 |
| 50 μM | 0.612* | 0.807** | 0.694** | 0.867** | 0.303 | 0.286 |
| CYP2C9 | ||||||
| 5 μM | 0.742** | 0.629** | 0.873** | 0.759** | 0.502* | 0.499* |
| 50 μM | 0.763** | 0.616* | 0.888** | 0.809** | 0.537* | 0.528* |
| CYP2C19 | ||||||
| 5 μM | 0.429 | 0.152 | 0.145 | 0.239 | 0.500* | 0.502* |
| 50 μM | 0.379 | 0.152 | 0.122 | 0.255 | 0.424 | 0.428 |
| CYP2D6 | ||||||
| 5 μM | 0.110 | 0.167 | 0.436 | 0.190 | 0.032 | 0.045 |
| 50 μM | 0.167 | 0.190 | 0.481 | 0.251 | 0.063 | 0.055 |
| CYP2E1 | ||||||
| 5 μM | 0.110 | 0.032 | 0.226 | 0.000 | 0.190 | 0.192 |
| 50 μM | 0.197 | 0.000 | 0.167 | 0.000 | 0.237 | 0.251 |
| CYP2J2 | ||||||
| 5 μM | 0.510* | 0.089 | 0.438 | 0.390 | 0.548* | 0.571* |
| 50 μM | 0.540* | 0.095 | 0.514* | 0.518* | 0.573* | 0.571* |
| CYP3A4/5 | ||||||
| 5 μM | 0.853** | 0.148 | 0.399 | 0.509* | 0.991** | 0.990** |
| 50 μM | 0.905** | 0.138 | 0.466 | 0.632* | 0.994** | 0.995** |
| CYP4A11 | ||||||
| 5 μM | 0.389 | 0.512* | 0.458 | 0.489 | 0.164 | 0.179 |
| 50 μM | 0.423 | 0.544* | 0.511* | 0.511* | 0.217 | 0.219 |
P < 0.05; **P < 0.01.
Inhibition studies were conducted to corroborate the identities of the relevant enzymes involved in the formation of TMP N-oxides. As shown in Fig. 3C, the formation of 1-NO-TMP was inhibited markedly (greater than 20% inhibition) and in a concentration-dependent manner by thiotepa (CYP2B6 inhibitor) and ketoconazole (CYP3A inhibitor). However, cDNA-expressed CYP2B6 did not appear to catalyze 1-NO-TMP formation, nor was CYP2B6 activity correlated significantly (P < 0.01) with 1-NO-TMP formation in the panel of HLMs, suggesting that CYP2B6 may not be a major catalyst of this reaction. α-Naphthoflavone (CYP1A inhibitor) was the only inhibitor examined that was found to inhibit 3-NO-TMP formation. The other competitive P450 inhibitors resulted in little or no inhibition of either 1-NO-TMP or 3-NO-TMP formation. Cumulatively, this evidence suggests that in HLMs, 1-NO-TMP and 3-NO-TMP formation is catalyzed primarily by CYP3A4 and CYP1A, respectively.
TMP Demethylation (3′- and 4′-Desmethyl-TMP).
Several of the heterologously expressed P450 isoforms examined in this study demonstrated the capability to catalyze the formation of 3′ or 4′-desmethyl-TMP at substrate concentrations of 5 µM (Fig. 4A) and 50 µM TMP (data not shown). Unlike other P450 enzymes, CYP2B6, CYP2C19, CYP2D6, CYP2J2, and CYP3A4 were capable of catalyzing both the 3′- and 4′-demethylation of TMP at the concentrations examined. At a substrate concentration of 5 µM, there was nearly 3-fold variation in 3′-desmethyl-TMP formation (range: 1.5 ± 0.14 to 4.8 ± 0.49 pmol/mg protein/min) and 5-fold variation in 4′-desmethyl-TMP formation (range: 0.4 ± 0.02 to 2.3 ± 0.35 pmol/mg protein/min) in the panel of HLMs (Figs. 2B and Fig. 4B). The sample-to-sample variation in the rates of formation were mostly highly correlated (P < 0.01) with CYP2C8 and CYP2C9 activities for both 3′- and 4′-desmethyl-TMP formation, although several other P450 activities, including CYP2J2 and CYP3A4/5 activities, were observed to have significant correlations with TMP 3′- or 4′-demethylation (Table 2). Interestingly, CYP2C8 and CYP2C9 activities were significantly correlated with one another (r = 0.668; P < 0.01), as were CYP2J2 and CYP3A activities (r = 0.648; P < 0.01) among the panel of HLMs.
Fig. 4.
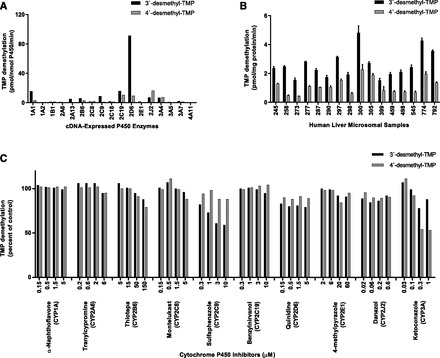
Demethylation of TMP to primary metabolites. (A) 3′- and 4′-desmethyl-TMP formation by human cDNA-expressed P450 enzymes. TMP (5 µM) was incubated with cDNA-expressed P450 enzymes as described under Materials and Methods. Rates shown are corrected for rates from microsomes containing vectors alone. Each bar represents the mean of the duplicate incubations. (B) Sample-to-sample variation in the rates of TMP 3′- and 4′-desmethyl-TMP by HLMs at a substrate concentration of 5 µM. Each bar represents the mean of the triplicate experiments ± S.D. (C) Effects of various P450 isoform–selective inhibitors on the formation of 3′- and 4′-desmethyl-TMP by pooled HLMs at a substrate concentration of 50 µM, as described under Materials and Methods. Each bar represents the mean of the triplicate incubations. Uninhibited rates of 3′- and 4′-desmethyl-TMP formation were 48.1 and 29.2 pmol/mg protein/min, respectively.
The CYP2C9 inhibitor, sulfaphenazole, markedly inhibited TMP 3′-demethylation in a concentration-dependent manner, but had little or no effect on the 4′-demethylation of TMP. In contrast, the CYP3A inhibitor, ketoconazole, had little or no effect on TMP 3′-demethylation but markedly inhibited TMP 4′-demethylation in a concentration-dependent manner. All of the other competitive P450 inhibitors tested resulted in little or no inhibition of TMP 3′- or 4′-demethylation.
TMP α-Bridge Oxidation (Cα-OH-TMP and Cα-NAC-TMP).
Incubations containing cDNA-expressed human P450 enzymes and TMP (5 µM) revealed that CYP1A1, CYP3A4, CYP3A5, and CYP3A7 had the capability to catalyze Cα-OH-TMP formation, whereas Cα-NAC-TMP formation appeared to be exclusively catalyzed by CYP3A4 (Fig. 5A). Similar results were obtained when heterologously expressed P450s were incubated with 50 µM TMP (results not shown). Considerable variability in the rates of oxidation of the α-bridge carbon of TMP was observed among the 16 different preparations of HLMs at a substrate concentration of 5 µM (Figs. 2C and Fig. 5B); nearly 10-fold variation in Cα-OH-TMP formation (range: 0.02 ± 0.004 to 0.18 ± 0.03 pmol/mg protein/min) and 17-fold variation in Cα-NAC-TMP formation (range: 0.007 ± 0.006 to 0.12 ± 0.07 pmol/mg protein/min) were observed. The sample-to-sample variation in the rates of Cα-OH-TMP and Cα-NAC-TMP formation by the panel of HLMs was most highly correlated with CYP3A4/5 activity (P < 0.01), but was significantly correlated with the activities of a number of other P450 enzymes (Table 2). Inhibition studies revealed that the formation of both Cα-OH-TMP and Cα-NAC-TMP was inhibited markedly by the CYP3A inhibitor, ketoconazole, and moderately by the CYP2B6 inhibitor, thiotepa, but not by any other selective P450 inhibitors (Fig. 5C).
Fig. 5.
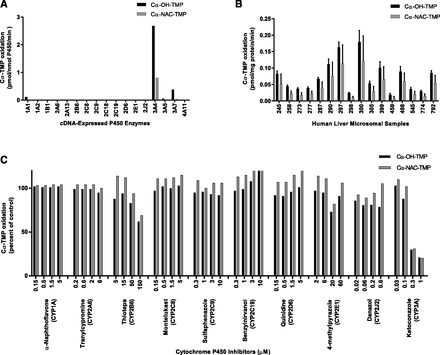
Cα-oxidation of TMP to primary metabolites. (A) Cα-OH-TMP and Cα-NAC-TMP formation by human cDNA-expressed P450 enzymes. TMP (5 µM) was incubated with cDNA-expressed P450 enzymes as described under Materials and Methods. Rates shown are corrected for rates from microsomes containing vectors alone. Each bar represents the mean of the duplicate incubations. (B) Sample-to-sample variation in the rates of Cα-OH-TMP and Cα-NAC-TMP formation by HLMs at a substrate concentration of 5 µM. Each bar represents the mean of the triplicate experiments ± S.D. (C) Effects of various P450 isoform–selective inhibitors on the formation of Cα-OH-TMP and Cα-NAC-TMP by pooled HLMs at a substrate concentration of 50 µM, as described under Materials and Methods. Each bar represents the mean of the triplicate incubations. Uninhibited rates of Cα-OH-TMP and Cα-NAC-TMP formation were 4.6 and 2.1 pmol/mg protein/min, respectively.
Discussion
Trimethoprim undergoes biotransformation to a number of metabolites by humans, and Fig. 1 depicts the primary oxidative metabolites formed from TMP. Many TMP metabolites are stable, but a few reactive intermediates, which can be trapped with NAC, such as an iminoquinone methide species, have also been identified (Damsten et al., 2008; van Haandel et al., 2014). To date, little information is available on the roles of individual oxidative enzymes, including P450s, involved in the formation of these metabolites. Only one previous in vitro study (Damsten et al., 2008) has provided insight into the enzymes involved in the formation of TMP metabolites. Based on their experiments with heterologously expressed human P450s and the average expression content of these enzymes in human livers, Damsten et al. (2008) inferred the identities of the P450 enzymes they believed were responsible for TMP biotransformation. Unfortunately, the activities of heterologously expressed P450 enzymes do not necessarily reflect the activities of native P450s, even when relative abundance is taken into consideration. Differences in P450 reductase and cytochrome b5 expression levels as well as differences in lipid membrane composition of the used expression system can contribute to alterations in P450 activities (Parkinson, 1996). By combining additional kinetic, correlation, and inhibition experimental approaches with experiments utilizing cDNA-expressed enzymes, a more accurate assessment of the identities and roles of P450s involved in TMP biotransformation can be obtained. Therefore, we undertook these in vitro studies to further characterize the roles of P450s involved in those TMP primary metabolite biotransformation pathways observed in vivo.
Although there were some differences, many of the results from our study corroborate observations by Damsten et al. (2008). 3′-Desmethyl-TMP and 4′-desmethyl-TMP were the most abundant metabolites formed by HLMs and accounted for approximately 65 and 25% of total metabolite formation, respectively, whereas TMP N-oxides constituted minor metabolites (≤5%) and the benzylic metabolites, Cα-OH-TMP and Cα-NAC-TMP, were in even lower abundance. Damsten et al. (2008) concluded that CYP2C9 and CYP3A4 were primarily responsible for the formation of 3′-desmethyl-TMP and 4′-desmethyl-TMP, respectively. On the basis of results from our correlation, inhibition, and cDNA-expressed enzyme experiments, we reached the same conclusions.
CYP3A4 appears to play a major role in the formation of 4′-desmethyl-TMP. Although several heterologously expressed P450s, including CYP2C19, CYP2D6, CYP2J2, and CYP3A4, demonstrated the capability to catalyze TMP 4′-demethylation, the only effective inhibitor of TMP 4-demethylation examined in HLMs was the CYP3A inhibitor, ketoconazole, which inhibited ∼50% of TMP 4′-demethylase. The highest concentration of inhibitor used in this study should eliminate >90% of the targeted P450 isoform activity, which suggests that, although CYP3A4 appears to be a major contributor to 4′-desmethyl-TMP formation, it is likely that other P450 enzymes may contribute to the formation of this metabolite at therapeutically relevant concentrations of TMP.
Heterologously expressed CYP2D6 was the most prolific enzyme catalyzing TMP 3′-demethylation, corroborating the observation by Damsten et al. (2008), but the CYP2D6 inhibitor quinidine caused no appreciable inhibition of TMP 3′-demethylation, and interindividual CYP2D6 activity in HLMs correlated poorly with rates of TMP 3′-demethylation (r = 0.481). The low abundance of CYP2D6 in HLMs is likely responsible for the lack of apparent contribution by CYP2D6 to the formation of 3′-desmethyl-TMP and the lack of inhibition in product formation by quinidine. These results suggest that, in vivo, CYP2D6 will make little or no contribution to the formation of 3′-desmethyl-TMP.
In contrast, heterologously expressed CYP2C9 catalyzed a much lower rate of TMP 3′-demethylation than did CYP2D6, but CYP2C9 appeared to play a major role in TMP 3′-demethylation in HLMs based on the observations that the CYP2C9 inhibitor, sulfaphenazole, substantially inhibited the formation of 3′-desmethyl-TMP (up to 40%) in pooled HLMs, and the rate of 3′-desmethyl-TMP formation significantly correlated with CYP2C9 and CYP2C8 activities (enzymes that are coregulated) in the panel of HLMs (Chen and Goldstein, 2009). Similar to the case with TMP 4′-demethylation, it is likely that other P450 enzymes may contribute to the formation of 3′-desmethyl-TMP.
Human liver microsomes formed only small amounts (≤5%) of TMP 1- and 3-N-oxides, and 3-NO-TMP was found to be primarily catalyzed by CYP1A2, observations also previously noted (Damsten et al., 2008). Damsten et al. concluded that 1-NO-TMP was also primarily formed by CYP1A2. However, the results of our study suggest that CYP3A4 is the enzyme primarily responsible for catalyzing TMP 1-N-oxidation. It is important to note that both the 1- and 3-N-oxides are found in much greater abundance in human urine (approximately 30% of total recovered metabolites), suggesting that these metabolites are formed by other processes, potentially by extrahepatic P450 enzymes or by other oxidative enzymes (Sigel et al., 1973).
We also identified CYP3A4 as the primary catalyst forming both Cα-NAC-TMP and Cα-OH-TMP. Heterologously expressed CYP3A4 catalyzed high rates of Cα-NAC-TMP and Cα-OH-TMP formation, CYP3A4/5 activity was significantly correlated with rates of formation for Cα-NAC-TMP and Cα-OH-TMP (r2 ≥ 0.990), and the potent CYP3A inhibitor ketoconazole inhibited formation of both metabolites in a concentration-dependent manner. Interestingly, CYP3A5 and CYP3A7, enzymes that are expressed in hepatic tissue (Hakkola et al., 1998), also catalyzed Cα-OH-TMP formation. CYP3A7 is the predominant P450 expressed in fetal and neonatal livers but rapidly declines postnatally, whereas CYP3A5 is expressed at relatively low levels in fetal tissue but is expressed at constant amounts postnatally (Hines, 2007). Thiotepa, a CYP2B6 inhibitor, inhibited Cα-OH-TMP formation at the highest concentration used and to a much lesser extent than did the CYP3A inhibitor, ketoconazole. However, heterologously expressed CYP2B6 did not appear to catalyze Cα-OH-TMP formation. Hence, it is possible that the inhibition caused by the highest concentration of thiotepa was not due to inhibition of CYP2B6 but a loss in selectivity of the inhibitor at this concentration.
It is conceivable that Cα-NAC-TMP and Cα-OH-TMP arise from a single intermediate, but the factors that determine whether the intermediate forms a reactive species capable of forming an adduct or a stable metabolite are currently unknown. It is possible that these two metabolites arise via different mechanisms or pathways. P450s in addition to CYP3A4, such as CYP3A5 and CYP3A7, are capable of catalyzing the formation of Cα-OH-TMP but not Cα-NAC-TMP. In addition, we observed that the ratio of Cα-OH-TMP to Cα-NAC-TMP varied between individual HLMs nearly 2-fold, suggesting that other factors play a role in the balance of formation of the two metabolites.
Our results suggest that CYP2C9 and CYP3A4 are key enzymes in the biotransformation pathways responsible for metabolic clearance of TMP and the formation of both stable metabolites and reactive intermediates. Genotypic variation in CYP2C9 and CYP3A4 alleles can lead to alterations in the activity of these enzymes (Kirchheiner and Brockmoller, 2005; Werk and Cascorbi, 2014), which has the potential to alter the balance between bioactivation and detoxification, and thus alter the relative abundance of reactive and stable TMP metabolites. Further studies assessing in vivo TMP bioactivation are needed to better understand the extent and variability of TMP bioactivation in humans.
Interestingly, previous data propose that SMX undergoes CYP2C9-mediated bioactivation to a hydroxylamine. This hydroxylamine undergoes subsequent auto-oxidation, resulting in a reactive nitroso intermediate that can form haptenated cellular proteins capable of triggering a cellular immune response (Naisbitt et al., 2002; Callan et al., 2009). The results of the present study demonstrate that CYP2C9 contributes to formation of the most abundant primary TMP metabolite (3′-desmethyl-TMP) in vitro. Consequently, the potential exists for either TMP or SMX to directly influence the extent of metabolism and possible bioactivation of the other component when administered concomitantly at a combined fixed dose. For example, an individual who extensively metabolizes TMP to the 3′-desmethyl-TMP metabolite could conceivably experience a relative decrease in the amount of SMX hydroxylamine that is formed. There is no evidence of mechanism-based inhibition for either trimethoprim or sulfamethoxazole; however, the impact of simultaneous biotransformation of the two substrates on each other, and the potential impact on bioactivation of one component on the other, has not been evaluated (Wen et al., 2002).
The work presented here characterizes only the role that P450s play in the primary biotransformation pathways of TMP. Hence, reactive metabolites that were previously observed (van Haandel et al., 2014) but that are formed via multiple biotransformation reactions, such as the iminoquinone or paraquinone-methides, were not detected in our incubations, nor were they expected to be produced under the conditions used in these experiments. Identification of the determinants leading to the formation of the iminoquinone methide intermediate is crucial, as this may serve as a potential risk factor for IADR development. It is possible that variability in CYP3A4 activity determines the extent of primary TMP bioactivation in vivo, and further studies are underway to assess the extent of primary TMP metabolism variability in vivo. Hence, factors modulating activity of these contributing enzymes may affect the risk of adverse drug reactions (Gill et al., 1999).
Detection of 3′- and 4′-demethylated TMP glutathione or NAC adducts in vitro and in vivo implies that 3′-desmethyl- and 4′-desmethyl-TMP undergo further oxidation, leading to reactive intermediates (Leblanc et al., 2010; van Haandel et al., 2014). Subsequent bioactivation of stable, primary metabolites to reactive intermediates has been demonstrated with other drugs commonly associated with IADRs, such as phenytoin and carbamazepine (Cuttle et al., 2000; Pearce et al., 2008). Hence, additional studies are needed to investigate the enzymes responsible for converting 3′-desmethyl- and 4′-desmethyl-TMP to reactive species capable of forming protein adducts.
In summary, our in vitro studies implicate CYP3A4 as the predominant cytochrome P450 enzyme involved in the direct formation of a reactive metabolite (i.e., Cα-NAC-TMP) from TMP. In addition, CYP 3A4 plays a major role in the generation of 4′-desmethyl-TMP, which is believed to be a precursor in a second bioactivation pathway. Characterization of the isoforms responsible for secondary TMP bioactivation is critical to understand all potential sources of interindividual variation involved in the generation of reactive intermediates capable of haptenating proteins. Although TMP has historically been overlooked as a probable contributor of IADRs, evidence of TMP bioactivation suggests that it too may contribute to these well recognized, unpredictable reactions associated with TMP-SMX. Therefore, a better understanding of bioactivation may provide insight into means of identifying individuals at risk for developing an IADR prior to its occurrence.
Abbreviations
- Cα-OH-TMP
benzylic alcohol
- FMO
flavin-containing monooxygenase
- HLM
human liver microsome
- IADR
idiosyncratic adverse drug reaction
- LC
liquid chromatography
- MS
mass spectrometry
- NAC
N-acetyl cysteine
- 1-NO-TMP
TMP 1-N-oxide
- 3-NO-TMP
TMP 3-N-oxide
- P450
cytochrome P450
- SMX
sulfamethoxazole
- TMP
trimethoprim
Authorship Contributions
Participated in research design: Goldman, Pearce.
Conducted experiments: Goldman, Van Haandel, Pearce.
Performed data analysis: Goldman, Van Haandel, Leeder, Pearce.
Wrote or contributed to the writing of the manuscript: Goldman, Van Haandel, Leeder, Pearce.
Footnotes
References
- Brooks MA, De Silva JA, D’Aroconte L. (1973) Determination of trimethoprim and its N-oxide metabolites in urine of man, dog, and rat by differential pulse polarography. J Pharm Sci 62:1395–1397. [DOI] [PubMed] [Google Scholar]
- Callan HE, Jenkins RE, Maggs JL, Lavergne SN, Clarke SE, Naisbitt DJ, Park BK. (2009) Multiple adduction reactions of nitroso sulfamethoxazole with cysteinyl residues of peptides and proteins: implications for hapten formation. Chem Res Toxicol 22:937–948. [DOI] [PubMed] [Google Scholar]
- Chen Y, Goldstein JA. (2009) The transcriptional regulation of the human CYP2C genes. Curr Drug Metab 10:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson A, Choonara I. (2002) Surveillance for fatal suspected adverse drug reactions in the UK. Arch Dis Child 87:462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp HL, Shapiro DJ, Hersh AL. (2011) National ambulatory antibiotic prescribing patterns for pediatric urinary tract infection, 1998-2007. Pediatrics 127:1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle L, Munns AJ, Hogg NA, Scott JR, Hooper WD, Dickinson RG, Gillam EM. (2000) Phenytoin metabolism by human cytochrome P450: involvement of P450 3A and 2C forms in secondary metabolism and drug-protein adduct formation. Drug Metab Dispos 28:945–950. [PubMed] [Google Scholar]
- Damsten MC, de Vlieger JS, Niessen WM, Irth H, Vermeulen NP, Commandeur JN. (2008) Trimethoprim: novel reactive intermediates and bioactivation pathways by cytochrome p450s. Chem Res Toxicol 21:2181–2187. [DOI] [PubMed] [Google Scholar]
- Gill HJ, Tjia JF, Kitteringham NR, Pirmohamed M, Back DJ, Park BK. (1999) The effect of genetic polymorphisms in CYP2C9 on sulphamethoxazole N-hydroxylation. Pharmacogenetics 9:43–53. [DOI] [PubMed] [Google Scholar]
- Hakkola J, Tanaka E, Pelkonen O. (1998) Developmental expression of cytochrome P450 enzymes in human liver. Pharmacol Toxicol 82:209–217. [DOI] [PubMed] [Google Scholar]
- Hawkins T, Carter JM, Romeril KR, Jackson SR, Green GJ. (1993) Severe trimethoprim induced neutropenia and thrombocytopenia. N Z Med J 106:251–252. [PubMed] [Google Scholar]
- Hersh AL, Chambers HF, Maselli JH, Gonzales R. (2008) National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med 168:1585–1591. [DOI] [PubMed] [Google Scholar]
- Hines RN. (2007) Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol 21:169–175. [DOI] [PubMed] [Google Scholar]
- Hoppu K, Arjomaa P. (1984) Difference in trimethoprim pharmacokinetics between children and adults. Chemotherapy 30:283–287. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Brockmöller J. (2005) Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther 77:1–16. [DOI] [PubMed] [Google Scholar]
- Lai WG, Zahid N, Uetrecht JP. (1999) Metabolism of trimethoprim to a reactive iminoquinone methide by activated human neutrophils and hepatic microsomes. J Pharmacol Exp Ther 291:292–299. [PubMed] [Google Scholar]
- Leblanc A, Shiao TC, Roy R, Sleno L. (2010) Improved detection of reactive metabolites with a bromine-containing glutathione analog using mass defect and isotope pattern matching. Rapid Commun Mass Spectrom 24:1241–1250. [DOI] [PubMed] [Google Scholar]
- Manchanda T, Hess D, Dale L, Ferguson SG, Rieder MJ. (2002) Haptenation of sulfonamide reactive metabolites to cellular proteins. Mol Pharmacol 62:1011–1026. [DOI] [PubMed] [Google Scholar]
- Mofenson LM, Brady MT, Danner SP, Dominguez KL, Hazra R, Handelsman E, Havens P, Nesheim S, Read JS, Serchuck L, et al. Centers for Disease Control and Prevention. National Institutes of Health. HIV Medicine Association of the Infectious Diseases Society of America. Pediatric Infectious Diseases Society. American Academy of Pediatrics (2009) Guidelines for the Prevention and Treatment of Opportunistic Infections among HIV-exposed and HIV-infected children: recommendations from CDC, the National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics. MMWR Recomm Rep 58 (RR-11):1–166. [PMC free article] [PubMed] [Google Scholar]
- Mortimer NJ, Bermingham MR, Chapple SJ, Sladden MJ. (2005) Fatal adverse drug reaction to trimethoprim. Aust Fam Physician 34:345–346. [PubMed] [Google Scholar]
- Naisbitt DJ, Farrell J, Gordon SF, Maggs JL, Burkhart C, Pichler WJ, Pirmohamed M, Park BK. (2002) Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex-restricted antigen presentation. Mol Pharmacol 62:628–637. [DOI] [PubMed] [Google Scholar]
- Nwokolo C, Byrne L, Misch KJ. (1988) Toxic epidermal necrolysis occurring during treatment with trimethoprim alone. Br Med J (Clin Res Ed) 296:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson A.(1996) Biotransformation of xenobiotics, in Casarett and Doull's Toxicology (Klaassen CD. ed), McGraw-Hill, New York: 113–186. [Google Scholar]
- Pearce RE, Lu W, Wang Y, Uetrecht JP, Correia MA, Leeder JS. (2008) Pathways of carbamazepine bioactivation in vitro. III. The role of human cytochrome P450 enzymes in the formation of 2,3-dihydroxycarbamazepine. Drug Metab Dispos 36:1637–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roujeau JC, Kelly JP, Naldi L, Rzany B, Stern RS, Anderson T, Auquier A, Bastuji-Garin S, Correia O, Locati F, et al. (1995) Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. N Engl J Med 333:1600–1607. [DOI] [PubMed] [Google Scholar]
- Rylance GW, George RH, Healing DE, Roberts DG. (1985) Single dose pharmacokinetics of trimethoprim. Arch Dis Child 60:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel CW, Grace ME, Nichol CA.(1973) Metabolism of trimethoprim in man and measurement of a new metabolite: a new fluorescence assay. J Infect Dis 128 (Suppl):580–583. [DOI] [PubMed] [Google Scholar]
- van Haandel L, Goldman JL, Pearce RE, Leeder JS. (2014) Urinary biomarkers of trimethoprim bioactivation in vivo following therapeutic dosing in children. Chem Res Toxicol 27:211–218. [DOI] [PubMed] [Google Scholar]
- Wen X, Wang JS, Backman JT, Laitila J, Neuvonen PJ. (2002) Trimethoprim and sulfamethoxazole are selective inhibitors of CYP2C8 and CYP2C9, respectively. Drug Metab Dispos 30:631–635. [DOI] [PubMed] [Google Scholar]
- Werk AN, Cascorbi I. (2014) Functional gene variants of CYP3A4. Clin Pharmacol Ther 96:340–348. [DOI] [PubMed] [Google Scholar]