

Abstract
Bupropion’s metabolism and the formation of hydroxybupropion in the liver by cytochrome P450 2B6 (CYP2B6) has been extensively studied; however, the metabolism and formation of erythro/threohydrobupropion in the liver and intestine by carbonyl reductases (CR) has not been well characterized. The purpose of this investigation was to compare the relative contribution of the two metabolism pathways of bupropion (by CYP2B6 and CR) in the subcellular fractions of liver and intestine and to identify the CRs responsible for erythro/threohydrobupropion formation in the liver and the intestine. The results showed that the liver microsome generated the highest amount of hydroxybupropion (Vmax = 131 pmol/min per milligram, Km = 87 μM). In addition, liver microsome and S9 fractions formed similar levels of threohydrobupropion by CR (Vmax = 98–99 pmol/min per milligram and Km = 186–265 μM). Interestingly, the liver has similar capability to form hydroxybupropion (by CYP2B6) and threohydrobupropion (by CR). In contrast, none of the intestinal fractions generate hydroxybupropion, suggesting that the intestine does not have CYP2B6 available for metabolism of bupropion. However, intestinal S9 fraction formed threohydrobupropion to the extent of 25% of the amount of threohydrobupropion formed by liver S9 fraction. Enzyme inhibition and Western blots identified that 11β-dehydrogenase isozyme 1 in the liver microsome fraction is mainly responsible for the formation of threohydrobupropion, and in the intestine AKR7 may be responsible for the same metabolite formation. These quantitative comparisons of bupropion metabolism by CR in the liver and intestine may provide new insight into its efficacy and side effects with respect to these metabolites.
Introduction
Bupropion is a norepinephrine/dopamine reuptake inhibitor (Stahl et al., 2004) clinically used for treatment of major depressive disorder (MDD) and smoking cessation (Fava et al., 2005). Currently, over 11 million prescriptions annually of bupropion have been issued to more than 40 million patients (Reese et al., 2008; Desmarais et al., 2011). Bupropion hydrochloride (HCl) extended release (ER) tablets, which is marketed as Wellbutrin XL by Biovail, has many generic manufacturers, such as Teva Pharmaceutical Industries/Impax Laboratories, Mylan, Actavis, and Par Pharmaceuticals, which use a current bioequivalence (BE) standard based on Cmax and AUC. The original approvals of the 300-mg generic bupropion HCl ER tablets were made on the basis of the demonstration of in vivo BE of the 150-mg generic bupropion HCl ER tablets compared to the brand name product, as well as other in vitro criteria. However, a follow up in vivo BE study on 300-mg Budeprion (bupropion HCl) ER tablets manufactured by Impax Laboratories and distributed by Teva Pharmaceuticals showed that the 300-mg strength failed to demonstrate BE (Woodcock et al., 2012). It is not clear if the failure of extrapolating the BE conclusion from 150-mg to 300-mg tablets was related to changes in metabolism of bupropion in the liver and intestine between different formulations and different strengths of bupropion. Therefore, it is important to study in detail the mechanisms of bupropion metabolism in the liver and intestine.
Bupropion is rapidly absorbed (Tmax 1.3–1.9 hours) and extensively distributed throughout the body (Vd = 19 l/kg), and less than 1% of the parent compound is eliminated in urine (Schroeder, 1983; Jefferson et al., 2005). The majority of bupropion is eliminated by metabolism. It is well known that bupropion forms three primary metabolites: hydroxybupropion (by CYP2B6) and the diastereoisomers threohydrobupropion and erythrohydrobupropion [by carbonyl reductase (CR)] (Loboz et al., 2005) (Fig. 1). Different metabolites of bupropion have significant impact on its efficacy, since these metabolites have 25–50% potency compared with bupropion on the basis of antidepressant screening tests in an animal model (Bondarev et al., 2003; Damaj et al., 2004). In addition, the plasma levels of hydroxybupropion are 5- to 10-fold higher than the parent drug bupropion after oral administration of bupropion HCl (Bondarev et al., 2003; Damaj et al., 2004; Jefferson et al., 2005; Damaj et al., 2010; Zhu et al., 2012).
Fig. 1.
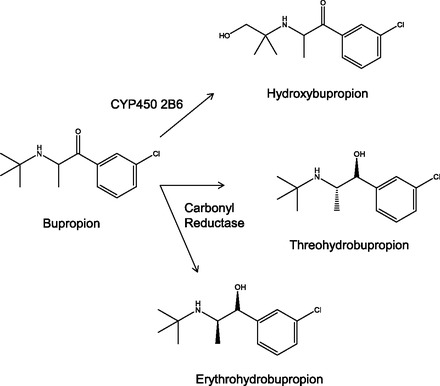
Bupropion and metabolism. Bupropion is metabolized by CYP2B6 to form hydroxybupropion and by carbonyl reductase to form the diastereoisomers threohydrobupropion and erythrohydrobupropion.
The metabolism of bupropion by CYP2B6 in the liver to form hydroxybupropion, the major metabolite of bupropion, has been extensively studied. Studies have shown that the kinetic formation of hydroxybupropion in liver microsome occurs to a high extent, with Vmax ranging from 85–254 pmol/mg per minute and Km ranging from 103 to 198 μM (Coles and Kharasch, 2008; Molnari and Myers, 2012; Skarydova et al., 2014). However, the metabolism of bupropion by CR has not been well characterized. For example, what metabolic pathways (CYP2B6 and CR) play a more important role in the liver and intestine for bupropion metabolism? Which subcellular fraction in the liver and intestine are responsible for metabolism of bupropion? Are there any differences in how bupropion is metabolized in the liver and in the intestines? Which CR is responsible for bupropion metabolism in the liver and the intestine?
To date, 11 CR enzymes are known, and they are categorized into two superfamilies: short-chain dehydrogenase/reductase (SDR) and aldo-keto reductase (AKR) (Rosemond and Walsh, 2004; Matsunaga et al., 2006). The SDR family has five CR enzymes: CBR1, CBR3, 11 β-dehydrogenase isozyme 1 (11β-HSD), DHRS4, and L-xylulose reductase. AKR family has six CR enzymes: AKR7A2, AKR7A3, AKR1C1, AKR1C2, AKR1C3, and AKR1C4. The subcellular locations of most CR enzymes are in the cytoplasm, except for 11β-HSD, which is localized in the microsomes (Matsunaga et al., 2006).
In this study we investigated the metabolism of bupropion in subcellular fractions (microsome, cytosolic, and S9 fractions) of the liver and intestine to compare the extent of formation of all three metabolites in the different subcellular fractions. In addition, we conducted inhibition studies with these subcellular fractions to determine which CR enzymes are important for bupropion metabolism. These results confirm that CYP2B6 in microsome is mainly responsible for hydroxybupropion. In comparison, in the liver microsome and S9 fractions, levels of threohydrobupropion formed by CR were similar to those for hydroxybupropion’s formation. This suggests that the metabolism of bupropion by CYP2B6 and its metabolism by CR in the liver are equally important. In contrast, in none of the intestinal fractions was hydroxybupropion detected, which suggests that the intestines do not contribute to the CYP2B6 metabolism of bupropion. Intestinal S9 fraction indeed generated threohydrobupropion; in fact, the amount was 25% of that formed from liver S9 fraction. Furthermore, the enzyme inhibition studies and Western blotting assay suggest that 11β-HSD is responsible for the formation of threohydrobupropion in the liver microsome, and aldo-keto reductase 7 may be responsible for the same metabolite in the intestine. These results quantitatively compare bupropion's metabolism by CR in liver and intestine, which may provide new insight into the contribution of the metabolites to bupropion’s efficacy.
Materials and Methods
Chemicals and Reagents.
Bupropion HCl and venlafaxine HCl (internal standard) were purchased from Sigma-Aldrich (St. Louis, MO). Hydroxybupropion was purchased from Caymen Chemicals (Ann Arbor, MI) and a racemic mixture of both erythrohydrobupropion and threohydrobupropion were purchased from Toronto Research Chemicals (Toronto, Canada). β-Nicotinamide adenine dinucleotide 2′-phosphate (NADPH) was also purchased from Sigma-Aldrich. Acetonitrile [high-performance liquid chromatography (HPLC) grade] and methanol (HPLC grade) were purchased from Fisher Scientific (Pittsburgh, PA). Water was purified with a Milli-Q water system (Bedford, MA). Mixed Gender Pooled Human Live Microsomes and cytosolic and S9 fractions for both liver and intestines (duodenum and jejunum) were purchased from Xenotech (Lenexa, KS). The following CR inhibitors were purchased from Sigma-Aldrich: rutin, fluefenamic acid, and carbenozolone. The following antibodies were used in the Western blot; AKR1C1/2 (Abcam, Cambridge, England; cat. no. ab131375), carbonyl reductase 1/2/3 (Santa Cruz Biotechnology, Dallas, TX; cat. no. sc-292143), 11β-hydroxysteroid dehydrogenase (Type 1; Cayman Chemical, Ann Arbor, MI; cat. no. 10004303), AKR7A antibody (Santa Cruz Biotechnology; cat. no. sc-32944), CYP2B6 antibody (Santa Cruz Biotechnology; cat. no. sc-67224), goat anti-mouse secondary antibody (Santa Cruz Biotechnology; cat. no. sc2005), and anti-rabbit IgG antibody (Cell Signaling Technology, Danvers, MA; cat. no. 7074).
Liquid Chromatography–Tandem Mass Spectrometry Method.
The liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis was conducted using an Agilent 1200 HPLC system coupled to an API 3200 mass spectrometer (Applied Biosystems/MDS Sciex, Toronto, Canada) equipped with an API electrospray ionization (ESI) source. Quantitative analysis was accomplished on a Supelco C18 (150 × 4.6 mm i.d., 5 μm). The mobile phases used were purified water + 0.04% formic acid (A) and methanol + 0.04% formic acid (B). The LC was run isocratic at 35% methanol + 0.04% and a flow rate of 0.8 ml/min. The LC-MS/MS was operated at positive ESI. The multiple reaction monitoring (MRM) transitions and collision energies were determined for bupropion, hydroxybupropion, threohydrobupropion, erythrohydrobupropion, and venlafaxine. The analytical data were processed by Analyst software (version 1.2; Applied Biosystems, Foster City, CA). The quantitation of bupropion, hydroxybupropion, threohydrobupropion, and erythrohydrobupropion in these in vitro assay were performed by MRM of the [M-H]+ ion, using an internal standard to establish peak area ratios. The method development was derived and optimized from previous studies that monitored bupropion and metabolites by HPLC or LC-MS/MS (Cooper et al., Glassman, 1984; Borges et al., 2004; Loboz et al., 2005; Wang et al., 2012).
Subcellular Kinetic Assay.
Liver and intestinal microsome, cytosolic, and S9 fractions were conducted with concentrations of bupropion as the substrate from 1 to 4000 μM dissolved in phosphate-buffered aline (PBS; 3.3 mM MgCl2 + 100 mM K2HP04 + 100 mM KH2HPO4 buffer pH 7.4) (no organic solvent was used in this system). The master mix consisted of microsome, cytosolic, or S9 fractions at a final concentration of 1 mg/ml, 4 μl of corresponding substrate, and PBS (3.3 mM MgCl2 + 100 mM K2HP04 + 100 mM KH2HPO4 buffer pH 7.4). A fresh sample of the cofactor nicotinamide adenine dinucleotide phosphate (NADPH) was prepared at 16.7 mg/ml in PBS buffer. Both master mix and NADPH were heated for 3 minutes at 37°C. Following, NADPH was added to master mix to initiate reaction. Sample was collected at 30 minutes; sample was spiked into ice-cold methanol containing 500 nM of internal standard (venlafaxine).
Carbonyl Reductase Inhibition Study.
For inhibition studies, bupropion substrate was used at the corresponding Km for threohydrobupropion (since this was the dominant metabolite formed by CR) determined from the subcellular kinetic analysis (liver microsome = 186 μM, liver S9 = 265 μM, liver cytosolic = 90 μM, intestinal microsome = 150 μM, intestinal cytosolic = 5.6 μM, and intestinal S9 = 573 μM). The master mix consisted of microsome, cytosolic, or S9 at a final concentration of 1 mg/ml, bupropion, PBS (3.3 mM MgCl2 + 100 mM potassium phosphate buffer pH 7.4), and CR inhibitor at 3-fold higher than the IC50 (the 3-fold IC50 values were: rutin 6.1 μM, flufenamic acid 60 μM, and carbenoxolone 250 nM). A fresh sample of the cofactor NADPH was prepared at 16.7 mg/ml in PBS buffer. Both master mix and NADPH was heated for 3 minutes at 37°C. Following, NADPH was added to master mix to initiate reaction. Samples were collected at 0, 30, 60, and 90 minutes; samples were spiked into ice-cold methanol containing 500 nM of internal standard (venlafaxine).
Standards and Sample Preparation.
Stock solutions of bupropion, hydroxybupropion, threohydrobupropion, or erythrohydrobupropion at 2 mg/ml were prepared in methanol to generate a working solution of 100 μg/ml. An aliquot of this solution was diluted in 1:1 MeOH/Milli-Q water to get a series of working standard solutions of 5, 10, 25, 50, 100, 250, 500, 1000, 2500, and 5000 ng/ml. Internal standard solution was prepared by diluting the stock solution of venlafaxine to yield a final concentration of 500 nM in 1:1 MeOH/Milli-Q water. After preparation of working standards, 50 μl of the appropriate concentrations of analyte was added to 150 μl of internal standard solution (500 nM of venlafaxine in 1:1 MeOH/Milli-Q water), and 50 μl of PBS. Fifty μl of sample from microsome, cytosolic, or S9 reaction at each time point was spiked into 150 μl of internal standard solution (500 nM of venlafaxine in 1:1 MeOH/Milli-Q water and 50 μl 1:1 MeOH/Milli-Q water). Samples were vortexed for 1 minute, followed by centrifugation for 15 minutes at 14,000 rpm in an Eppendorf centrifuge. The supernatant was transferred to vials and 5 μl was injected for LC-MS/MS analysis.
Western Blot.
Subcellular fractions—liver and intestinal microsome, S9, and cytosolic fractions—were lysed using radioimmunoprecipitation assay buffer (RIPA; 50 mM Tris-HCL, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, pH 7.4 ± 0.2; Boston BioProducts, Ashland, MA; BP-115) with 1% protease inhibitor and 1% EDTA. Approximately 200 μl of RIPA buffer was used to resuspend each subcellular fraction, which was incubated on ice for 30 minutes. Each sample was then centrifuged at 14,000 rpm for 15 minutes at 4°C. The protein concentration of the supernatant of each sample was quantified using Pierce BCA Protein Assay Kit (23225). All samples were diluted to have a protein concentration of 750 μg/ml. Laemmli sample buffer (Bio-Rad, Hercules, CA; 161-0737) was used according to protocol: Mix 950 μl of sample buffer with 50 μl of β-mercaptoethanol. Each sample was prepared by using 50 μl of protein sample with 25 μl of sample buffer and boiled at 95°C for 10 minutes. All samples were loaded on an SDS-PAGE gel (25 μl) and run at 200 V for about 2 hours. The SeeBlue Pre-Stained Protein Standard (Life Technologies, Grand Island, NY; LC5625) was used to determine protein molecular weights. The running buffer for the SDS-PAGE gel consisted of 3.0 g of Tris base, 14.4 g of glycine, and 1 g of SDS with ddH2O to 1 liter. A wet transfer was performed using transfer buffer (3.03 g of Tris base, glycine 14.4 g, 200 μl of methanol, and ddH2O to 1 liter). The transfer was done using a polyvinylidene fluoride immune-blot membrane (Bio-Rad; 162-0177) at 250 mV for 3 hours. The membrane was blocked for 1.5 hours using 5% of milk in Tris-buffered saline and Tween-20 [TBST buffer: 2.4 g of Tris, 8 g of NaCl, pH adjusted to 7.6 with HCl, 0.1% Tween-20 (v/v), and 1 liter of ddH2O]. Primary antibody was added to membrane at various dilutions according to manufacturer’s protocol: AKR1C1/2 antibody (1:500), CRB1/2/3 (1:500), 11β-hydroxysteroid dehydrogenase (1:200), AKR7 (1:200), and CYP2B6 (1:200) and incubated at 4°C overnight. Membrane was washed with TBST (3×) before the corresponding secondary antibody was added: AKR1C1/2 (dilution 1:2000), CRB1/2/3 (1:2000), 11β-hydroxysteroid dehydrogenase (1:5000), AKR7 (1:5000), and CYP2B6 (1:5000) for 1.5 hours at room temperature. The membrane was washed again with TBST (3×). Stripping buffer (Thermo Scientific, Sunnyvale, CA; cat. no. 21059) was used to remove previous antibody; we confirmed that the antibody was washed out each time. Proteins were detected using X-ray development; 5 ml of substrate (2.5 ml of reagent 1 and 2.5 ml of reagent 2) was added to the membrane before detection (Thermo Scientific; cat. nos. 1859701 and 1859698).
Data Analysis.
For microsome, cytosolic, and S9 kinetics, all data were converted into pmol/min per milligram and plotted against concentration of bupropion. Graphpad Prism 5 was used to simulate the Km and Vmax with the Michaelis-Menten model using the following equation.
| (1) |
The intrinsic clearance for S9 formation of each metabolite was calculated using the following equation.
| (2) |
For statistical analysis, R version 3.0.3 was run with a t test.
Results
LC-MS/MS Development for Bupropion and Metabolites
An LC-MS/MS method was developed to quantify bupropion, hydroxybupropion, erythrohydrobupropion, and threohydrobupropion. Since the fragmentations of the diastereoisomers (threohydrobupropion and erythrohydrobupropion) were the same and bupropion also had very similar fragmentation, it was necessary for all analytes to be separated by LC. Figure 2A shows the MRM chromatograms of the successful separation of all analytes. The MS parameters are highlighted in Fig. 2B for each analyte.
Fig. 2.
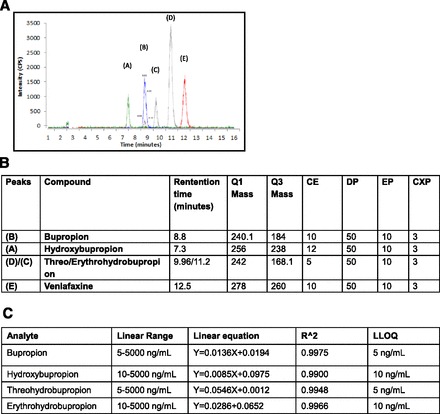
Method development for bupropion and metabolites. (A) Separation of bupropion and metabolite for detection using LC-MS/MS. (B) LC/MS parameters for bupropion and metabolites. (C) Validation with standards for bupropion and metabolites. All analytes had a good linear range with acceptable coefficient of determination.
Calibration curves for each analyte were performed to quantify samples in later studies. A wide linear range was achieved for each analyte (Fig. 2C). In addition, the coefficient of determination for each analyte was greater than or equal to 0.99. The lower limit of detection was either 5 or 10 ng/ml (noted in Fig. 2C) depending on which analyte was being monitored.
Metabolism in Liver Subcellular Fractions
To begin with, we used liver microsome, cytosolic, and S9 fractions to look at bupropion’s metabolism. Bupropion was used as the substrate at concentrations ranging from 1 to 4000 μM. Samples were analyzed by LC-MS/MS to establish the kinetics, and bupropion, hydroxybupropion, threohydrobupropion, and erythrohydrobupropion were monitored. Figure 3, A–C shows the formation of hydroxybupropion in liver microsome, S9 fraction, and cytosolic fraction respectively. Hydroxybupropion was formed to the highest extent in liver microsome (Km = 87.98 ± 20.2 μM and Vmax = 131.2 ± 5.6 pmol/min per milligram), which was expected since microsomes typically contain concentrated amounts of P450s. In the S9 fraction, hydroxybupropion formation was still apparent but the formation occurred to a lower extent (Km = 99.53 ± 18.91 μM and Vmax = 51.45 ± 1.9 pmol/min per milligram). Hydroxybupropion formation in the cytosolic fraction was almost negligible (Km = 71.35 ± 127 μM and Vmax = 1.594 ± 0.52 pmol/min per milligram). These results suggest that P450 enzymes that are subcellularly localized in microsomes are responsible for the formation of hydroxybupropion in the liver.
Fig. 3.
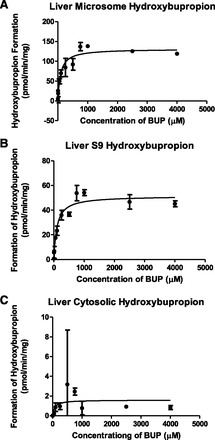
Hydroxybupropion metabolite formation in liver subcellular fractions. Hydroxybupropion formation is indicated in (A) liver microsome, (B) liver S9, and (C) liver cytosolic fractions. Data are presented as mean ± S.D. (n = 3).
Threohydrobupropion was also formed in all subcellular liver fractions (Fig. 4, A–C). The extent of formation in both microsome and S9 fractions were about the same; however, the affinity differed slightly (microsome: Km = 186.3 ± 53.48 μM and Vmax = 98.37 ± 6.6 pmol/min per milligram; S9: Km = 265.7 ± 77.79 μM and Vmax = 99 ± 7.5 pmol/min per milligram). In the cytosolic fraction, threohydrobupropion was formed to a lesser extent (Vmax: 14.56 ± 0.714 pmol/min per milligram and Km: 89.82 ± 22 μM). These results suggested that the CR enzyme that is localized subcellularly in the microsomes, 11β-hydroxysteroid dehydrogenase, plays a major role in the conversion of bupropion to threohydrobupropion. In addition, since the cytosolic fraction still forms threohydrobupropion to some extent, this suggested there may be multiple CR enzymes responsible for this metabolism.
Fig. 4.
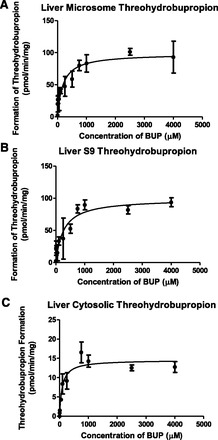
Threohydrobupropion metabolite formation in liver subcellular fractions. Threohydrobupropion formation is indicated in (A) liver microsome, (B) liver S9, and (C) liver cytosolic fractions. Data are presented as mean ± S.D. (n = 3).
Finally, we saw that erythrohydrobupropion was also formed in liver microsome, S9, and cytosolic fractions (Fig. 5, A–C, respectively); however, the extent of formation was very small in all subcellular fractions (microsome: Km = 41.45 ± 26.62 μM and Vmax = 2.649 ± 0.3 pmol/min per milligram; cytosolic: Km= 274.4 ± 254 μM and Vmax = 3.654 ± 1.2 pmol/min per milligram; S9: Km = 107 ± 32.14 μM and Vmax = 4.23 ± 0.286 pmol/min per milligram). These results suggest that hydroxybupropion and threohydrobupropion are the dominant metabolites in the liver subcellular fraction. Yet, as with threohydrobupropion, since formation of erythrohydrobupropion occurred in both the microsome and cytosolic fractions, this again suggested that multiple CR enzymes may be involved in erythrohydrobupropion’s formation. Liver kinetics are summarized in Table 1.
Fig. 5.
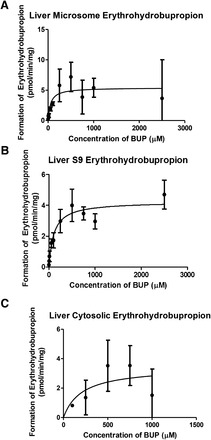
Erythrohydrobupropion metabolite formation in liver subcellular fractions. Erythrohydrobupropion formation is indicated in (A) liver microsome, (B) liver S9, and (C) liver cytosolic fractions. Data are presented as mean ± S.D. (n = 3).
TABLE 1.
Summary of subcellular kinetics: Vmax and Km for each metabolite in liver microsome, S9, and cytosolic fractions
| Liver Microsome |
Liver S9 |
Liver Cytosolic |
||||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax | Km | Vmax | Km | |
| pmol/min per milligram | μM | pmol/min per milligram | μM | pmol/min per milligram | μM | |
| HBUP | 131.2 ± 5.8 | 87.9 ± 20.2 | 51.4 ± 1.9 | 99.5 ± 18.9 | 1.5 ± 0.5 | 71.3 ± 127 |
| TBUP | 98.4 ± 6.6 | 186.3 ± 53.5 | 99 ± 7.5 | 265.7 + 77.7 | 14.5 + 0.7 | 89.8 ± 22 |
| EBUP | 2.6 ± 0.3 | 41.4 ± 26.6 | 4.2 ± 0.28 | 107 ± 32.1 | 3.65 ± 1.2 | 274 ± 254 |
HBUP, hydroxybupropion; TBUP, threohydrobupropion; EBUP, erythrohydrobupropion.
Using the Michaelis-Menten kinetic parameters (Vmax and Km) we were able to calculate the intrinsic clearance for each metabolite using the S9 fraction (since this contains both microsome and cytosolic fractions) in the liver (Table 3). After adding each metabolite, intrinsic clearance, we calculated that the liver contributes a CLint of 931.8 μl/min per milligram.
TABLE 3.
Estimated intrinsic clearance
Using eq. (2) the intrinsic clearance for both liver and intestinal S9 fractions was calculated on the basis of the Michaelis Menten equation, assuming the linear portion of the curve. The relative contribution for each metabolite in S9 liver or intestines is indicated in the table.
| Subcellular Fraction | Metabolite | Clint |
|---|---|---|
| Liver S9 fraction | Hydroxybupropion | 519 μl/min per milligram |
| Threohydrobupropion | 372 μl/min per milligram | |
| Erythrohydrobupropion | 39 μl/min per milligram | |
| Total Clint from liver | 931.8 μl/min per milligram | |
| Intestinal S9 fraction | Hydroxybupropion | N/A |
| Threohydrobupropion | 45 μl/min per milligram | |
| Erythrohydrobupropion | N/A | |
| Total Clint from intestines | 45 μl/min per milligram | |
| Total contribution from liver and intestinal S9 fractions | 976 μl/min per milligram |
Metabolism in Intestinal Subcellular Fractions
We continued to evaluate the metabolism of bupropion using intestinal microsome, cytosolic, and S9 fractions. As in liver metabolism, we used bupropion at concentrations from 1 to 4000 μM and analyzed samples by LC-MS/MS to establish the kinetics of hydroxybupropion, erythrohydrobupropion, and threohydrobupropion. However, unlike the liver fractions, where all metabolites were detected, the only metabolite that formed through the intestinal metabolism was threohydrobupropion. Both hydroxybupropion and erythrohydrobupropion were undetectable in both intestinal microsome, cytosolic, and S9 fractions. This suggested that CYP2B6 metabolism does not occur in the intestines since hydroxybupropion was unable to form.
The extent in which threohydrobupropion was formed was less than its formation in the liver (Fig. 6, A–C) (microsome: Km = 149.9 ± 28.8 μM and Vmax = 5.55 ± 0.4 pmol/min per milligram; cytosolic: Km = 569 ± 64.89 μM and Vmax = 5.649 ± 0.214 pmol/min per milligram and S9: Km = 573.4 ± 188.9 μM and Vmax = 25.87 ± 2.8 pmol/min per milligram). The formation of threohydrobupropion was 25% of the formation in the liver S9 fraction. Similar to results for the liver, these data suggest that multiple CR enzymes are involved in the formation of this metabolite. The intestinal S9 kinetics formation of each metabolite are summarized in Table 2.
Fig. 6.
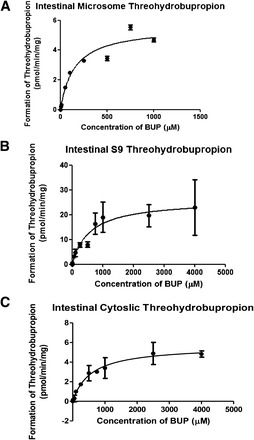
Threohydrobupropion metabolite formation in intestinal subcellular fractions. Threohydrobupropion formation is indicated in (A) intestinal microsome, (B) intestinal S9, and (C) intestinal cytosolic fractions. Data are presented as mean ± S.D. (n = 3).
TABLE 2.
Summary of subcellular kinetics: Vmax and Km for threohydrobupropion metabolite in intestinal microsome, S9, and cytosolic fractions
| Intestinal Μicrosome |
Intestinal S9 |
Intestinal Cytosolic |
||||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax | Km | Vmax | Km | |
| pmol/min per milligram | μM | pmol/min per milligram | μM | pmol/min per milligram | μM | |
| HBUP | NF | NF | NF | NF | NF | NF |
| TBUP | 5.55 ± 0.3 | 149.9 ± 28.8 | 25.87 ± 2.8 | 573.4 ± 188 | 5.6 ± 0.2 | 569 ± 64.8 |
| EBUP | NF | NF | NF | NF | NF | NF |
HBUP, hydroxybupropion; TBUP, threohydrobupropion; EBUP, erythrohydrobupropion; NF, no metabolite formation occurred.
As with the liver, Michaelis-Menten kinetic parameters (Vmax and Km) enabled us to calculate the intrinsic clearance for threohydrobupropion using the S9 fraction in the intestines (Table 3). This was about 20-fold lower than the liver CLint clearance since two of the metabolites did not form in the intestines and threohydrobupropion formation was 25% of the formation in the liver S9 fraction. Nevertheless, the CLint in the intestinal S9 fraction was estimated to be 45 μl/min per milligram.
Metabolite Inhibition by Carbonyl Reductase Inhibitors
Next we went on to evaluate which CR enzymes are important for the reduction of bupropion and whether there might be multiple enzymes involved in this process. Using the microsome, cytosolic, and S9 fractions assay, we added various CR inhibitors and analyzed the reduction of metabolite formation. The inhibitors that were chosen were the following: rutin, which has been shown to target the CRB family of CR at an IC50 of 2.1 μM; flufenamic acid, which has been reported to inhibit AKR family 67% at concentrations of 20 μM; and carbenoxolone, which targets the microsomal CR 11β-hydroxysteroid dehydrogenase at IC50 values in the nanomolar range (Rosemond et al., 2004; Su et al., 2007; Carlquist et al., 2008). We monitored all metabolite formation with each inhibitor and compared these results to a control with no inhibitor.
For the formation of hydroxybupropion, none of the three inhibitors had a significant effect compared with control on any liver subcellular fraction (Fig. 7A), as expected since formation of this metabolite occurs via CYP2B6. In addition, this metabolite was again not detected in any intestinal fraction. These results suggested no CRs are involved in formation of hydroxybupropion.
Fig. 7.
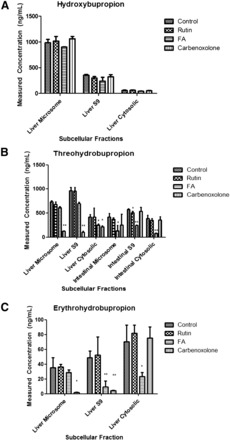
Metabolite inhibition by carbonyl reductase inhibitors. (A) Hydroxybupropion’s direct measured concentration. (B) Threohydrobupropion’s direct measured concentration. (C) Erythrohydrobupropion’s direct measured concentration. FA, flufenamic acid. Data are presented as mean ± S.D. (n = 3). *P < 0.05, **P < 0.01.
However, for threohydrobupropion formation, inhibition was observed in both the liver and intestinal subcellular fractions. In the liver, carbenoxolone, the inhibitor of 11β-hydroxysteroid dehydrogenase, showed as much as 82.4% inhibition compared with control in the liver microsome. Flufenamic acid was shown to have about a 40% inhibition on threohydrobupropion formation in liver cytosolic fraction (Fig. 7B). These results suggested that in the liver, 11β-hydroxysteroid dehydrogenase was the dominant enzyme in the liver for reduction of bupropion to threohydrobupropion.
However, in the intestinal subcellular fractions, carbenoxolone seemed to have no significant effect on inhibiting threohydrobupropion formation. Furthermore, flufenamic acid showed inhibition on intestinal fractions ranging from 57.8 to 78.7% of threohydrobupropion formation (Fig. 7B). Minor inhibition was seen with rutin, implicating minor involvement of the CRB family of CR enzymes in the formation of threohydrobupropion. Altogether, the liver and intestinal data for the formation of threohydrobupropion suggest that both 11β-hydroxysteroid dehydrogenase and the AKR family of CR enzymes are the major CR enzymes responsible for threohydrobupropion formation.
In the same way, erythrohydrobupropion formation was inhibited by both carbenoxolone and flufenamic acid (Fig. 7C). Carbenoxolone inhibited the formation of erythrohydrobupropion by 95% in liver microsome and 91.6% in liver S9 fraction yet had no effect on liver cytosolic fraction. Flufenamic acid showed about 67–88% inhibition in the liver S9 and cytosolic fractions. These results suggested that 11β-hydroxysteroid dehydrogenase and AKR family are the dominant enzymes that form erythrohydrobupropion in the liver.
Western Blot.
Finally, we went on to confirm whether these various CR enzymes are expressed in different subcellular fractions. Analysis of protein expression was performed using an immunoblot after separation by SDS-PAGE gel (Fig. 8). Microsome, cytosolic, and S9 fractions of both liver and intestines were examined for CYP2B6, 11β-hydroxysteroid dehydrogenase, CRB1/2/3, AKR7 family, and AKR1A family.
Fig. 8.
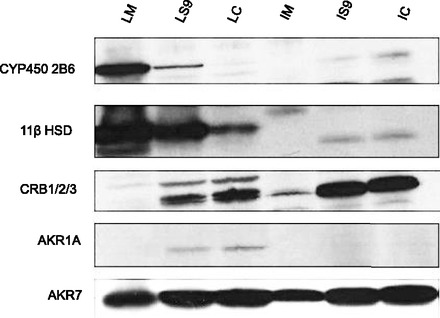
Enzyme expression in human subcellular fractions. Subcellular fractions were run on an SDS-polyacrylamide gradient (4–12% w/v) gel to detect various carbonyl reductase enzymes and CYP2B6. Lane 1: liver microsome (LM). Lane 2: liver S9 (LS9). Lane 3: liver cytosolic (LC). Lane 4: intestinal microsome (IM). Lane 5: intestinal S9 (IS9). Lane 3: intestinal cytosolic (IC).
It was observed that CYP2B6 was primarily expressed in liver microsome with minor expression in the liver S9 fraction (lane 1 and 2). CYP2B6 was absent in liver cytosolic (lane 3) and all intestinal fractions (Fig. 8, lane 4–6). This is consistent with the metabolite formation data suggesting that hydroxybupropion is formed predominantly in liver microsome and S9 fractions and does not participate in intestinal metabolism of bupropion. 11β Hydroxysteroid dehydrogenase was highly expressed in liver microsome and S9 fractions (lanes 1 and 2); its expression in the intestines was almost nonexistent, supporting our results with the inhibition data that 11β-hydroxysteroid dehydrogenase activity is dominant in the liver. The CRB1/2/3 enzymes were primarily found to be expressed in both liver and intestinal S9 and cytosolic fractions (lanes 2, 3, 5, 6); however, these enzymes may not be important in bupropion’s metabolism, as suggested by the inhibition data. The AKR1A family had very little expression in any of the subcellular fractions except minor expression in liver S9 and liver cytosolic (Fig. 8, lanes 2 and 3). Finally, the AKR7 family enzymes were found to be expressed in all subcellular fractions. This supported the CR inhibition data seen with flufenamic acid in both liver and intestines. Altogether, the enzyme expression data verified the results seen in the formation and inhibition studies as those enzymes were expressed in the corresponding subcellular fraction.
Discussion
In our studies, we show that the formation of hydroxybupropion and threohydrobupropion in the liver (microsome and S9 fractions) occur to a similar extent. In addition, we show that no CYP2B6 expression or metabolism to form hydroxybupropion occurs in the intestines. However, the only metabolite that forms in the intestines is threohydrobupropion. Its formation in the intestinal S9 fraction is 25% of that seen in the liver S9 fraction. Furthermore, inhibition studies prove that there are multiple CR enzymes involved in the metabolism of bupropion to threohydrobupropion; and the CR activity may have a gastrointestinal-region dependency that influences the metabolism of the parent compound. Western blots confirmed that the CR enzymes important for metabolizing bupropion are consistent with the expression in subcellular fractions.
Previous studies have shown that CYP2B6 metabolism of bupropion forms hydroxybupropion (Kirchheiner et al., 2003; Coles and Kharasch, 2008; Kharasch et al., 2008; Benowitz et al., 2013; Ilic et al., 2013). In addition, studies have also suggested that other P450s, such as CYP2C19, CYP2E1, and CYP3A4, might have minor roles in the hydroxybupropion formation, but this still needs to be confirmed (Chen et al., 2010). Therefore, on the basis of liver microsome stability assays, it was thought that hydroxybupropion was the major metabolite. Several studies failed to realize that the CR pathway to form threohydrobupropion and erythrohydrobupropion may not occur extensively in liver microsomes since most of the CR enzymes involved are located subcellularly within the cytosol (Coles and Kharasch, 2008; Meyer et al., 2013).
Therefore, examining all subcellular fractions—microsome, cytosolic, and S9 fractions—will help explain more broadly which enzymes are responsible for bupropion’s metabolism and at what rate its metabolites are formed. Typically, P450 enzymes are located in microsomes. On the other hand, most CRs are located subcellularly in cytosolic fractions, expect for 11β-HSD, one of the few CR enzymes located subcellularly in microsomes. Using an S9 fraction, which contains both cytosol and microsomes, allowed us to compare metabolite formations across the three metabolites. We found that in the S9 fractions threohydrobupropion has a 2-fold higher formation compared with hydroxybupropion, suggesting that many CR enzymes have been overlooked as contributors to bupropion’s metabolism. Although hydroxybupropion formation in microsomes is slightly higher than that in S9 fractions, this difference in activity between microsomal and S9 fractions is normal, P450 enzymes are concentrated in microsomes, whereas S9 fraction contains both P450s and cytosolic fraction (Brandon et al., 2003; Jia and Liu, 2007).
In Molnari et al. (2012), the authors found that threohydrobupropion was the major metabolite in liver microsome, which disagrees with the results presented here and many previous studies that identified hydroxybupropion as the major metabolite. Although in our studies threohydrobupropion was the major metabolite formed in liver S9 fraction, hydroxybupropion still forms to the highest extent in liver microsomes. Moreover, Molnari et al. saw no change with flufenamic acid in inhibition studies in the liver, whereas we did. However, this inhibitor seemed to have a greater effect in intestinal fractions, which was not pursued in the Molnari study. Likewise in Meyer et al. (2013) the authors showed that 11 β-HSD was the CR enzyme important for metabolizing bupropion to form threohydrobupropion. Although our data agrees with this, a broader analysis of subcellular fractions was possible. In the Meyer study, the authors examined only liver microsomes, so only 11 β-HSD activity could have been observed. Instead, cytosolic fractions also needed examination in order to determine if multiple CR enzymes were contributing to bupropion metabolism.
Our results suggest that both hydroxybupropion and threohydrobupropion are important metabolites for elucidating the metabolism of bupropion. This is consistent with in vivo studies that looked at the pharmacokinetic levels of bupropion and metabolites and showed plasma concentrations of both hydroxybupropion and threohydrobupropion higher than that of the parent drug bupropion (erythrohydrobupropion concentration was minor or undetectable) (Laizure et al., 1985).
To the best of our knowledge, no authors have studied the formation of bupropion’s metabolites in any intestinal fractions. The intestines have been shown to be involved in both phase I and phase II metabolism, which might influence the metabolism of bupropion. Although P450 enzyme expression is typically lower in the intestines than in liver (20 pmol/mg of microsome compared with 300 pmol/mg of microsome) (Peters and Kremers, 1989), metabolism in this region of the gastrointestinal tract should still be investigated. Likewise, the expression of CR enzymes has been found to be highly concentrated in both liver and small intestines (Peters and Kremers, 1989; Gervot et al., 1999). We did not observe any hydroxybupropion (CYP2B6 metabolism) in the intestines, and our finding is consistent with another study that looked for various P450 expression in intestinal microsomes and likewise saw no CYP2B6 (Paine et al., 2006). However, in this study threohydrobupropion metabolized by CR was able to form in all three subcellular intestinal fractions (microsome, S9, and cytosolic), again showing how previous studies have discounted the CR pathway for metabolism of bupropion. The intestinal subcellular fractions used in these studies were taken from the duodenum and jejunum. The intestinal metabolism is an important concept to understand since these metabolites are active. However, a more thorough analysis would be needed to disprove or prove the hypothesis, and this would also be true in vivo.
In conclusion, these results suggest that bupropion metabolism can differ depending on the subcellular localization and tissue type, and that different metabolites are formed by multiple enzymes (both P450s and CRs).
Acknowledgments
The authors thank Ruijuan Luo and Ting Zhao for their advice on method development.
Abbreviations
- ARK
aldo-keto reductase
- BE
bioequivalence
- CR
carbonyl reductase
- ER
extended release
- HPLC
high-performance liquid chromatography
- 11β-HSD
11 β-dehydrogenase isozyme 1
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MRM
multiple reaction monitoring
- P450
cytochrome P450
- PBS
phosphate-buffered saline
- TBST
Tris-buffered saline and Tween-20
Authorship Contributions
Participated in research design: Connarn, Zhang, Babiskin, Sun.
Conducted experiments: Connarn.
Performed data analysis: Connarn.
Wrote or contributed to the writing of the manuscript: Connarn.
Footnotes
Funding was provided by the Food and Drug Administration (FDA) [HHSF223201310183C]
Disclaimer: The views expressed in this article are those of the authors and not necessarily those of the Food and Drug Administration (FDA).
References
- Benowitz NL, Zhu AZ, Tyndale RF, Dempsey D, Jacob P., 3rd (2013) Influence of CYP2B6 genetic variants on plasma and urine concentrations of bupropion and metabolites at steady state. Pharmacogenet Genomics 23:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarev ML, Bondareva TS, Young R, Glennon RA. (2003) Behavioral and biochemical investigations of bupropion metabolites. Eur J Pharmacol 474:85–93. [DOI] [PubMed] [Google Scholar]
- Borges V, Yang E, Dunn J, Henion J. (2004) High-throughput liquid chromatography-tandem mass spectrometry determination of bupropion and its metabolites in human, mouse and rat plasma using a monolithic column. J Chromatogr B Analyt Technol Biomed Life Sci 804:277–287. [DOI] [PubMed] [Google Scholar]
- Brandon EF, Raap CD, Meijerman I, Beijnen JH, Schellens JH. (2003) An update on in vitro test methods in human hepatic drug biotransformation research: pros and cons. Toxicol Appl Pharmacol 189:233–246. [DOI] [PubMed] [Google Scholar]
- Carlquist M, Frejd T, Gorwa-Grauslund MF. (2008) Flavonoids as inhibitors of human carbonyl reductase 1. Chem Biol Interact 174:98–108. [DOI] [PubMed] [Google Scholar]
- Chen Y, Liu HF, Liu L, Nguyen K, Jones EB, Fretland AJ. (2010) The in vitro metabolism of bupropion revisited: concentration dependent involvement of cytochrome P450 2C19. Xenobiotica 40:536–546. [DOI] [PubMed] [Google Scholar]
- Coles R, Kharasch ED. (2008) Stereoselective metabolism of bupropion by cytochrome P4502B6 (CYP2B6) and human liver microsomes. Pharm Res 25:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TB, Suckow RF, Glassman A. (1984) Determination of bupropion and its major basic metabolites in plasma by liquid chromatography with dual-wavelength ultraviolet detection. J Pharm Sci 73:1104–1107. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. (2004) Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol 66:675–682. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Grabus SD, Navarro HA, Vann RE, Warner JA, King LS, Wiley JL, Blough BE, Lukas RJ, Carroll FI. (2010) Effects of hydroxymetabolites of bupropion on nicotine dependence behavior in mice. J Pharmacol Exp Ther 334:1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmarais JE, Beauclair L, Margolese HC. (2011) Switching from brand-name to generic psychotropic medications: a literature review. CNS Neurosci Ther 17:750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M, Rush AJ, Thase ME, Clayton A, Stahl SM, Pradko JF, Johnston JA. (2005) 15 years of clinical experience with bupropion HCl: from bupropion to bupropion SR to bupropion XL. Prim Care Companion J Clin Psychiatry 7:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervot L, Rochat B, Gautier JC, Bohnenstengel F, Kroemer H, de Berardinis V, Martin H, Beaune P, de Waziers I. (1999) Human CYP2B6: expression, inducibility and catalytic activities. Pharmacogenetics 9:295–306. [PubMed] [Google Scholar]
- Ilic K, Hawke RL, Thirumaran RK, Schuetz EG, Hull JH, Kashuba AD, Stewart PW, Lindley CM, Chen ML. (2013) The influence of sex, ethnicity, and CYP2B6 genotype on bupropion metabolism as an index of hepatic CYP2B6 activity in humans. Drug Metab Dispos 41:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson JW, Pradko JF, Muir KT. (2005) Bupropion for major depressive disorder: Pharmacokinetic and formulation considerations. Clin Ther 27:1685–1695 . [DOI] [PubMed] [Google Scholar]
- Jia L, Liu X. (2007) The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr Drug Metab 8:822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharasch ED, Mitchell D, Coles R. (2008) Stereoselective bupropion hydroxylation as an in vivo phenotypic probe for cytochrome P4502B6 (CYP2B6) activity. J Clin Pharmacol 48:464–474. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Klein C, Meineke I, Sasse J, Zanger UM, Mürdter TE, Roots I, Brockmöller J. (2003) Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 13:619–626 . [DOI] [PubMed] [Google Scholar]
- Laizure SC, DeVane CL, Stewart JT, Dommisse CS, Lai AA. (1985) Pharmacokinetics of bupropion and its major basic metabolites in normal subjects after a single dose. Clin Pharmacol Ther 38:586–589. [DOI] [PubMed] [Google Scholar]
- Loboz KK, Gross AS, Ray J, McLachlan AJ. (2005) HPLC assay for bupropion and its major metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 823:115–121. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Shintani S, Hara A. (2006) Multiplicity of mammalian reductases for xenobiotic carbonyl compounds. Drug Metab Pharmacokinet 21:1–18. [DOI] [PubMed] [Google Scholar]
- Meyer A, Vuorinen A, Zielinska AE, Strajhar P, Lavery GG, Schuster D, Odermatt A. (2013) Formation of threohydrobupropion from bupropion is dependent on 11β-hydroxysteroid dehydrogenase 1. Drug Metab Dispos 41:1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnari JC, Myers AL. (2012) Carbonyl reduction of bupropion in human liver. Xenobiotica 42:550–561. [DOI] [PubMed] [Google Scholar]
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. (2006) The human intestinal cytochrome P450 “pie”. Drug Metab Dispos 34:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters WH, Kremers PG. (1989) Cytochromes P-450 in the intestinal mucosa of man. Biochem Pharmacol 38:1535–1538. [DOI] [PubMed] [Google Scholar]
- Reese MJ, Wurm RM, Muir KT, Generaux GT, St John-Williams L, McConn DJ. (2008) An in vitro mechanistic study to elucidate the desipramine/bupropion clinical drug-drug interaction. Drug Metab Dispos 36:1198–1201. [DOI] [PubMed] [Google Scholar]
- Rosemond MJ, Walsh JS. (2004) Human carbonyl reduction pathways and a strategy for their study in vitro. Drug Metab Rev 36:335–361. [DOI] [PubMed] [Google Scholar]
- Rosemond MJ, St John-Williams L, Yamaguchi T, Fujishita T, Walsh JS. (2004) Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: role of human liver cytosolic aldo-keto reductases. Chem Biol Interact 147:129–139. [DOI] [PubMed] [Google Scholar]
- Schroeder DH. (1983) Metabolism and kinetics of bupropion. J Clin Psychiatry 44:79–81. [PubMed] [Google Scholar]
- Skarydova L, Tomanova R, Havlikova L, Stambergova H, Solich P, Wsol V. (2014) Deeper insight into the reducing biotransformation of bupropion in the human liver. Drug Metab Pharmacokinet 29:177–184. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned-Coughlin S. (2004) A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion J Clin Psychiatry 6:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Vicker N, Lawrence H, Smith A, Purohit A, Reed MJ, Potter BV. (2007) Inhibition of human and rat 11beta-hydroxysteroid dehydrogenase type 1 by 18beta-glycyrrhetinic acid derivatives. J Steroid Biochem Mol Biol 104:312–320. [DOI] [PubMed] [Google Scholar]
- Wang X, Vernikovskaya DI, Abdelrahman DR, Hankins GD, Ahmed MS, Nanovskaya TN. (2012) Simultaneous quantitative determination of bupropion and its three major metabolites in human umbilical cord plasma and placental tissue using high-performance liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 70:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock J, Khan M, Yu LX. (2012) Withdrawal of generic budeprion for nonbioequivalence. N Engl J Med 367:2463–2465. [DOI] [PubMed] [Google Scholar]
- Zhu AZ, Cox LS, Nollen N, Faseru B, Okuyemi KS, Ahluwalia JS, Benowitz NL, Tyndale RF. (2012) CYP2B6 and bupropion’s smoking-cessation pharmacology: the role of hydroxybupropion. Clin Pharmacol Ther 92:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]