

Abstract
There are currently no registered drugs that slow the progression of neurodegenerative diseases, in part because translation from animal models to the clinic has been hampered by poor distribution to the brain. The present studies examined a selected series of para-phenyl–substituted diindolylmethane (C-DIM) compounds that display anti-inflammatory and neuroprotective efficacy in vitro. We postulated that the pharmacokinetic behavior of C-DIM compounds after oral administration would correlate with neuroprotective efficacy in vivo in a mouse model of Parkinson’s disease. Pharmacokinetics and metabolism of 1,1-bis(3′-indolyl)-1-(p-methoxyphenyl)methane (C-DIM5), 1,1-bis(3′-indolyl)-1-(phenyl)methane, 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (C-DIM8), and 1,1-bis(3′-indolyl)-1-(p-chlorophenyl)methane (C-DIM12) were determined in plasma and brain of C57Bl/6 mice after oral and intravenous administration at 10 and 1 mg/Kg, respectively. Putative metabolites were measured in plasma, liver, and urine. C-DIM compounds given orally displayed the highest area under the curve, Cmax, and Tmax levels, and C-DIM12 exhibited the most favorable pharmacokinetics of the compounds tested. Oral bioavailability of each compound ranged from 6% (C-DIM8) to 42% (C-DIM12). After pharmacokinetic studies, the neuroprotective efficacy of C-DIM5, C-DIM8, and C-DIM12 (50 mg/Kg per oral) was examined in mice exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and probenecid for 14 days, a model of progressive neurodegeneration with a strong neuroinflammatory component. C-DIM5 and C-DIM12 given orally once daily after one week of exposure to MPTP and probenecid prevented further loss of dopaminergic neurons in the substantia nigra pars compacta and striatal dopamine terminals, indicating that these compounds could be effective therapeutic agents to prevent neurodegeneration.
Introduction
Efficient delivery of chemotherapeutics to the brain represents one of the main challenges in the development of effective treatments for neurodegenerative disorders. In addition to the difficulty of brain distribution, toxicity and lack of clinical efficacy limit the use of many potential compounds that demonstrate efficacy in animal models. Small molecule therapeutics with a central nervous system (CNS) indication have lower approval rates and longer development periods, compared with compounds targeted to peripheral tissues (Glass et al., 2010), contributing to the current lack of neuroprotective modalities for treating neurodegenerative disorders. Furthermore, poor performance of translational animal models of neurodegeneration in predicting clinical outcomes has limited the progress of therapeutic development for CNS disorders (Schintu et al., 2009; Carta et al., 2011). Thus, a great need exists for orally bioavailable drugs for treatment of diseases, such as Alzheimer’s, Parkinson’s disease (PD), amyotrophic lateral sclerosis, and multiple sclerosis (Polazzi and Monti, 2010).
One class of compounds with potential neuroprotective efficacy is based on 3-3′-diindolylmethane, an acidic condensation product of indole-3-carbinol, a phytochemical found in cruciferous vegetables. 3-3′-Diindolylmethane has been extensively studied as a potential cancer chemotherapeutic compound (Safe et al., 2008), and several novel para-phenyl–substituted diindolylmethanes (C-DIMs) demonstrate effective inhibition of tumor cell growth through activation or inactivation of nuclear receptors, including Nur77/NR4A1 (Lee et al., 2010) and Nurr1/NR4A2 (Inamoto et al., 2008). In the CNS, Nurr1 is expressed during the development of dopamine neurons in the sustantia nigra (SN), the portion of the ventral midbrain affected in PD (Saucedo-Cardenas et al., 1998). Of interest, Nurr1 also has important homeostatic effects in the adult brain, where it positively regulates expression of trophic genes in dopaminergic neurons and tonically suppresses inflammatory activation of microglia and astrocytes by inhibiting activation of NF-κB (Saijo et al., 2009).
Activation of microglia and astrocytes can result in persistent neuroinflammation that is associated with the progression of neuronal injury in neurodegenerative diseases (Rossi and Volterra, 2009; Polazzi and Monti, 2010). Signaling pathways responsible for initiating neuroinflammatory responses are dynamically suppressed in the absence of extracellular stimuli but are rapidly induced in response to stress, injury, or pathogens. Inflammatory mediators produced by activated glial cells, such as cytokines and reactive oxygen species, may progress to neurotoxic levels during states of chronic neuroinflammation (Lambertsen et al., 2009; Brown and Neher, 2010). It has been shown that selected nuclear orphan receptors, such as the nerve growth factor responsive NR4A family (NR4A1/Nur77, NR4A2/Nurr1, and NR4A3/Nor1) can downregulate pro-inflammatory genes through transcriptional repression or transrepression mechanisms by stabilizing binding of nuclear co-repressor proteins to regulatory elements in the promoters of inflammatory genes (Saijo et al., 2009). The discovery of this transrepressive property of nuclear receptors suggests that these proteins could be viable targets for developing neuroprotective therapeutics involving ligand-induced blockade of neuroinflammatory signaling pathways in glial cells.
The anti-inflammatory and neuroprotective efficacy of C-DIM compounds in neural cells was demonstrated by the capacity of selected para-phenyl–substituted analogs in this series to inhibit NF-κB–dependent expression of NOS2 in astrocytes and subsequent neuronal injury after inflammatory stimulation with cytokines and neurotoxins (Tjalkens et al., 2008; Carbone et al., 2009). On the basis of these in vitro findings, we generated pharmacokinetic data for each of the C-DIM compounds to determine their potential neuroprotective use in vivo. To determine initial pharmacokinetic parameters for selected C-DIM compounds in adult male C57Bl/6 mice, intravenous (1 mg/kg) and oral (10 mg/kg) administration was used for 1,1-bis(3′-indolyl)-1-(p-methoxyphenyl)methane (C-DIM5); 1,1-bis(3′-indolyl)-1-(phenyl)methane (C-DIM7); 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (C-DIM8); and 1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (C-DIM12). Levels of each compound were measured in plasma and brain tissue for both administration routes; tissue levels of parent compound were quantified, and putative metabolites were analyzed in urine, liver, and plasma samples. On the basis of the results of these pharmacokinetic studies, we then examined the effectiveness of selected C-DIM compounds in attenuating loss of dopaminergic neuronal cell bodies, terminals, and dopamine levels in C57Bl/6 mice after subacute administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine (MPTP) and probenecid (MPTPp). Two of the three C-DIM compounds examined in efficacy studies significantly reduced additional loss of dopaminergic neurons in the substantia nigra of mice treated with MPTPp, with C-DIM12 displaying the highest activity. These results suggest that selected C-DIM compounds could be effective neuroprotective agents for attenuating progressive loss of neurons in neurodegenerative diseases.
Materials and Methods
Chemicals and Reagents.
C-DIM 5, 7, 8, and 12 (Fig. 1) were synthesized and characterized as described elsewhere (Qin et al., 2004). All other reagents were of analytical grade and were purchased from Sigma-Aldrich (St. Louis, MO).
Fig. 1.
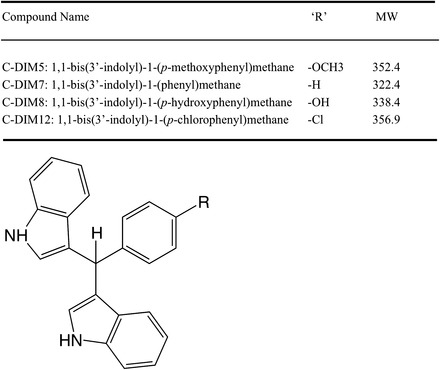
Structure and molecular weight of C-DIM compounds.
Compound Administration for Pharmacokinetic Studies.
C-DIMs were prepared for intravenous administration by dissolving in 25% solutol for a final dose of 1 mg/kg, sterile filtered, and administered through tail vein injection. Oral gavage preparations of C-DIMs were made as a suspension in corn oil for a final dose of 10 mg/kg; controls were given saline or corn oil for intravenous or gavage administration, respectively. Adult male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were randomly placed into groups for each C-DIM, with n=3 each for intravenous and oral gavage. Each mouse was weighed and treated with a single dose of the appropriate volume of C-DIM (10–15 µl for intravenous and 90–130 µl for oral gavage). After administration, three mice per treatment group were sacrificed at 10, 30, 60, 120, 240, 360, 480, and 720 minutes by cardiac exsanguination under deep isoflurane anesthesia, and plasma and brain samples were collected. Brain samples were quickly frozen in liquid nitrogen and stored at −80°C for analysis. For tissue distribution studies, mice (n=3 per C-DIM) were treated as described above, using only oral gavage, and were housed in metabolic cages to collect urine samples over 4 hours. At 4 hours, mice were sacrificed as described, and plasma and brain samples were collected along with peripheral organs (heart, lung, liver, kidney, intestine, fat, and brain) and rinsed in phosphate-buffered saline before freezing in liquid nitrogen and stored at −80°C. All studies were conducted under an approved Institutional Animal Care and Use Committee protocol at Colorado State University in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health.
MPTP Treatment of C-DIM Efficacy Studies.
Male and female transgenic reporter mice (C57 background) expressing an enhanced green fluorescent protein reporter under control of an NF-κB reporter construct (cis-NF-κBenhanced green fluorescent protein; Magness et al., 2004; generously provided by Dr. Christian Jobin, University of North Carolina at Chapel Hill) were aged to 12 weeks and then randomly divided into treatment groups (Miller et al., 2011). A subacute administration strategy with MPTPp was used to create a progressive lesion in the substantia nigra, characterized by more gradual loss of dopamine neurons and a prominent neuroinflammatory response in glial cells (Schintu et al., 2009). Mice were injected every other day with MPTP (25 mg/kg s.c.) and probenecid (250 mg/kg i.p.) for 7 days for a total of 4 injections (MPTP prepared in saline as free base; Sigma-Aldrich). Control mice received saline and probenecid (250 mg/kg i.p.) only on the same administration schedule. After the final exposure to MPTPp, mice were either terminated at day 7 (MPTPp7d) or at day 14 (MPTPp14d). Mice terminated at day 14 were gavaged once daily with either corn oil (vehicle control) or 50 mg/kg of each respective C-DIM compound dissolved in corn oil (MPTPp14d + C-DIM5, MPTPp14d + C-DIM8, MPTPp14d + C-DIM12). Treatment with MPTPp in this model causes initial loss of dopamine neurons and a sustained neuroinflammatory response that causes further loss of dopamine neurons even after treatment with MPTPp has ceased (Schintu et al., 2009; Carta et al., 2011). All animal procedures were performed in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Colorado State University Institutional Animal Care and Use Committee. Every effort was made to minimize pain and discomfort. Terminal procedures were performed under deep isofluorane anesthesia.
Stereological Assessment of Dopamine Neurons in the SN and Nerve Terminals in the Striatum (ST).
Tissue processing and stereologic deterimination of dopamine neuron numbers in the SN were performed as reported previously (Miller et al., 2011). In brief, 7 or 14 days after treatment, animals were terminally anesthetized with isoflurane and transcardially perfused. After perfusion fixation, the brains were carefully removed from the skull and immersion fixed in the same fixative at 4°C for 3 hours. The brains were then cryoprotected in cacodylate–phosphate-buffered saline containing 15% sucrose overnight, followed by 30% sucrose. The tissue was then embedded and sectioned at 40-μm thickness on a cryostat microtome producing a mean of 60 sections through the anatomic SN nucleus. Sections were stored at –20°C, free floating, in cryoprotectant (30% w/v sucrose, 30% v/v ethylene glycol; 0.5 M phosphate buffer; pH 7.2) until staining.
Free-floating serial sections used for tyrosine hydroxylase (TH; Millipore) staining were obtained using systematic sampling from all sections encompassing the entire length of the SNpc, where every third tissue was selected and counted, for a total of 20 sections per animal. Slides were stored at 4°C until imaged for stereological counting. Stereological counts of TH-positive cells were performed using Slidebook software (version 5.0; Intelligent Imaging Innovations, Denver, CO) with use of the optical fractionator method (West and Gunderson, 1990). Images were captured using a Zeiss Axiovert 200M inverted fluorescence microscope equipped with a Hammatsu ORCA-ER-cooled charge-coupled device camera (Hammamatsu Photonics, Hamamatsu City, Japan). The boundary of the SNpc was determined using low-magnification (10×) montage imaging. Total numbers of TH-positive cells were obtained through imaging (40×) uniform randomly placed counting frames (100 × 100 μm) with use of an optical dissector of 30 μm with 5 μm upper and lower guard zones. Representative montage images were generated for each treatment group with use of a 20× objective and displayed using inverted monochrome. Anatomic landmarks were used to select striatal sections for TH intensity staining in an identical process as described above and in Miller et al. (2011), staining all treatment groups simultaneously. Montage images of the ST were created using a 10× objective and a mask generated to outline the striatum with use of Slidebook software, and mean fluorescence intensity in relative florescence units was obtained. Representative montage images were generated for each treatment group with use of a 10× objective and were displayed using inverted monochrome.
Striatal Dopamine and Dopamine Metabolite Content.
Catecholamine levels were determined in the striatum with use of high-performance liquid chromatography (HPLC) coupled with electrochemical detection. In brief, animals were anesthetized using isoflurane and were immediately decapitated, followed by rapid brain removal and isolation of the striatal brain region with use of a mouse brain matrix block for reference. Striatal brain tissues were flash frozen in liquid nitrogen and stored at –80°C until they were sent for analysis. The Neurochemistry Core Laboratory at Vanderbilt University’s Center for Molecular Neuroscience Research (Nashville, TN) analyzed the samples with coded treatment groups for unbiased analysis of the concentration of dopamine and its metabolites. For HPLC analysis details, see Perez and Palmiter (2005).
Liquid Chromatography–Tandem Mass Spectrometry Analysis of C-DIM Metabolites.
Standard dilutions of C-DIMs were prepared in 1:1 acetonitrile (ACN)/10 mM ammonium acetate (pH 7.5). For the analysis of each C-DIM in plasma and tissue homogenate, C-DIMs standards (1–1000 ng/ml) were added to control plasma, urine, or tissue homogenate (100 mg/ml in water, using a PowerGen 700 tissue homogenizer; Fisher Scientific). Samples were prepared using 100 µl of plasma, urine, or homogenate. Each sample was spiked with 10 µl of 1:1 ACN/10 mM ammonium acetate (pH 7.5) or 10 µl of the appropriate C-DIM standard, 10 µl of 250 ng/ml naringenin as an internal standard, and vortex briefly, and then 600 µl of 1:1 ACN/methyl tert-butyl ether was added. Samples were vortexed continuously for 10 minutes, followed by centrifugation for 10 minutes at 20,800g at 4°C. Then, 500 µl of each supernatant was collected and transferred to a fresh 2-ml microcentrifuge tube. The samples were then dried on a Savant Automatic Environmental SpeedVac AES 1000 (Farmingdale, NY) using medium heat for approximately 1.25 hours. The samples were then resuspended in 200 µl of 1:1 ACN/10 mM ammonium acetate (pH 7.5), vortexed for 5 minutes, followed by centrifugation for 5 minutes, then transferred to HPLC vials with inserts for analysis. The in vitro formation of the glucuronide metabolite for each C-DIM was assessed using the method previously described (Guo et al., 2007).
Negative ion electrospray ionization mass spectra were obtained using an MDS Sciex 3200 Q-TRAP triple quadrupole mass spectrometer (Applied Biosystems, Inc., Foster City, CA) with a turbo ionspray source interfaced to a Shimadzu LC-20AD High-Performance Liquid Chromatograph system (Shimadzu Corporation, Kyoto, Japan). Samples were chromatographed using a Waters Sunfire C8 (5 µm, 4.6 × 50 mm column; Waters Corp.) protected by a C18 guard cartridge (4.0 × 2.0 mm; Phenomenex, Torrance, CA). A liquid chromatography gradient was used with mobile phase A consisting of 10 mM ammonium acetate and mobile phase B consisting of acetonitrile. Chromatographic resolution was achieved by linearly holding the B solvent at 40% for 1 minute. The solvent mixture was then altered by increasing mobile phase B linearly from 40 to 98% at 1–2 minutes, maintaining at 98% at 2–4 minutes, and then decreasing linearly from 98 to 40% at 4–4.5 minutes, followed by re-equilibration of the column at 40% mobile phase B at 4.5–6 minutes. The liquid chromatogrphy flow rate was 750 µl/min, the sample injection volume was 60 µl, and the analysis run time was 6 minutes.
The mass spectrometer settings were optimized as follows: turbo ionspray temperature, 600°C; ion spray voltage, −4500 V; curtain gas, N2, (CUR), 30 units; collision gas, N2, (CAD), medium; nebulizer gas, N2, 60 units; and auxiliary gas, N2, 60 units. The compound-specific parameters for each C-DIM are shown in Supplemental Table 1. The product ion scans obtained after infusion of 1 µg/ml each C-DIM (in beginning mobile phase, 10 µl/min) into a 0.750 ml/min flow of starting mobile phase and suggested formation of the precursor ion, product ion, and neutral loss can be seen in Fig. 2A. The predominant product ions were m/z (mass-to-charge ratio) 116.0, 220.0, and 335.0 amu for C-DIM5; m/z 116.0, 204.0, and 243.0 amu for C-DIM7; m/z 116.0, and 220.0 amu for C-DIM8; and m/z 116.0 and 243.1 amu for C-DIM12. Samples were quantified in the multiple reaction monitoring (MRM) mode monitoring the relevant ion transitions and then summing the counts for each C-DIM. Each ion transition was integrated for 50 milliseconds. Q1 and Q3 were both operated in unit resolution mode. All C-DIMs eluted at 2.0–4.0 minutes. Peaks were detected by monitoring the appropriate ion transitions for each compound. No interfering peaks were detected at the monitored ion transitions in extracted matrix (Fig. 2B). Chromatographic conditions were optimized for peak shape. Representative chromatographs of each C-DIM are shown at 1 ng/ml in plasma (Fig. 2B). Quantitation of each C-DIM was based on linear standard curves in spiked matrix with 1/x2 weighting of linear regression.
Fig. 2.
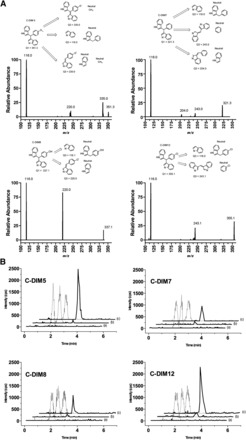
Analytical determination of C-DIM compounds in plasma by liquid chromatography–tandem mass spectrometry (LC-MS/MS). (A) Product ion spectra and proposed structures for each C-DIM compound identified by LC-MS/MS. (B) Blank mouse plasma spiked with 25 ng/ml of naringenin, the internal standard (A). C-DIM 5, 7, 8, and 12 were spiked into blank plasma at 1 ng/ml with internal standard (B). A representative mouse plasma sample spiked with 25 ng/ml naringenin internal standard (C). The solid line represents the summed m/z transitions for each C-DIM compound. The retention times for C-DIM5, C-DIM7, C-DIM8, and C-DIM12 were 3.0 minutes, 3.0 minutes, 2.8 minutes, and 3.0 minutes, respectively. The dashed line represents the internal standard for the m/z transition 271.0 → 119.0 (Rt = 1.9 minutes).
Mouse Microsomal Metabolism Assay.
Incubation of C-DIM8 and C-DIM12 with microsomes was conducted as previously described (Fisher et al., 2000). In brief, 0.25 mg of mouse liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1), and 12.5 mg of alamethicin were mixed and placed on ice for 15 minutes. MgCl2 (3.3 mM in incubation) and C-DIMs (1 µM C-DIM8 or 300 nM C-DIM12 in incubation) were added, and the mixture was preincubated at 37°C for 3 minutes. To initiate the reaction, UDPGA (5 mM in incubation) was added to give a 100 µl final volume. Control incubations were performed with heat-inactivated microsomes and immediate quenching of reactions after addition of UDPGA by adding 200 µl of ice-cold acetonitrile. After addition of acetonitrile, samples were centrifuged to pellet-precipitated protein and supernatant was analyzed as indicated above for metabolites in tissue and plasma samples. Relative abundance of metabolites was determined measuring peak areas of theoretical MRMs and comparing to parent peak areas for each C-DIM compound.
Plasma and Urine incubation with β-Glucuronidase.
Plasma and urine were collected from animals 4 hours after a single dose of C-DIM5, C-DIM7, C-DIM8, or C-DIM12, delivered by oral gavage at 10 mg/kg in corn oil. Plasma and urine samples were then incubated with 300 µM saccharic acid or 500 units β-glucuronidase (Karunairatnam and Levvy, 1949; Guo et al., 2007) and analyzed as described above, using mass spectrometry for the quantitation of parent C-DIM compound in each sample.
Pharmacokinetic and Statistical Analysis.
Pharmacokinetic parameters were calculated from plasma or brain homogenate concentration-time data with standard noncompartmental methods using Microsoft Excel. Descriptive statistics were used for pharmacokinetic variables (including Cmax, Tmax, areas under the curve [AUC]0-t, and t1/2). Statistical analyses were performed using GraphPad Prism software (version 5.0; GraphPad Prism, La Jolla, CA); data sets were analyzed using one-way analysis of variance with a Tukey-Kramer posttest (significance is denoted at *P < 0.05). Oral bioavailability was calculated using Eq. 1:
| (1) |
Results
Analytical Characterization of Diindolylmethane Compounds.
Structures and masses of the four para-phenyl–substituted diindolymethane compounds examined in this study are described in Fig. 1. The relative abundance of the C-DIM parent compounds and the respective product ions under the mass spectrometry conditions used for analysis are presented in Fig. 2A. MRM analyses detecting the most abundant product ions for each drug were measured to ensure accuracy and sensitivity for detection of each compound. Representative chromatographs used for analytical determination of each C-DIM compound is described in Fig. 2B, which depicts chromatograms of either blank spiked with internal standard, C-DIM–spiked plasma, or unknown sample. Chromatograms shown in Fig. 2B for plasma are similar to the standards measured for each C-DIM compound in the various tissues examined in this study. Using this analytical method, we were able to detect all four C-DIM compounds in biologically relevant matrices with a high level of sensitivity.
Plasma Levels of C-DIM Compounds after Intravenous and Oral Administration.
The plasma concentration over time for each substituted diindolylmethane analog is shown in Fig. 3, with the corresponding calculated noncompartmental pharmacokinetic values in Table 1. AUC values for each C-DIM compound varied on the basis of route of administration and individual C-DIM structure. Half-life (t1/2) in plasma for intravenous administration ranged from 71 (C-DIM8) to 144 minutes (C-DIM5); oral gavage t1/2 values ranged from 55 (C-DIM5) to 131 minutes (C-DIM7). The time for each C-DIM to reach Cmax (Tmax) after oral gavage administration was 60 minutes for C-DIM8 and 120 minutes for all others. Clearance as a function of bioavailability, extrapolated volume of distribution, and mean resonance time were dependent on C-DIM structure and route of administration; C-DIM8 was rapidly cleared when given orally, displaying a low AUC via this route.
Fig. 3.
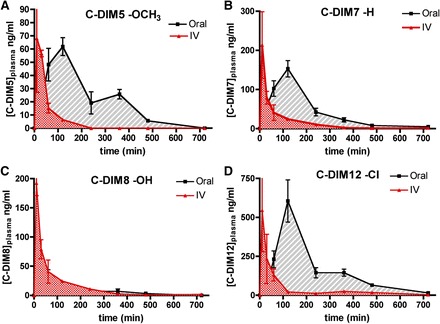
Plasma pharmacokinetic distribution of C-DIM compounds. Plasma concentrations for C-DIM compounds and relationship between the route of exposure and plasma concentration. Points represent mean plasma level for either intravenous (red) or oral gavage (black) of C-DIM5 (A), C-DIM 7 (B), C-DIM8 (C), or C-DIM12 (D) over a period of 12 hours (n = 3 animals per time point for each route of exposure) data are expressed as mean ± S.E.M.
TABLE 1.
Plasma pharmacokinetic values of C-DIM compounds
Data representing the mean of three animals per time point per C-DIM compound were used for the construction of plasma concentration versus time curves (Fig. 3). Noncompartmental modeling was used for the calculation of pharmacokinetic parameters based on the composite data.
| C-DIM (Plasma) | AUC0->720 min | t1/2λ | CL(area)/kg | AUMC0->t | Vss | MRT | Oral Bioavailability |
|---|---|---|---|---|---|---|---|
| (ng/ml) × min | min | ml/min× kg | (μg/ml) × min2 | l/kg | min | % | |
| C-DIM5 I.V. | 3,838 | 144 | 261 | 356 | 24.2 | 93 | – |
| C-DIM5 Oral | 14,828 | 55 | – | 2,971 | – | 200 | 39 |
| C-DIM7 I.V. | 9,727 | 139 | 103 | 993 | 10.5 | 102 | – |
| C-DIM7 Oral | 29,577 | 131 | – | 5,167 | – | 175 | 30 |
| C-DIM8 I.V. | 9,129 | 71 | 109 | 602 | 7.13 | 66 | – |
| C-DIM8 Oral | 5,151 | 114 | – | 1,012 | – | 196 | 6 |
| C-DIM12 I.V. | 27,391 | 133 | 36.5 | 4,184 | 5.58 | 153 | – |
| C-DIM12 Oral | 115,891 | 110 | – | 24,648 | – | 213 | 42 |
AUC, area under the curve; AUMC, area under the first-moment curve; C-DIM, para-phenyl subsitiuted diindolylmethane; CL, clearance limit; Kel, elimination rate; MRT, mean residence time.
The percentage of oral bioavailability of each C-DIM compound was calculated on the basis of their total exposure (AUC; Table 1). C-DIM5 and C-DIM12 exhibited the highest bioavailability at 38 and 42%, respectively. It is likely that C-DIM8 (6% oral bioavailability) undergoes rapid first-pass hepatic metabolism when given orally because of glucuronidation of the phenolic group; this was further examined after incubation of C-DIM8 and C-DIM12 with liver microsomes (Table 3) and treatment of plasma and urine collected from treated mice with β-glucuronidase (Fig. 5).
TABLE 3.
Identification of putative C-DIM metabolites using theoretical MRM(s)
Putative metabolites are listed for each C-DIM, indicating the structural position to which metabolism occurs. Metabolites were detected in several tissues, plasma and urine.
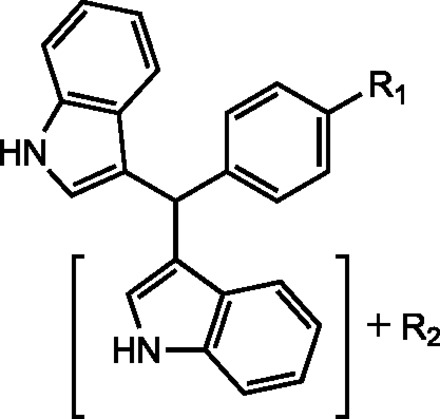 |
|||||
|---|---|---|---|---|---|
| Compound | Theoretical Metabolism | Tissue/Microsome Observed in | R1 | R2/Addition to Indole Ring | MRM(s) / Theoretical MRM(s) used to ID Putative Metabolite |
| C-DIM5 | OCH3 | — | 351.3 → 335, 116, 220 | ||
| Oxidation | U ± β-gluc; L; B | OCH3 | O | 367.3 → 132, 116 | |
| Demethylation | L; M; H; K; B | OH | — | 337.3 → 116 | |
| Demethylation and addition of glucose | U | O-glucose | — | 513.4 → 116 | |
| C-DIM7 | H | — | 321.3 → 116, 243, 204 | ||
| Oxidation | P ± β-gluc; U ± β-gluc; L; K; B | H | O | 337.3 → 132, 116 | |
| C-DIM8 | OH | — | 337.1 → 116, 220 | ||
| Addition of glucose | P; U | O-glucose | — | 513.1 → 116 | |
| Oxidation | P; U ± β-gluc; L; Micro | OH | O | 353.1 → 132, 116 | |
| Glucuronidation | U; L | O-Gluc | — | In source decay to parent MRMs with RT=1.08 min | |
| C-DIM12 | Cl | — | 355.1 → 116, 243 | ||
| Oxidation | P ± β-gluc; U + β-gluc; L; Micro; K; B; Lung | Cl | O | 371.1 → 132, 116 | |
| Dehalogenation oxidation and glucuronidation | U + β-gluc | O-Gluc | — | 337.1 → 220 |
β-gluc, β-glucuronidase treated; C-DIM, para-phenyl subsitiuted diindolylmethane; Gluc, glucuronic acid; H, heart; K, kidney; L, liver; M, muscle; Micro, liver microsome incubation; MRM, multiple reaction monitoring; P, plasma; U, urine.
Fig. 5.
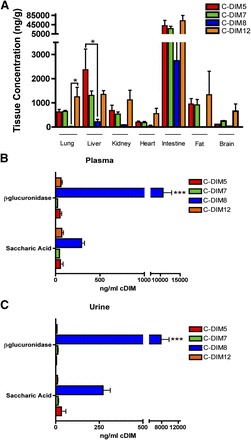
Tissue accumulation and metabolism of C-DIM compounds after oral administration. (A) Mice were administered a single oral dose (10 mg/kg) of C-DIM5, C-DIM7, C-DIM8, or C-DIM12 and examined for tissue distribution of each C-DIM compound 4 hours after administration, the time at which peak plasma and brain concentrations of drug were measured in pharmacokinetic studies. Colors denote different C-DIM compounds, error bars represent standard deviation, and significance is indicated by * (P < 0.05, n = 3 animals per group). Plasma (B) and urine (C) samples were also collected and treated with β-glucuronidase or saccharic acid (control), and the concentration of each C-DIM parent compound was determined by liquid chromatograpjy–tandem mass spectrometry. Mice were housed in metabolic cages for urine sample collection. Colors denote different C-DIM compounds, error bars represent standard deviation, and significance is indicated by *** (P > 0.001, n = 3 animals per group); data are expressed as mean ± S.E.M.
Brain Levels of C-DIM Compounds after Intravenous and Oral Administration.
Brain levels of each C-DIM were determined to investigate the suitability of these compounds for use as potential neuroprotective agents in the CNS. Concentrations of each compound in brain over time for each C-DIM analog are shown in Fig. 4, with the corresponding calculated noncompartmental pharmacokinetic values in Table 2. Values reported in Table 2 for brain to plasma AUC suggest very reasonable distribution to the brain for each C-DIM compound evaluated, with brain to plasma AUC ratios for oral delivery of 3.2 (C-DIM8), 4.1 (C-DIM7 and C-DIM12), and 6.0 (C-DIM5). Corresponding Cmax values in brain tissue ranged from 31.2 (C-DIM8) to 1173 ng/ml (C-DIM12). Among the compounds evaluated, C-DIM12 had the highest values for AUC in brain tissue after oral administration. The t1/2 measurements in brain tissue are similar to t1/2 plasma values for each respective C-DIM compound, with the exception of C-DIM8, which displayed a significantly shorter plasma t1/2 (71 minutes in the plasma versus 248 minutes in the brain after intravenous administration). Tmax, clearance, and Kel (elimination rate) across all C-DIM compounds varied on the basis of compound and route of administration but, in general, were similar to their respective plasma pharmacokinetic measurements. These data indicate that each C-DIM reaches the brain via both intravenous and oral gavage routes of administration, with selected compounds, such as C-DIM12, exhibiting increased partitioning to the brain.
Fig. 4.
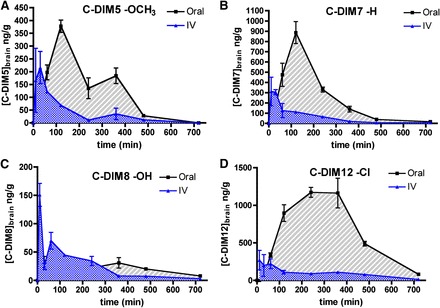
Brain pharmacokinetic distribution of C-DIM compounds. Brain tissue concentrations for C-DIM compounds and relationship between route of exposure and concentration in brain. Points represent mean brain tissue level for either IV (blue) or oral gavage (black) of C-DIM5 (A), C-DIM7 (B), C-DIM8 (C), or C-DIM12 (D) over a period of 12 hours (n = 3 animals per time point for each route of exposure); data are expressed as mean ± S.E.M.
TABLE 2.
Brain pharmacokinetic values of C-DIM compoundsData representing the mean of three animals per time point per C-DIM compound were used for the construction of brain tissue concentration versus time curves (Fig. 4).
Noncompartmental modeling was used for the calculation of pharmacokinetic parameters based on the composite data.
| C-DIM (brain) | AUC0->720 min | Cmax | t1/2λ | Tmax | CL(area)/kg | Kelλ | Brain AUC: Plasma AUC |
|---|---|---|---|---|---|---|---|
| (ng/ml) × min | ng/ml | min | min | ml/min×kg | (μg/ml) × min2 | ||
| C-DIM5 I.V. | 23,192 | 216 | 47 | 30 | 261 | 0.0146 | 6.0 |
| C-DIM5 Oral | 88,325 | 378 | 44 | 120 | 647 | 0.0157 | 6.0 |
| C-DIM7 I.V. | 39,397 | 313 | 147 | 10 | 103 | 0.0047 | 4.1 |
| C-DIM7 Oral | 171,596 | 887 | 112 | 120 | 338 | 0.0062 | 5.8 |
| C-DIM8 I.V. | 16,165 | 151 | 248 | 10 | 109 | 0.0028 | 1.8 |
| C-DIM8 Oral | 16,322 | 31.2 | 187 | 360 | 1,941 | 0.0037 | 3.2 |
| C-DIM12 I.V. | 68,042 | 272 | 117 | 10 | 36.5 | 0.0059 | 2.4 |
| C-DIM12 Oral | 465,845 | 1,173 | 95 | 240 | 86.3 | 0.0073 | 4.1 |
AUC, area under the curve; C-DIM, para-phenyl subsitiuted diindolylmethane; CL, clearance limit; Kel, elimination rate.
Tissue Distribution of C-DIM Compounds and Identification of Glucuronide Metabolites in Plasma and Urine.
The pharmacokinetic analysis profiled plasma and brain levels of each compound, based on CNS as the primary intended target tissue, but we also determined the initial tissue distribution for each C-DIM compound and for the major metabolites in plasma and urine (Fig. 5). Because AUC and half-life values for orally administered C-DIMs peaked at approximately 4 hours for brain tissue, we investigated the tissue distribution of each C-DIM at 4 hours after oral gavage (10 mg/kg). As shown in Fig. 5A, C-DIM levels are highest in the intestine for oral administration and displayed similar distribution profiles, as suggested by pharmacokinetic data; C-DIM8 levels were lowest in the peripheral tissues, and C-DIM12 were highest, with the exception of the liver, where C-DIM5 levels were increased. No significant differences were observed between C-DIM levels in most tissues, except for the lung and liver, where C-DIM8 was significantly lower than C-DIM5 and 12 (P < 0.05).
The structure of C-DIM8 contains a phenolic R–group substituent, which suggested that is was very likely to undergo glucuronide conjugation. To investigate this possibility for each C-DIM compound examined, we obtained plasma samples from mice four hours after a single 10 mg/kg oral dose of each compound and then measured metabolites by liquid chromatography–tandem mass spectrometry after incubation with β-glucuronidase (three mice per group). The addition of β-glucuronidase resulted in significantly increased levels of C-DIM8 parent compound, consistent with glucuronidation of the phenolic substituent (P < 0.0001; Fig. 5B). β-glucuronidase treatment of plasma from mice treated with other C-DIM compounds did not result in a significant increase in concentrations of parent compound. Plasma samples were also treated with saccharic acid, an inhibitor of β-glucuronidase, but the levels of parent compound for C-DIM5, C-DIM7, and C-DIM12 remained unchanged relative to the increase observed in C-DIM8, indicating a possible metabolite other than a glucuronide that is converted back to parent C-DIM8. Similar metabolite profiles were observed in urine samples collected over four hours from mice housed in metabolic cages that were given a single oral dose of each C-DIM compound at 10 mg/kg (Fig. 5C). Excreted C-DIM parent compounds measured in the urine were not affected by the addition of β-glucuronidase inhibitors, with the exception of C-DIM8, which was significantly increased in the presence of β-glucuronidase, suggesting that the glucuronidated form of C-DIM8 is abundant in the urine four hours after a single oral dose of this compound.
Determination of Oxidative Metabolites of C-DIM Compounds.
Extracts from plasma, urine, and various peripheral tissue samples were analyzed for putative metabolites of each C-DIM after a 10-mg/kg oral dose for each drug. Mice were housed in metabolic cages, and plasma, urine, and liver samples were collected 4 hours after a single 10-mg/kg oral dose of each C-DIM compound, based on the calculated Tmax values (Table 1). Without valid reference standards for each metabolite, it was not possible to establish a complete quantitative profile of oxidative metabolites for each compound, but theoretical MRM for various metabolic reactions that resulted in an identifiable mass signature are shown in Table 3. Metabolites were detected in plasma, urine, and muscle, heart, and kidney tissues, as well as in reactions containing murine hepatic microsomes incubated with C-DIM8 and C-DIM12 for comparison of possible glucuronide formation. Microsomal preparations treated with 1 µM of C-DIM8 and 300 nM C-DIM12 revealed minor levels of oxidation (max abundance of 15% at 40 minutes of the parent AUC at 0 minute) and di-oxidation (max abundance of 3% at 70 minutes of the parent AUC at 0 minute). Oxidation of the indole ring (R2) was observed in plasma and urine for all C-DIM compounds, with the exception of C-DIM5, for which oxidation was only detected in urine. These similarities in metabolism are not surprising, given the 1,1-bis (3′-indolyl) moiety, common to all C-DIM compounds. Measurement of C-DIM12 in urine incubated with β-glucuronidase indicated that oxidative dehalogenation occurred, resulting in generation of C-DIM8 parent compound, which is then metabolized to a glucuronide conjugate. A summary of the metabolism of each C-DIM compound is presented in Fig. 6, which demonstrates that dealkylation of C-DIM5 and oxidative dehalogenation of C-DIM12 generate the C-DIM8 parent compound. After analysis of liver and urine of C-DIM8–treated mice, we observe an extensive peak corresponding to parent MRMs, with a retention time of 1.08 minute, which suggests in-source decay of a C-DIM8–glucuronide.
Fig. 6.
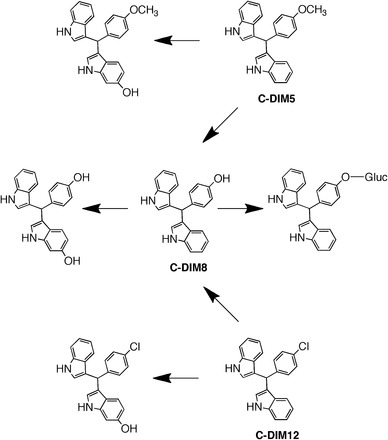
Metabolic scheme for C-DIM compounds. Identified metabolites of parent C-DIMs reported in Table 3 show the common oxidative metabolism observed for C-DIM5, C-DIM8, and C-DIM12.
Neuroprotective Efficacy of C-DIM Compounds.
On the basis of the oral bioavailability and brain concentrations of C-DIMs, in vivo efficacy studies were conducted to determine the capacity of each C-DIM compound to prevent loss of dopamine neurons in the SN subacute MPTPp model of PD (Fig. 7; Table 4). Stereologic counting of dopamine neurons numbers in the SN indicated that mice treated for 7 days with MPTPp (MPTPp7d) experienced modest but significant loss of TH–positive neurons, compared with saline-treated animals (Table 4). Numbers of TH-positive neurons continued to decrease on days 7–14, even after cessation of MPTPp treatment on day 7, leading to approximately 50% loss of TH-positive neurons by day 14 (P < 0.05 and P < 0.05 for 14 days and 7 days, respectively, compared with saline control; n=4). The number and integrity of dopaminergic neurons was assessed using immunofluorescence staining for TH, presented as the negative grayscale image for each representative brain specimen (Fig. 7). On the basis of these data, the neuroprotective efficacy of each C-DIM compound was evaluated using a postlesion strategy for delivery, in which C-DIMs were administered by oral gavage during the 7-day period after MPTPp treatment (50 mg/kg in corn oil vehicle, once daily on days 7–14). Thus, MPTPp14 and C-DIM groups represent animals that have sustained injury to the SN before treatment with each C-DIM compound. Loss of dopaminergic soma, axons and dendrites was evident at both day 7 and day 14 in mice exposed to MPTPp (Fig. 7, A–C). This loss was attenuated in mice treated orally with C-DIM5, 8, and 12 (Fig. 7, D–F), with C-DIM12 resulting in the most obvious preservation of dopamine neuron number and structure. Overt behavioral changes were not observed between any groups of mice by open-field activity measurements with use of this relatively moderate administration strategy with MPTP and probenecid (unpublished data). Stereological assessment of TH-positive neurons in the SN (Table 4) revealed that the number of dopamine neurons in mice treated with MPTPp plus C-DIM5 and C-DIM12 was the same as that for MPTPp-treated mice at day 7, indicating that there was no further loss of dopaminergic neurons in the SN during days 7–14 in these groups. Moreover, the number of TH-positive neurons in the SN in mice treated with MPTPp plus C-DIM5 and C-DIM12 was also significantly greater than that in the MPTPp 14 day group. The number of TH-positive neurons in C-DIM8–treated mice was not different from that in the MPTPp 7 day group or that in the MPTPp 14 day group, despite an obvious trend toward a greater number of neurons in the SN.
Fig. 7.
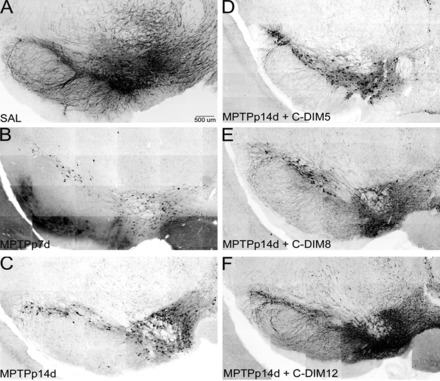
C-DIM compounds protect against progressive loss of TH-positive neurons in the SN after subacute exposure to MPTP and probenecid. Representative montage images of TH-positive neurons in the SN indicate that, compared with saline (SAL)-treated controls (A), MPTPp treatment causes loss of dopaminergic cell bodies and processes by 7 days (B) that progresses by 14 days (C), even after cessation of MPTPp treatment on day 7. (A–C) Progressive loss of dopamine neurons expressing TH in the SN during MPTPp treatment regimen. (D–F) The SN of C-DIM–treated animals given daily oral gavage (50 mg/kg) of C-DIM5, 8, or 12 on days 7–14, after administration with MPTPp. Dopamine neuronal cell bodies and axons are preserved in these animals at levels similar to those in MPTPp 7 days treatment, indicating protection against loss of TH-positive cells when C-DIMs are administered after the onset of neuronal injury. Montages were reconstructed from each series of 20× individual images. TH immunofluorescence images were converted to inverted monochrome for presentation. Scale bar = 500 μm.
TABLE 4.
Stereological assessment of dopamine neuron numbers in the SN and TH staining intensity in the ST
Mice were treated with saline or MPTPp (25 mg/kg MPTP subcutaneous, 250 mg/kg probenecid intraperitoneal) every other day for 7 days (total four injections). MPTPp 7 day mice were terminated at 7 days and assessed using stereology for TH-positive neurons in the SN. Remaining groups were either given corn oil (MPTPp 14 days), C-DIM5 (MPTPp 14 days plus C-DIM5), C-DIM8 (MPTPp 14 days plus C-DIM8), or C-DIM12 (MPTPp 14 days plus C-DIM12) via daily oral gavage (50 mg/kg) for the following 7 days. After a total of 14 days, mice were used for stereology studies in the SN. Mean fluorescence intensity of TH expression in the ST as a representation of dopamine neuron terminal survival is presented for each treatment group as a percentage of control (SAL). Data are expressed as mean ± S.E.M. (n = 4 animals per group).
| Treatment | SAL | MPTPp 7 Days | MPTPp 14 Days | MPTPp 14 Days Plus C-DIM5 | MPTPp 14 Days Plus C-DIM8 | MPTPp 14 Days Plus C-DIM12 |
|---|---|---|---|---|---|---|
| TH + Neurons (SN) | 11,434 ± 566 | 8,061 ± 1010* | 5,689 ± 406*,† | 8,556 ± 699*,‡ | 7,408 ± 2,587* | 9,572 ± 823*,‡ |
| TH Intensity (%Control) (ST) | 100.0 ± 5.3 | 81.6 ± 3.5* | 65.4 ± 2.6* | 88.5 ± 3.9*,‡ | 89.3 ± 1.9*,‡ | 88.4 ± 0.6*,‡ |
C-DIM, para-phenyl subsitiuted diindolylmethane; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine; MPTPp. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine with probenecid; SN, substantia nigra; ST, striatum; TH, tyrosine hydroxylase.
P < 0.05 compared with saline control.
P < 0.05 compared with MPTPp 7 days.
P < 0.05 compared with MPTPp 14 days.
After stereological assessment of dopaminergic neuronal cell bodies in the SN, preservation of dopamine nerve terminals in the ST was determined for all treatment groups. Treatment with MPTPp caused a progressive loss of TH-immunoreactive fibers in the striatum, expressed as percentage control of mean fluorescence intensity (Table 4). Striatal TH intensity in MPTPp 7 day animals decreased to approximately 81.6% of control, whereas MPTPp 14 day animals degenerated further to 65.4% of control TH fluorescence, even after cessation of MPTPp treatments. In contrast to dopamine cell body counts in the SN, striatal intensity levels representing dopamine neuron projections were significantly preserved in all the C-DIM–treated animals, with 88.5, 89.3, and 88.4% of control mean fluorescence in MPTPp 14 days plus C-DIM5, MPTPp 14 days plus C-DIM8, and MPTPp 14 days plus C-DIM12 groups, respectively. Representative montages (inverted monochrome, 10×) of the striatum show the TH intensity changes seen in the MPTPp-treated animals and the preservation of dopamine terminals in the C-DIM–treated animals (Fig. 8).
Fig. 8.
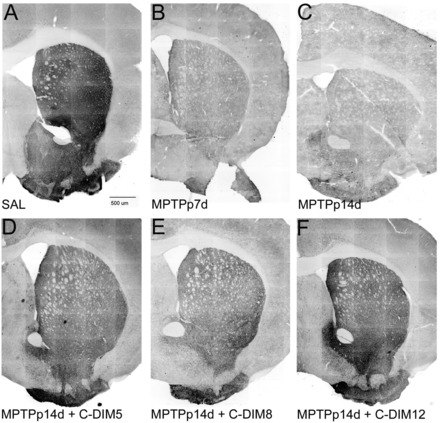
Neuroprotective effects of C-DIM compounds on TH expression in the ST. Representative montage images of TH immunostaining in the striatum of mice treated with saline (SAL) (A), MPTPp 7 days (B), MPTPp 14 days (C), MPTPp 14 days plua C-DIM5 (D), MPTPp 14 days plus C-DIM8 (E), and MPTPp 14 days plus C-DIM12 (F). Loss of TH intensity in the ST was evident 7 days after treatment with MPTPp and continued to decrease through 14 days after MPTPp treatment. All three C-DIM compounds prevented this progressive decrease in striatal TH intensity on days 7–14. Representative montages were reconstructed from each series of 10× individual images stained for TH immunofluoresence and converted to inverted monochrome for presentation. Scale bar = 500 μm.
To determine changes in neurochemistry in this model, levels of dopamine and its primary metabolite, 3,4-dihydroxyphenylacetic acid (DOPAC), were measured in striatal tissue from mice treated with MPTPp in the presence and absence of C-DIM compounds (Table 5). Dopamine content was significantly decreased after 7 days of MPTPp treatment, with MPTPp 14 day dopamine levels apparently further decreased despite the lack of statistical significance. Dopamine levels in C-DIM–treated mice were not statistically different from those in the MPTPp 7 day or MPTPp 14 day groups, although a trend toward perseveration of striatal dopamine levels was consistently noted, particularly for C-DIM12. DOPAC levels showed a similar trend (Table 5).
TABLE 5.
Dopamine and DOPAC levels in the striatum
A separate set of mice from the same MPTPp study used in stereological and TH intensity assessment (Table 4) were also analyzed for striatal levels of dopamine and its primary metabolite DOPAC with use of HPLC. Data are expressed as mean ± S.E.M. (n = 5 animals per group).
| Treatment | SAL | MPTPp 7 Days | MPTPp 14 Days | MPTPp 14 Days Plus C-DIM5 | MPTPp 14 Days Plus C-DIM8 | MPTPp 14 Days Plus C-DIM12 |
|---|---|---|---|---|---|---|
| Dopamine, ng/mg | 49.0 ± 6.7 | 20.3 ± 2.3* | 7.0 ± 3.9* | 6.4 ± 1.4* | 8.4 ± 2.3* | 11.9 ± 3.9* |
| DOPAC, ng/mg | 3.9 ± .81 | 2.5 ± .32 | 0.9 ± .14* | 0.8 ± .13* | 1.1 ± .14* | 1.4 ± .35* |
DOPAC, 3,4-dihydroxyphenylacetic acid; HPLC, high performance liquid chromatography; MPTPp, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine with probenecid; SAL, control; TH, tyrosine hydroxylase.
P < 0.05 compared with saline control.
Discussion
The objective of this study was to characterize the pharmacokinetic behavior of selected C-DIM compounds and to assess the comparative efficacy of each compound in vivo in attenuating the progressive loss of dopamine neurons in the subacute MPTPp model of PD. Determination of levels of each C-DIM compound and major metabolites by liquid chromatography–tandem mass spectrometry in plasma, urine, and tissue samples indicated bioavailability and pharmacokinetic values after oral administration that are acceptable for a potential small molecule therapeutic. The pharmacokinetic behavior of the parent 3-3′-diindolylmethane molecule was previously studied in mice after administration of a single oral dose, which indicated that levels of diindolylmethane rapidly increased in tissue samples, followed by a relatively long elimination phase (Anderton et al., 2004). A single oral dose of para-phenyl–substituted C-DIM compounds (triaryl methanes) in the present study gave a similar result, with a rapid increase in the plasma concentration of each compound and distribution into brain and peripheral tissues. Each of the four C-DIM compounds tested displayed variable distribution in the plasma and brain based on structure and route of administration (Tables 1 and 2). Oral administration yielded greater accumulation in brain than did intravenous administration (except for C-DIM8) and significantly increased the Cmax in brain for C-DIM12 from 272 ng/ml (intravenous) to 1,173 ng/ml (oral; Table 2). Calculated values for the brain to plasma AUC ratio further support oral administration as an acceptable route of administration for C-DIM compounds targeted to the CNS, with markedly increased brain AUC to plasma AUC values over intravenous injection (Table 2).
On the basis of plasma pharmacokinetics, we determined which C-DIM compound had the most biologically relevant kinetic properties for in vivo efficacy studies. C-DIM12 given orally has the largest AUC (115,891 ng/ml/min) and Cmax (606 ng/ml) and the slowest clearance rate (86.3 ml/min×kg) in plasma. In contrast, orally administered C-DIM8 has the smallest AUC (9129 ng/ml/min) and Cmax (28.0 ng/ml) and the most rapid rate of clearance (1,941 ml/min×kg; Table 1) in plasma. Because C-DIM8 administered by oral gavage was more rapidly metabolized than after intravenous administration, we postulated that glucuronide conjugation likely occurred during first-pass metabolism in the liver. Urine and plasma measurements showed a significant increase in C-DIM8 parent compound when treated with β-glucuronidase, an enzyme that degrades glucuronic acid conjugates (Fig. 5, B and C). C-DIM5 and 12 were not significantly affected after incubation with this enzyme, suggesting that only the C-DIM8 parent compound undergoes direct glucuronide conjugation, which is consistent with its phenolic R-group. Because of its rapid metabolism and clearance, C-DIM8 may be less effective as a neuroprotective agent in chronic disease studies; however, initial in vitro screening studies conducted in our laboratory indicated that C-DIM8 had activity similar to C-DIM5 and 12 in suppressing cytokine-induced expression of inflammatory genes, such as NOS2, TNFα family member 10, and IL-1β mRNA, in activated astrocytes (unpublished data). Thus, the presence of a small, relatively polar R-group on the aryl ring substituent may correlate with neuroprotective activity, although more extensive structure-activity studies will be needed to determine the precise structural components contributing to this effect.
Putative metabolites of C-DIM compounds were identified using theoretical MRM and revealed that the shared indole structure undergoes common oxidative metabolism for all compounds (Fig. 7; Table 3). Despite the similar parent structure, differential metabolism of the substituent group for each C-DIM clearly plays a role in the pharmacokinetics of C-DIMs, and this was particularly evident for glucuronide conjugation to the hydroxyl group of C-DIM8 (Fig. 7). Although more detailed metabolic studies need to be performed for each compound, identification of major metabolites is helpful in establishing an initial profile for further examination of therapeutic efficacy.
The failure of small molecule therapeutics to modulate neurodegenerative disease is largely blamed on unsuccessful translation of rodent studies to human trials (Glass et al., 2010). This may be attributable in part to poor CNS pharmacokinetic behavior after oral administration and incongruities among routes of administration in rodent studies, compared with human clinical trials. Oral bioavailability is a desirable property of any putative therapeutic agent, and calculated oral bioavailability for the C-DIM compounds ranged from 6% (C-DIM8) to 42% (C-DIM12; Fig. 5), similar to Levodopa (41% ± 16%), the most widely used symptomatic treatment of PD. In comparison, the monamine oxidase B inhibitor Selegiline indicated for PD has an oral bioavailability of less than 10% (Robertson et al., 1989; Azzaro et al., 2007). On the basis of these data, it is reasonable to postulate that orally delivered C-DIM compounds, particularly C-DIM12, would distribute adequately to the CNS and could therefore be suitable agents for translational efficacy studies.
To determine the neuroprotective efficacy of C-DIM compounds in a relevant animal model of PD, we used a subacute administration strategy with MPTP and probenecid in mice to induce modest loss of dopamine neurons over a 7-day period, after which neuronal loss continued for an additional 7 days in the absence of MPTPp exposure, indicating a progressive neuroinflammatory lesion (Hirsch and Hunot, 2009; Schintu et al., 2009; Carta et al., 2011). Administration of C-DIM5 or C-DIM12 attenuated this progressive loss of neurons in the SN after the initial neurotoxin-induced lesion (Fig. 7; Table 4). In contrast, C-DIM8 showed less neuroprotection after MPTPp exposure, with neuronal counts in the SN remaining statistically unchanged from the MPTPp 14-day time point, although counts trended toward those determined at the 7-day time point (Table 4). The relatively lower efficacy of C-DIM8 may be the result of its greater clearance because of high first-pass metabolic elimination through rapid formation of the glucuronide conjugate. Of interest, oxidative metabolism of both C-DIM5 and C-DIM12 generates the phenolic metabolite corresponding to the C-DIM8 parent compound, which could extend the potential window of efficacy of these compounds by forming a metabolite that retains a degree of neuroprotective activity.
The integrity of dopaminergic nerve terminals is another important measure of neuroprotective efficacy (Kreitzer and Malenka, 2008). MPTPp 14 day animal groups treated with all three C-DIM compounds experienced significantly increased TH fluorescence intensity in the striatum, compared with their untreated counterparts (Fig. 8; Table 4). However, treatment with C-DIM compounds only slightly increased striatal dopamine and DOPAC levels, compares with animals in the MPTPp 14 day group (Table 5). However, it is likely that the robust oxidative effects of MPTP on dopamine nerve terminals may preclude the detection of more subtle differences of dopamine and DOPAC between MPTPp- and C-DIM–treated animals (Przedborski et al., 2000). This also could explain the apparent lack of structure-related differences among C-DIM5, 8, and 12 in preventing loss of TH expression in the striatum, which relies on immunohistochemical analysis of a large region of tissue and lacks the cellular specificity of stereological counting.
The treatment regimen used in these studies was established to mimic what is clinically relevant for individuals with early stages of PD, when dopamine neuron loss in the SN has already occurred at the time of diagnosis (Meredith et al., 2008). The selected C-DIM compounds examined in this model, particularly C-DIM5 and 12, had excellent pharmacokinetic behavior after oral administration and were effective at inhibiting progressive loss of dopamine neurons in a postlesioning model of PD. In previous in vitro studies with two other C-DIM analogs in this series, 1,1-bis(3′-indolyl)-1-(p-trifluoromethylphenyl)methane (C-DIM1) and 1,1-bis(3′-indolyl)-1-(p-t-butylphenyl)methane (C-DIM4), we reported that these compounds suppressed activation of NF-κB in mixed glial cultures exposed to inflammatory stimuli that prevented expression of inducible nitric oxide synthase (NOS2) and protected cocultured neurons from apoptosis (Tjalkens et al., 2008; Carbone et al., 2009). The mechanism underlying this anti-inflammatory activity in glia involved stabilization of nuclear corepressor proteins that prevented DNA binding of NF-κB at cis elements in the Nos2 promoter. Because C-DIM5 and C-DIM12 are activators in cancer cell lines of the NR4A family nuclear receptors Nur77 and Nurr1, respectively (Inamoto et al., 2008; Lee et al., 2010, 2012; Li et al., 2012), they may exert neuroprotective effects by broadly suppressing NF-κB–dependent expression of inflammatory genes in glia, as suggested in recent studies characterizing the neuroprotective role of Nurr1 in PD, and warrants further study (Saijo et al., 2009). Examination of the three C-DIM compounds used in the present studies indicate that, although these compounds may have similar neuroprotective activities in vitro, the differing substituents on the aryl ring yield distinct pharmacokinetic profiles, which may in part be responsible for the differences in efficacy observed in vivo. These data should be useful for designing additional pharmacodynamic efficacy studies to further characterize mechanisms underlying the neuroprotective efficacy of these and related C-DIM compounds and the role of NR4A receptors in mediating these effects.
Abbreviations
- ACN
acetonitrile
- AUC
area under the curve
- CID
collision-induced dissociation
- C-DIM
para-phenyl subsitiuted diindolylmethane
- C-DIM5
1,1-bis(3′-indolyl)-1-(p-methoxyphenyl)methane
- C-DIM7
1,1-bis(3′-indolyl)-1-(phenyl)methane
- C-DIM8
1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane
- C-DIM12
1,1-bis(3′-indolyl)-1-(p-chlorophenyl)methane
- CNS
central nervous system
- DIM
diindolylmethane
- DOPAC
3,4-dihydroxyphenylacetic acid
- HPLC
high-performance liquid chromatography
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine
- MPTPp
1-methyl-4-phenyl-1,2,3,6-tetrahydropyradine with probenecid
- MRM
multiple reaction monitoring
- m/z
mass-to-charge ratio
- PD
Parkinson’s disease
- SN
substantia nigra
- ST
striatum
- TH
tyrosine hydroxylase
Authorship Contributions
Participated in research design: De Miranda, Miller, Hansen, Lunghofer, Gustafson, Colagiovanni, Tjalkens.
Conducted experiments: De Miranda, Miller, Hansen, Lunghofer, Colagiovanni, Tjalkens.
Contributed new reagents or analytic tools: Safe.
Performed data analysis: De Miranda, Hansen, Lunghofer, Tjalkens.
Wrote or contributed to the writing of the manuscript: De Miranda, Miller, Hansen, Lunghofer, Safe, Gustafson, Tjalkens.
Footnotes
This work was supported by the Michael J. Fox Foundation for Parkinson’s Disease Research (to R.B.T.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, Mager DE. (2004) Physiological modeling of formulated and crystalline 3,3′-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos 32:632–638. [DOI] [PubMed] [Google Scholar]
- Azzaro AJ, Ziemniak J, Kemper E, Campbell BJ, VanDenBerg C. (2007) Pharmacokinetics and absolute bioavailability of selegiline following treatment of healthy subjects with the selegiline transdermal system (6 mg/24 h): a comparison with oral selegiline capsules. J Clin Pharmacol 47:1256–1267. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41:242–247. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Popichak KA, Moreno JA, Safe S, Tjalkens RB. (2009) Suppression of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced nitric-oxide synthase 2 expression in astrocytes by a novel diindolylmethane analog protects striatal neurons against apoptosis. Mol Pharmacol 75:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR, Frau L, Pisanu A, Wardas J, Spiga S, Carboni E. (2011) Rosiglitazone decreases peroxisome proliferator receptor-γ levels in microglia and inhibits TNF-α production: new evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 194:250–261. [DOI] [PubMed] [Google Scholar]
- Fisher MB, Campanale K, Ackermann BL, VandenBranden M, Wrighton SA. (2000) In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab Dispos 28:560–566. [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Lu P, Farrell E, Zhang X, Weller P, Monshouwer M, Wang J, Liao G, Zhang Z, Hu S, et al. (2007) In silico and in vitro pharmacogenetic analysis in mice. Proc Natl Acad Sci USA 104:17735–17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397. [DOI] [PubMed] [Google Scholar]
- Inamoto T, Papineni S, Chintharlapalli S, Cho SD, Safe S, Kamat AM. (2008b) 1,1-Bis(3′-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol Cancer Ther 7:3825–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunairatnam MC, Levvy GA. (1949) The inhibition of beta-glucuronidase by saccharic acid and the role of the enzyme in glucuronide synthesis. Biochem J 44:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. (2008) Striatal plasticity and basal ganglia circuit function. Neuron 60:543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, et al. (2009) Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci 29:1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-O, Li X, Khan S, Safe S. (2011a) Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin Ther Targets 15:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, Safe S. (2010) Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res 70:6824–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Andey T, Jin U-H, Kim K, Singh M, Safe S. (2012) The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 31:3265–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lee SO, Safe S. (2012) Structure-dependent activation of NR4A2 (Nurr1) by 1,1-bis(3′-indolyl)-1-(aromatic)methane analogs in pancreatic cancer cells. Biochem Pharmacol 83:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. (2004) In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol 173:1561–1570. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Potashkin JA, Surmeier DJ. (2008) Modeling PD pathogenesis in mice: advantages of a chronic MPTP protocol. Parkinsonism Relat Disord 14 (Suppl 2):S112–S115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Trout BR, Sullivan KA, Bialecki RA, Roberts RA, Tjalkens RB. (2011) Low-dose 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine causes inflammatory activation of astrocytes in nuclear factor-κB reporter mice prior to loss of dopaminergic neurons. J Neurosci Res 89:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. (2005) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA 102:2174–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polazzi E, Monti B. (2010) Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92:293–315. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Djaldetti R, Liberatore G, Vila M, Vukosavic S, Almer G. (2000) The parkinsonian toxin MPTP: action and mechanism. Restor Neurol Neurosci 16:135–142. [PubMed] [Google Scholar]
- Qin C, Morrow D, Stewart J, Spencer K, Porter W, Smith R, Phillips T, Abdelrahim M, Samudio I, Safe S. (2004) A new class of peroxisome proliferator-activated receptor gamma (PPARgamma) agonists that inhibit growth of breast cancer cells: 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes. Molecular Cancer Therapeutics 3:247–260. [PubMed] [Google Scholar]
- Robertson DR, Wood ND, Everest H, Monks K, Waller DG, Renwick AG, George CF. (1989) The effect of age on the pharmacokinetics of levodopa administered alone and in the presence of carbidopa. Br J Clin Pharmacol 28:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Volterra A. (2009) Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull 80:224–232. [DOI] [PubMed] [Google Scholar]
- Safe S, Papineni S, Chintharlapalli S. (2008) Cancer chemotherapy with indole-3-carbinol, bis(3′-indolyl)methane and synthetic analogs. Cancer Lett 269:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach J., Conneely OM. (1998) Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci 95:4013–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E, Carta AR. (2009) PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur J Neurosci 29:954–963. [DOI] [PubMed] [Google Scholar]
- Tjalkens RB, Liu X, Mohl B, Wright T, Moreno JA, Carbone DL, Safe S. (2008) The peroxisome proliferator-activated receptor-γ agonist 1,1-bis(3′-indolyl)-1-(p-trifluoromethylphenyl)methane suppresses manganese-induced production of nitric oxide in astrocytes and inhibits apoptosis in cocultured PC12 cells. J Neurosci Res 86:618–629. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. (1990) Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296:1–22. [DOI] [PubMed] [Google Scholar]