

Abstract
(E)-Methyl 2-((2S,3S,7aS,12bS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (7-hydroxymitragynine), a main active constituent of the traditional herbal medicine Mitragyna speciosa, is an indole alkaloid that is structurally different from morphine. 7-Hydroxymitragynine induces a potent antinociceptive effect on mouse acute pain through μ-opioid receptors. In this study, we developed dual-acting μ- and δ-opioid agonists MGM-15 and MGM-16 from 7-hydroxymitragynine for the treatment of acute and chronic pain. MGM-16 showed a higher potency than that of 7-hydroxymitragynine and MGM-15 in in vitro and in vivo assays. MGM-16 exhibited a high affinity for μ- and δ-opioid receptors, with Ki values of 2.1 and 7.0 nM, respectively. MGM-16 showed μ- and δ-opioid full agonistic effects in a guanosine 5′-O-(3-[35S]thiotriphosphate) binding assay and in a functional test using electrically elicited guinea pig ileum and mouse vas deferens contractions. Systemic administration of MGM-16 produced antinociceptive effects in a mouse acute pain model and antiallodynic effects in a chronic pain model. The antinociceptive effect of MGM-16 was approximately 240 times more potent than that of morphine in a mouse tail-flick test, and its antiallodynic effect was approximately 100 times more potent than that of gabapentin in partial sciatic nerve-ligated mice, especially with oral administration. The antinociceptive effect of MGM-16 was completely and partially blocked by the μ-selective antagonist β-funaltrexamine hydrochloride (β-FNA) and by the δ-selective antagonist naltrindole, respectively, in a tail-flick test. The antiallodynic effect of MGM-16 was completely blocked by β-FNA and naltrindole in a neuropathic pain model. These findings suggest that MGM-16 could become a class of a compound with potential therapeutic utility for treating neuropathic pain.
Introduction
Morphine has been proposed for the treatment of neuropathic pain, but it has incomplete efficacy and dose-limiting adverse effects (Gilron et al., 2005). To develop additional analgesics, derivatives have been synthesized by simplification and introduction of substituents into morphine’s chemical structure (Corbett et al., 2006). Thus, most morphine-derived opioid analgesics used clinically have μ-receptor agonist profiles. Although all three major types of opioid receptors (μ, δ, and κ) are able to mediate analgesia and antinociception, they have different pharmacological activities. Recently, a prominent role of δ-opioid receptors in chronic pain such as neuropathic pain and inflammatory pain was reported from studies using mutant animals and selective agonists (Nadal et al., 2006; Nozaki et al., 2012). In a neuropathic pain rat model, δ-opioid receptor protein expression was increased compared with a control rat in the dorsal root ganglion (Kabli and Cahill, 2007). δ-Opioid receptor activation leads to decreased chronic pain but weakly influences acute pain, in contrast to μ-opioid receptor activation (Gaveriaux-Ruff et al., 2011). A novel strategy for pain management is to use dual-acting and/or mixed opioid agonists (Matsumoto et al., 2008; Cremeans et al., 2012). Therefore, opioid agonists, which act on not only δ- but also μ-opioid receptor subtypes, might be broad-spectrum analgesics useful to treat a variety of painful conditions.
The traditional herbal medicine Mitragyna speciosa has long been used in Thailand for its opioid-like effects (Burkill, 1935) and as a replacement for opium (Suwanlert, 1975). This medicinal herb contains many indole alkaloids (Takayama, 2004). (E)-Methyl 2-((2S,3S,7aS,12bS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (7-hydroxymitragynine), a main active constituent of this plant, is an indole alkaloid that is structurally different from morphine (Horie et al., 2005). We have reported that 7-hydroxymitragynine induces a potent antinociceptive effect on mouse acute pain through the activation of μ-opioid receptors. It is approximately 14 times more potent than morphine when orally administered (Matsumoto et al., 2008). Furthermore, 7-hydroxymitragynine inhibited gastrointestinal transit (GIT) less potently than morphine at each equally antinociceptive dose in mice (Matsumoto et al., 2006a). We investigated the structural similarities between morphine and 7-hydroxymitragynine using molecular modeling techniques. However, we could not superimpose all three functional groups, i.e., a nitrogen atom, a benzene residue, and an oxygen atom on the benzene ring in their structures (Matsumoto et al., 2005a). Therefore, we developed novel opioid analgesics derived from 7-hydroxymitragynine that have different pharmacophore groups from morphine that bind to μ-opioid receptors. We found a novel dual-acting μ- and κ-opioid agonist, (E)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxyindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (MGM-9), which has stronger antinociceptive and weaker rewarding effects than morphine (Matsumoto et al., 2008). 7-Hydroxymitragynine-related indole alkaloids have interesting pharmacological characteristics such as high oral potency and few side effects. Therefore, further investigation of the development of novel analgesics against acute and chronic pain is warranted.
In this study, we hypothesized that a dual-acting μ- and δ-opioid agonist derived from 7-hydroxymitragynine could not only induce potent antinociceptive effects against acute pain but also induce an antiallodynic effect against neuropathic pain. We synthesized the novel μ/δ-opioid dual agonists (E)-methyl 2-((2S,3S,7aS,12aR,12bS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12,12a,12b-decahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (MGM-15) and (E)-methyl 2-((2S,3S,7aS,12aR,12bS)-3-ethyl-9-fluoro-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12,12a,12b-decahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (MGM-16) and clarified their pharmacological profiles using in vitro and in vivo experiments under physiologic conditions. Furthermore, we examined the antiallodynic effects of MGM-16 in a mouse sciatic nerve ligation model. We found that the orally active μ- and δ-opioid receptor agonist MGM-16 produces potent effects against acute and neuropathic pain.
Materials and Methods
Preparation of MGM-15.
NaBH4 (4.3 mg, 0.11 mmol) was added to a stirred solution of 7-hydroxymitragynine (37.5 mg, 0.091 mmol) in dry MeOH (1.1 ml) at 0°C under an argon atmosphere. After 30 minutes, H2O was added to the reaction mixture. The mixture was concentrated under reduced pressure and poured into saturated aqueous NaHCO3 solution. The whole mixture was extracted with 5% MeOH/CHCl3 three times. The combined extract was washed with brine, dried over MgSO4, and evaporated to give a residue that was purified by silica gel column chromatography (ethyl acetate/n-hexane = 35:65). The purified compound was then crystallized from ethyl acetate to give 26.3 mg of MGM-15 (y. 70%).
MGM-15; m.p.: 219–223°C (ethyl acetate). UV (MeOH) lmax nm (log e): 288 (3.13), 277 (3.06), 239 (4.16), 232 (4.14), 214 (4.50). Infrared spectroscopy (KBr) nmax cm−1: 3341, 2954, 1698, 1614, 1467, 1283. 1H NMR (500 MHz, CDCl3) d ppm: 7.40 (1H, s, H-17), 7.00 (1H, dd, J = 8.1, 8.1 Hz, H-11), 6.35 (1H, d, J = 7.9 Hz, H-12), 6.31 (1H, d, J = 8.2 Hz, H-10), 3.87 (1H, br.s, Na-H), 3.83 (3H, s, 9-OCH3), 3.80 (3H, s, 17-OCH3), 3.70 (3H, s, 22-OCH3), 3.47 (1H, br.s, H-2), 2.95 (2H, m, H-15 and H-21), 2.94 (1H, s, 7-OH), 2.53 (2H, m, H-5 and H-14), 2.25 (1H, ddd, J = 12.3, 12.3, 2.3 Hz, H-5), 2.16 (2H, m, H-3 and H-21), 2.03 (1H, d, J = 14.3 Hz, H-6), 1.92 (1H, ddd, J = 13.6, 13.6, 4.3 Hz, H-6), 1.79 (1H, m, H-19), 1.58 (1H, br.d, J = 11.3 Hz, H-20), 1.39 (1H, d, J = 12.8 Hz, H-14), 1.23 (1H, m, H-19), 0.85 (3H, dd, J = 7.3, 7.3 Hz, H3-18). 13C NMR (125 MHz, CDCl3) d ppm: 169.1 (C-22), 160.3 (C-17), 155.9 (C-9), 149.9 (C-13), 129.3 (C-11), 121.3 (C-8), 111.8 (C-16), 105.2 (C-12), 101.9 (C-10), 77.1 (C-7), 69.8 (C-2), 61.6 (17-OCH3), 61.6 (C-3), 58.5 (C-21), 55.1 (9-OCH3), 51.2 (22-OCH3), 50.7 (C-5), 40.7 (C-20), 40.2 (C-15), 35.2 (C-6), 28.5 (C-14), 19.1 (C-19), 13.1 (C-18). Electron ionization mass spectrometry (%) m/z: 416 (M+, 61), 400 (96), 399 (100), 398 (97), 397 (78), 383 (41), 256 (64), 214 (84). Anal. Calcd for C23H32O5N2: C, 66.32; H, 7.74; N, 6.73. Found: C, 66.08; H, 7.77; N, 6.71. CD (c = 0.29 mM, MeOH, 24°C), ∆ε (λ nm): 0 (318), +1.4 (293), 0 (257), −1.0 (248), 0 (233), +0.2 (231), 0 (227), −7.7 (215), +0.1 (208).
Preparation of MGM-16.
NaBH4 (2.1 mg, 0.056 mmol) was added to a stirred solution of 10-fluoro-7-hydroxymitragynine (23.7 mg, 0.055 mmol) in dry MeOH (0.5 ml) at 0°C under an argon atmosphere. The reaction mixture was stirred for 30 minutes. After adding H2O, the reaction mixture was poured into saturated aqueous NaHCO3 solution. The whole mixture was extracted with 5% MeOH/CHCl3 three times. The combined extract was washed with brine, dried over MgSO4, and evaporated to give a residue that was purified by amino-silica gel column chromatography (ethyl acetate/n-hexane = 1:1) to give 22.6 mg of MGM-16 (y. 95%).
MGM-16; UV (MeOH) lmax nm (log e): 297 (3.13), 276 (3.10), 240 (4.22), 226 (4.11), 205 (4.60). Infrared spectroscopy (KBr) nmax cm−1: 3364, 2947, 1701, 1626, 1490, 1284, 1240. 1H NMR (500 MHz, CDCl3) d ppm: 7.41 (1H, s, H-17), 6.76 (1H, dd, J = 12.8, 8.2 Hz, H-11), 6.28 (1H, dd, J = 8.5, 3.1 Hz, H-12), 4.01 (3H, d, J = 2.7 Hz, 9-OCH3), 3.79 (3H, s, 17-OCH3), 3.69 (3H, s, 22-OCH3), 3.46 (1H, d, J = 3.1 Hz, H-2), 2.95 (2H, m, H-15 and H-21), 2.77 (1H, br.s, 7-OH), 2.52 (2H, m, H-5 and H-14), 2.25 (1H, m, H-5), 2.15 (2H, m, H-3 and H-21), 1.95 (2H, m, H-6), 1.76 (1H, m, H-19), 1.58 (1H, br.d, J = 11.3 Hz, H-20), 1.37 (1H, d, J = 12.8 Hz, H-14), 1.23 (1H, m, H-19), 0.84 (3H, dd, J = 7.3, 7.3 Hz, H3-18). 13C NMR (125 MHz, CDCl3) d ppm: 169.0 (C-22), 160.3 (C-17), 148.4 (d, J = 237.4 Hz, C-10), 145.1 (C-13), 143.2 (d, J = 11.9 Hz, C-9), 126.8 (C-8), 116.4 (d, J = 21.5 Hz, C-11), 111.7 (C-16), 105.8 (d, J = 6.9 Hz, C-12), 77.4 (C-7), 70.0 (C-2), 61.6 (17-OCH3), 61.4 (C-3), 61.2 (d, J = 7.8 Hz, 9-OCH3), 58.4 (C-21), 51.2 (22-OCH3), 50.5 (C-5), 40.7 (C-20), 40.1 (C-15), 35.2 (C-6), 28.4 (C-14), 19.0 (C-19), 13.1 (C-18). CD (c = 0.26 mM, MeOH, 24°C), ∆ε (λ nm): 0 (337), +2.7 (297), 0 (255), −4.2 (236), −3.8 (227), −8.4 (214), +0.1 (207). Fast atom bombardment (nitrobenzyl alcohol) mass spectrometry m/z: 435 [M+H]+. High-resolution fast atom bombardment (nitrobenzyl alcohol/polyethylene glycol) mass spectrometry: calcd. for C23H32O5N2F: 435.2295, found: 435.2301.
Drugs.
The drugs used in this study were morphine hydrochloride (Takeda Chemical Industries, Osaka, Japan), naloxone hydrochloride (MP Biomedicals, Irvine, CA), gabapentin (Cayman Chemical Company, Ann Arbor, MI), naltrindole hydrochloride, nor-binaltorphimine dihydrochloride (norBNI), [d-Ala2, N-MePhe4, Gly-ol5]-enkephalin (DAMGO), [d-Pen2, d-Pen5]-enkephalin (DPDPE), (5α,7α,8β)-(+)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593), cyprodime hydrobromide, and [Met]-enkephalin (Sigma-Aldrich, St. Louis, MO) and β-funaltrexamine hydrochloride (β-FNA; Tocris-Cookson, Bristol, UK).
For experiments with guinea pig ileum and mouse vas deferens, 7-hydroxymitragynine-related indole alkaloids and cyprodime hydrobromide were first dissolved in 100% dimethylsulfoxide to yield a 5 mM solution, and then subsequently diluted with distilled water. The other drugs were dissolved in distilled water. For subcutaneous administration, 7-hydroxymitragynine-related indole alkaloids were dissolved in 0.01 N hydrochloric acid. Other drugs were dissolved in saline. For oral administration and receptor-binding assays, MGM-15, MGM-16, 7-hydroxymitragynine, and gabapentin were dissolved in 0.01 N hydrochloric acid.
In the antinociceptive test, naloxone hydrochloride (2 mg/kg), naltrindole (3 mg/kg), norBNI (20 mg/kg), or β-FNA (40 mg/kg) was administered subcutaneously 15 minutes, 30 minutes, 24 hours, or 24 hours before drug injection, respectively. These protocols were described by Matsumoto et al. (2005a).
Animals.
Male ddY strain mice (Japan SLC, Hamamatsu, Japan), male Hartley strain guinea pigs (Japan SLC), and male SD rats (Japan SLC) were used. Animals were housed in a temperature-controlled room at 24°C with lights on from 7:00 AM to 5:00 PM and free access to food and water. All experiments were performed in compliance with the Guiding Principles for the Care and Use of Laboratory Animals approved by the Japanese Pharmacological Society and the guidelines approved by the Ethics Committee on Animal Care and Animal Experimentation of Josai International University. The number of animals used was kept to the minimum necessary for a meaningful interpretation of the data, and animal discomfort was kept to a minimum.
Electrical Stimulation of Guinea Pig Ileum.
Guinea pig ileum was dissected and placed in Krebs-Henseleit solution (in mM: NaCl, 112.08; KCl, 5.90; CaCl2, 1.97; MgCl2, 1.18; NaH2PO4, 1.22; NaHCO3, 25.00; and glucose, 11.49). The ileum was placed under 1g tension in a 10-ml organ bath containing the nutrient solution. The bath was maintained at 37°C and continuously bubbled with a mixture of 95% O2 and 5% CO2. Tissues were stimulated by a platinum needle-ring electrode (Iwashiya Kishimoto Medical Instruments, Kyoto, Japan). After equilibration, the ileum was transmurally stimulated with monophasic pulses (0.2 Hz and 0.1-ms duration) by a stimulator (SEN-7203; Nihon Kohden, Tokyo, Japan). Contractions were isotonically recorded using a displacement transducer (TD-112S; Nihon Kohden). The effects of drug treatments on the twitch contractions evoked by transmural stimulation from the ring electrodes were examined. The height of the twitch response to transmural stimulation was measured before and after the drug challenge. Responses were expressed as contraction % of the twitch response to the transmural stimulation before the drug challenge as 100%.
Electrical Stimulation of Mouse Vas Deferens.
Mouse vas deferens was dissected and placed in Krebs-Henseleit solution without MgCl2. The tissues were placed under 0.2g tension in a 10-ml organ bath containing the nutrient solution. The bath was maintained at 37°C and continuously bubbled with a mixture of 95% O2 and 5% CO2. Tissues were stimulated by a platinum needle-ring (the ring was placed 20 mm above the base of a 5-mm-long needle) electrode. After equilibration, the tissues were transmurally stimulated with a train of 10 pulses of 0.5-ms duration with 2-ms intervals every 1 minute by a stimulator (SEN-7203). Contractions were isometrically recorded by using a displacement transducer (TB-651T; Nihon Kohden). The effects of drug treatments on the twitch contractions evoked by transmural stimulation from the ring electrodes were examined. The height of the twitch response to transmural stimulation was measured before and after the drug challenge. The responses were expressed as contraction percentage of the twitch response to the transmural stimulation before the drug challenge as 100%.
Receptor Binding.
Guinea pig brain membrane protein (without cerebellum, 500 μg) fractions were incubated with 1 nM [3H]DAMGO (50.0 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO) or 1 nM [3H]U69593 (55.0 Ci/mmol; GE Healthcare, Little Chalfont, Buckinghamshire, UK) in a final volume of 1 ml of 50 mM Tris-HCl buffer. Rat brain membrane protein fractions were incubated with 2 nM [3H]DPDPE (25.2 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA) in a final volume of 1 ml of 50 mM Tris-HCl buffer. Nonspecific binding was measured by the inclusion of 10 μM naloxone. The incubation periods were 1, 2.5, and 1 hour for [3H]DAMGO, [3H]DPDPE, and [3H]U69593, respectively, at 25°C. The reaction was terminated by rapid filtration under reduced pressure through glass fiber filters (Whatman GF/C, presoaked in 0.25% polyethyleneimine; Whatman, Clifton, NJ). After filtration, filters were washed three times with 3 ml of cold 50 mM Tris-HCl and counted in 4 ml of Ultima Gold scintillation cocktail (PerkinElmer Life and Analytical Sciences). The ability of unlabeled drugs to inhibit specific radioligand binding was expressed as the IC50 value, which is the molar concentration of the unlabeled drug necessary to displace 50% of the specific binding. Inhibition constants (Ki) of unlabeled compounds were calculated as described by Cheng and Prusoff (1973).
Guanosine 5′-O-(3-[35S]Thiotriphosphate) Binding Assay.
The agonist activities of the test compounds (MGM-15 and MGM-16) and DAMGO for μ-opioid receptor– or Met-enkephalin for δ-opioid receptor–positive controls were measured by radioactivities of guanosine 5′-O-(3-[35S]thiotriphosphate) ([35S]GTPγS). μ-Opioid receptor and δ-opioid receptor agonist activities were performed using a membrane fraction prepared from Chinese hamster ovary (CHO-K1) K1 cells expressing recombinant μ- and δ-opioid receptors. The membranes of cells stably expressing rat μ- and δ-opioid receptors were suspended with binding assay buffer [50 mM Tris-HCl (pH 7.4) containing EDTA (1 mM), MgCl2 (12.5 mM), NaCl (100 mM), bovine serum albumin (0.5%), and GDP (3 μM)]. The membranes (10 μg) were incubated at 25°C for 1 hour in 200 μl of binding assay buffer with various concentrations of the test compound or positive control (0.1–100,000 nM) and 50 pM [35S]GTPγS (specific activity, 1000 Ci/mmol; PerkinElmer Life and Analytical Sciences) in a 96-well plate. The reaction was terminated by filtration, and MicroScint-20 (PerkinElmer Life and Analytical Sciences) was added to the plate. Radioactivity in the samples was determined using a liquid scintillation analyzer. Nonspecific binding was measured in the presence of 10 μM unlabeled GTPγS. Agonist activation by each compound was expressed as the percentage stimulation, which was calculated as (T1 – T0)/(T2 – T0) × 100, where T0 is the nonspecific activity, T1 is the [35S]GTPγS activity in the presence of various concentrations of test compounds or positive control, and T2 is the maximal stimulation of [35S]GTPγS binding by the positive control.
All measurements were performed in duplicate. Three independent experiments were conducted in this study, and data are expressed as mean ± S.E.M. The EC50 values (a concentration that produces 50% of maximal stimulation of the [35S]GTPγS binding) and Emax (percentage of maximal stimulation in the [35S]GTPγS binding) were estimated from saturation analysis of agonist-stimulated [35S]GTPγS binding using program XLfit (Microsoft, Redmond, WA).
Tail-Flick Test.
Mice respond to a focused heat stimulus by flicking or moving their tail from the path of the stimulus, thereby exposing a photocell located in the tail-flick analgesia meter (Ugo Basile Tail-flick Unit 7360; Ugo Basile, Comerio, Italy) immediately below the tail. The reaction time is automatically recorded. Before treatment with drugs, vehicle, or saline, the nociceptive threshold was measured three times, and the mean of the reaction time was used as the predrug latency for each mouse. A cutoff time of 10 seconds was used to prevent tissue damage.
Antinociception in the tail-flick test was quantified using the percentage of maximum possible effect and calculated as: % maximum possible effect = [(test latency – predrug latency)/(cutoff time – predrug latency)] × 100.
GIT.
Mice were fasted, with water available ad libitum, for 18 hours before the experiments. Fifteen minutes after subcutaneous administration of 7-hydroxymitragynine, MGM-15, MGM-16, morphine, vehicle, or saline, a 0.25-ml charcoal meal (an aqueous suspension of 10% charcoal and 5% gum arabic) was orally administered. Fifteen minutes after oral administration of 7-hydroxymitragynine, MGM-15, MGM-16, or vehicle, and 30 minutes after oral administration of morphine or distilled water, a charcoal meal was orally administered. These time points were established by the time of peak effect for each drug and by the protocol as previously reported (Matsumoto et al., 2006a). Thirty minutes after administration of the charcoal meal, the animal was killed by cervical dislocation, and the small intestine from the pylorus to the ileocecal junction was carefully removed. Both the length of the small intestine from the pylorus to the ileocecal junction and the farthest distance to which the charcoal meal had traveled were measured. For each animal, the GIT was calculated as the percentage of the distance traveled by the charcoal meal relative to the total length of the small intestine. The inhibition of GIT (%) was calculated as: inhibition of GIT (%) = [(saline or vehicle GIT – drug GIT)/(saline or vehicle GIT)] × 100.
Neuropathic Pain Model.
The mice were anesthetized with 3% isoflurane. We produced a partial sciatic nerve injury by tying a tight ligature with an 8-0 silk suture around approximately one-third to one-half the diameter of the sciatic nerve on the right side (ipsilateral side) under a light microscope (SD30; Olympus, Tokyo, Japan), as described previously (Seltzer et al., 1990; Malmberg and Basbaum, 1998). In sham-operated mice, the nerve was exposed without ligation. The sciatic nerve–ligated mice exhibit thermal hyperalgesia and mechanical hyperalgesia on the ipsilateral side, indicating the state of neuropathic pain hypersensitivity (Narita et al., 2008). In the present study, the antinociceptive assay was performed 7 days after partial sciatic nerve ligation.
Measurement of Mechanical Allodynia.
The paw withdrawal threshold was determined as described by Chaplan et al. (1994). The mice were individually placed in suspended cages with wire mesh bottoms and allowed to acclimate to their environment for at least 30 minutes. The hind paw was stimulated with a series of calibrated (0.02–1.48 g) von Frey filaments applied to the plantar surface. The 50% paw withdrawal threshold that is nonparametrically distributed was determined using the up-down paradigm. Testing was initiated with 0.17-g hair, and a positive response was indicated by a withdrawal of the paw. The paw withdrawal threshold was measured repeatedly at the multiple time points indicated after administration of each drug.
Statistical Analysis.
The data are expressed as the mean ± S.E.M. Statistical analyses were performed with the two-tailed Student’s t test for comparison of two groups and by a one-way analysis of variance followed by a Bonferroni multiple-comparisons test for comparison of more than two groups. A P value < 0.05 was considered statistically significant. ED50 values and 95% confidence limits were determined using the Litchfield-Wilcoxon method (Litchfield and Wilcoxon, 1949). The statistical significance of differences between MGM-15 and MGM-16 in [35S]GTPγS binding assay was assessed using two-way analysis of variance followed by Bonferroni multiple-comparison tests.
Results
Effects of 7-Hydroxymitragynine-Related Indole Alkaloids on Electrically Stimulated Twitch Contraction in Guinea Pig Ileum.
The opioid agonistic activities of 7-hydroxymitragynine and semisynthetic compounds derived from 7-hydroxymitragynine (Fig. 1) were evaluated by measuring inhibition of the twitch contraction induced by electrical stimulation in the guinea pig ileum preparation (Table 1). MGM-15 and MGM-16 were prepared by NaBH4 reduction of 7-hydroxymitragynine and 10-fluoro-7-hydroxymitragynine, respectively. MGM-15 and MGM-16 showed a higher potency than 7-hydroxymitragynine, based on their pD2 values. The introduction of a fluoro group onto the C-10 position (MGM-16) led to a higher potency than MGM-15. MGM-16 showed an inhibitory effect as potent as that of the μ-selective agonist DAMGO.
Fig. 1.
Chemical structure and conformation of 7-hydroxymitragynine, MGM-15, and MGM-16.
TABLE 1.
pD2 values and maximum percentage inhibition of mitragynine derivatives in guinea pig ileum
Each value represents mean ± S.E.M. of data obtained from four to six animals.
| Compound | pD2 Value | Maximum Inhibition |
|---|---|---|
| % | ||
| Morphine | 7.16 ± 0.05 | 79.4 ± 5.7 |
| MGM-15 | 8.26 ± 0.05 | 78.2 ± 3.4 |
| MGM-16 | 8.81 ± 0.09 | 89.0 ± 2.3 |
| 7-Hydroxymitragynine | 7.58 ± 0.12 | 79.6 ± 3.0 |
| DAMGO | 8.46 ± 0.06 | 87.2 ± 1.8 |
Effects of MGM-15 and MGM-16 on Electrically Stimulated Twitch Contraction in Mouse Vas Deferens.
To clarify the effect of MGM-15 and MGM-16 on δ-opioid receptors, we investigated the effect of MGM-15 and MGM-16 using mouse vas deferens (Table 2). MGM-15 and MGM-16 inhibited the electrically elicited mouse vas deferens contraction in a dose-dependent manner, as did 7-hydroxymitragynine and the δ-selective agonist DPDPE. MGM-16 showed a higher potency than MGM-15. MGM-16 showed an inhibitory effect as potent as that of DPDPE. The concentration-response curves for MGM-15 and MGM-16 were shifted to the right in the presence of the μ-opioid receptor antagonist cyprodime and κ-opioid antagonist norBNI (Fig. 2). The curves of MGM-15 and MGM-16 were further shifted to the right in the presence of cyprodime, norBNI, and the δ-selective antagonist naltrindole (Table 3).
TABLE 2.
pD2 values and maximum inhibitory percent of DPDPE, MGM-15, MGM-16, and 7-hydroxymitragynine on electrical stimulation–induced contraction in mouse vas deferens in the absence or the presence of cyprodime (Cyp) and nor-BNI or Cyp, nor-BNI, and NTI
Each value represents mean ± S.E.M. of pD2 value (maximum inhibitory percent) obtained from four or five animals.
| Compound | pD2 Value of: |
||
|---|---|---|---|
| Compound Alone | Compound + Cyp and norBNI | Compound + Cyp, norBNI, and NTI | |
| DPDPE | 8.16 ± 0.20 (89.3 ± 1.1) | 7.80 ± 0.17 (90.7 ± 2.2) | 6.29 ± 0.10 (90.5 ± 1.6) |
| MGM-15 | 7.56 ± 0.06 (85.7 ± 2.3) | 7.10 ± 0.11 (82.3 ± 5.2) | 6.30 ± 0.08 (78.8 ± 3.3) |
| MGM-16 | 8.08 ± 0.11 (92.9 ± 1.8) | 7.50 ± 0.11 (95.8 ± 1.1) | 6.52 ± 0.22 (91.4 ± 3.0) |
| 7-Hydroxymitragynine | 7.22 ± 0.13 (82.7 ± 3.9) | 5.98 ± 0.16 (80.2 ± 3.3) | 5.55 ± 0.12 (78.2 ± 4.8) |
Fig. 2.
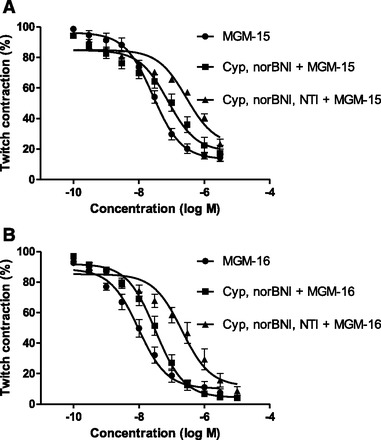
Concentration-response curves of MGM-15 (A) and MGM-16 (B) for electrical stimulation–induced contraction in mouse vas deferens in the absence or presence of cyprodime (Cyp) and norBNI or Cyp, norBNI, and naltrindole (NTI). Responses are expressed as contraction % of the twitch contraction before agonist addition. Data represent mean ± S.E.M. of five animals.
TABLE 3.
Ki values for the inhibition of μ- and κ-opioid binding to guinea pig brain membranes and δ-opioid binding to rat brain by test compounds
The μ-binding sites were labeled with [3H]DAMGO (1 nM), δ-binding sites with [3H]DPDPE (2 nM), and κ-binding sites with [3H]U69593 (1 nM). Data are expressed as the mean Ki value ± S.E.M. for three or four determinations performed in duplicate.
| Compound |
Ki of: |
||
|---|---|---|---|
| [3H]DAMGO (μ) | [3H]DPDPE (δ) | [3H]U69593 (κ) | |
| nM | |||
| Morphine | 2.7 ± 0.24 | 40 ± 6.0 | 55 ± 4.1 |
| MGM-15 | 6.4 ± 0.30 | 16 ± 1.0 | 55 ± 4.0 |
| MGM-16 | 2.1 ± 0.028 | 7.0 ± 0.23 | 29 ± 1.5 |
| DAMGO | 1.2 ± 0.092 | N.D. | N.D. |
| DPDPE | N.D. | 1.2 ± 0.13 | N.D. |
| U69593 | N.D. | N.D. | 0.66 ± 0.073 |
N.D., not determined.
Receptor Binding.
The affinities of MGM-15 and MGM-16 for the three opioid receptor types were determined by evaluating the inhibition of radioligand binding to μ-, δ-, and κ-opioid receptors (Table 3). MGM-15 and MGM-16 had a relatively high affinity for μ- and δ-opioid sites. The [3H]DAMGO and [3H]DPDPE binding was displaced by MGM-15 and MGM-16 in a concentration-dependent manner in the membrane samples from guinea pig and rat brain, respectively (data not shown).The Ki value for MGM-15 displacement of [3H]DAMGO and [3H]DPDPE binding to μ- and δ-opioid sites was 6.4 ± 0.30 and 16 ± 1.0 nM, respectively. The Ki value for MGM-16 displacement of [3H]DAMGO and [3H]DPDPE was 2.1 ± 0.028 and 7.0 ± 0.23 nM, respectively. MGM-15 and MGM-16 weakly displaced [3H]U69593 binding to κ-opioid sites.
[35S]GTPγS Binding Assay.
We next investigated the ability of MGM-15 and MGM-16 to activate G-proteins in CHO-K1 cells expressing recombinant μ- and δ-opioid receptors (Fig. 3; Table 4). The μ-opioid agonist DAMGO or δ-opioid agonist [Met]-enkephalin produced a concentration-dependent increase in [35S]GTPγS binding to the CHO-K1 cell membrane. MGM-16 also showed a concentration-dependent increase in [35S]GTPγS binding to CHO-K1 cell membranes expressing recombinant μ- and δ-opioid receptors. In contrast, MGM-15 showed a smaller increase in the binding of [35S]GTPγS than that of MGM-16.
Fig. 3.
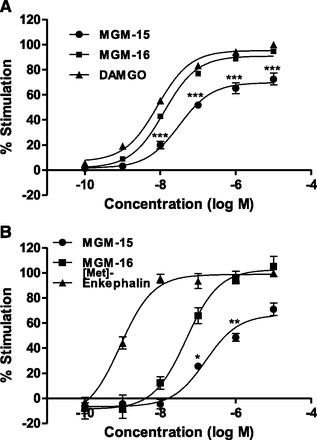
(A) Concentration-response curves of MGM-15, MGM-16, and DAMGO for [35S]GTPγS binding to CHO-K1 cells expressing recombinant μ-opioid receptors. (B) Concentration-response curves of MGM-15, MGM-16, and [Met]-enkephalin for [35S]GTPγS binding to CHO-K1 cells expressing recombinant δ-opioid receptors. The membrane-bound [35S]GTPγS was measured and expressed as % stimulation relative to the basal level. Each symbol represents the mean ± S.E.M. of three independent samples. *P < 0.05; **P < 0.01; ***P < 0.001 vs. MGM-16 group by Bonferroni correction.
TABLE 4.
[35S]GTPγS intrinsic efficacies and EC50 values of MGM-15, MGM-16, DAMGO, and [Met]-enkephalin in CHO-K1 cells expressing recombinant μ- and δ-opioid receptors
Each value represents the mean ± S.E.M. of three independent samples.
| Compound |
μ-Opioid Receptor |
δ-Opioid Receptor |
||
|---|---|---|---|---|
| EC50 | Emax | EC50 | Emax | |
| nM | nM | |||
| MGM-15 | 250 ± 61 | 72 ± 4.7 | 1492 ± 425 | 70 ± 4.9 |
| MGM-16 | 18 ± 0.055 | 94 ± 1.8 | 61 ± 3.2 | 105 ± 8.1 |
| DAMGO | 8.0 ± 0.69 | 100 | N.D. | N.D. |
| [Met]-enkephalin | N.D. | N.D. | 1.3 ± 0.26 | 100 |
N.D., not determined.
Antinociception of MGM-15 and MGM-16 in Mouse Tail-Flick Test by Subcutaneous and Oral Administration.
Antinociceptive effects of MGM-15 and MGM-16 were investigated in acute thermal pain tests in mice. MGM-15 and MGM-16 induced dose-related antinociceptive responses in tail-flick tests after subcutaneous and oral administration (Fig. 4). The effect peaked at 15–30 minutes after injection. The antinociceptive effect of MGM-15 was approximately 15 and 50 times more potent than that of morphine after subcutaneous and oral administration, respectively. The antinociceptive effect of MGM-16 was approximately 71 and 240 times more potent than that of morphine after subcutaneous and oral administration, respectively (Tables 5 and 6).
Fig. 4.
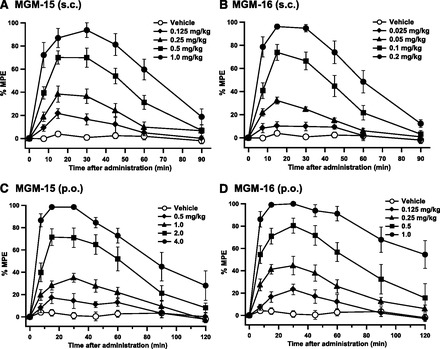
Time course of the antinociceptive effects produced by subcutaneous (A and B) or oral (C and D) administration of MGM-15 (A and C) and MGM-16 (B and D) in the tail-flick test in mice. Each value represents mean ± S.E.M. of data obtained from eight mice.
TABLE 5.
ED50 values of antinociceptive effects and IGIT produced by subcutaneous administration of morphine, 7-hydroxymitragynine (7-OHMG), MGM-15, and MGM-16 in mice
ED50 represents the median effective dose (mg/kg) (95% confidence limits).
| Test | ED50 of: |
|||
|---|---|---|---|---|
| Morphine | 7-OHMG | MGM-15 | MGM-16 | |
| mg/kg | ||||
| Tail-flick | 4.57 (3.12–6.69) | 0.80 (0.48–1.33) | 0.30 (0.18–0.50) | 0.064 (0.040–0.10) |
| IGIT | 1.07 (0.40–2.86) | 1.19 (0.54–2.63) | 0.35 (0.18–0.70) | 0.089 (0.040–0.188) |
TABLE 6.
ED50 values of antinociceptive effects and IGIT produced by oral administration of morphine, 7-hydroxymitragynine (7-OHMG), MGM-15, and MGM-16 in mice
ED50 represents the median effective dose (mg/kg) (95% confidence limits).
| Test | ED50 of: |
|||
|---|---|---|---|---|
| Morphine | 7-OHMG | MGM-15 | MGM-16 | |
| mg/kg | ||||
| Tail-flick | 63.0 (37.2–106.8) | 4.43 (1.57–6.93) | 1.26 (0.84–1.88) | 0.263 (0.165–0.420) |
| IGIT | 11.7 (5.6–24.6) | 7.50 (3.95–14.2) | 2.20 (0.18–0.70) | 0.451 (0.230–0.882) |
Next we investigated the effects of MGM-15 and MGM-16 on the inhibition of GIT (IGIT) and their antinociceptive effects in comparison with morphine and 7-hydroxymitragynine (Tables 5 and 6). The IGIT ED50 values of 7-hydroxymitragynine, MGM-15, and MGM-16 were slightly larger than their corresponding antinociceptive ED50 values. In the case of morphine, the IGIT ED50 value was much smaller than its antinociceptive ED50 value.
Effects of Selective Antagonists on MGM-15 and MGM-16 Antinociception in Mouse Tail-Flick Test.
To determine the opioid receptor type selectivity of MGM-15 and MGM-16 antinociception, mice were pretreated with selective opioid receptor antagonists in the tail-flick test (Fig. 5). The antinociceptive effect of MGM-15 and MGM-16 was completely blocked by the irreversible μ-opioid receptor selective antagonist β-FNA. These effects were partially and significantly blocked by the δ-opioid receptor selective antagonist naltrindole. The selective κ-opioid antagonist norBNI was ineffective against MGM-15- and MGM-16-induced antinociception. When these opioid antagonists were administered subcutaneously alone at the doses used in the present study, they did not produce any effects in the tail-flick test (data not shown).
Fig. 5.
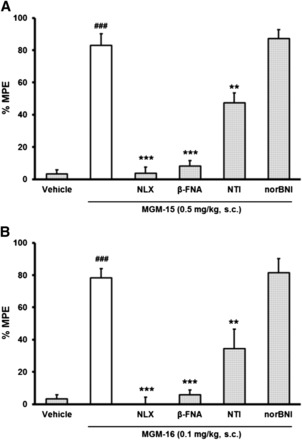
Effects of opioid receptor antagonists on antinociception by subcutaneous administration of MGM-15 (A) and MGM-16 (B). The antinociceptive effects of MGM-15 and MGM-16 were determined in mice by the tail-flick test after subcutaneous administration of the following antagonists: β-FNA (40 mg/kg), naloxonazine (NLZ; 35 mg/kg), NTI (3 mg/kg), and norBNI (20 mg/kg). Measurements were performed 15 minutes after subcutaneous administration of MGM-15 (0.5 mg/kg) or MGM-16 (0.1 mg/kg) in the tail-flick test. Each value represents mean ± S.E.M. of eight or nine mice. ###P < 0.001 vs. vehicle-treated mice by Student’s t test. **P < 0.01; ***P < 0.001 vs. MGM-15 or MGM-16 by Bonferroni correction.
Effect of MGM-16 on Thermal Hyperalgesia Induced by Sciatic Nerve Ligation in Mice.
MGM-16 showed potent μ/δ-opioid dual agonistic activities and further demonstrated higher potency than MGM-15 in vitro and in an in vivo acute pain model. Thus, we investigated the antiallodynic effect of MGM-16 in a neuropathic pain model. Mice with partial sciatic nerve ligation exhibited marked neuropathic pain-like behavior on the ipsilateral side 7 days after the nerve ligation. We evaluated the antiallodynic effects induced by subcutaneous and oral administration of MGM-16 in sciatic nerve–ligated mice using von Frey filaments (Fig. 6). MGM-16 (0.1, 0.2, and 0.4 mg/kg) dose-dependently increased the ipsilateral paw withdrawal threshold in sciatic nerve–ligated mice, and maximal antihyperalgesic responses were seen at 15 or 30 minutes after subcutaneous administration of MGM-16 (Fig. 6A). MGM-16 (0.2 mg/kg) reversed the threshold to control level in sciatic nerve–ligated mice. With oral administration, MGM-16 (0.5, 1, and 2 mg/kg) dose-dependently increased the ipsilateral paw withdrawal threshold in sciatic nerve–ligated mice, and maximal antihyperalgesic responses were seen 30 minutes after administration (Fig. 6B). MGM-16 at 1 mg/kg and gabapentin at 100 mg/kg reversed the threshold to control level in sciatic nerve–ligated mice.
Fig. 6.
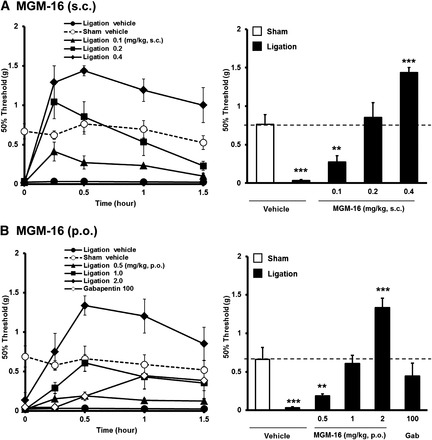
Effects of MGM-16 on mechanical allodynia in partial sciatic nerve–ligated mice. MGM-16 was administered subcutaneously (A) and orally (B), and the paw withdrawal thresholds on the ipsilateral side were determined repeatedly. Gabapentin (Gab; 100 mg/kg) was administered orally as a positive control. Several doses of MGM-16 and 100 mg/kg gabapentin are presented at 0.5 and 1 hour after administration, respectively. Each value represents mean ± S.E.M. of six or seven mice. **P < 0.01; ***P < 0.001 vs. vehicle by Bonferroni correction.
To investigate the contribution of the opioid receptor subtypes in the antiallodynic effect of MGM-16, sciatic nerve–ligated mice were pretreated with selective opioid receptor antagonists (Fig. 7). The antiallodynic effect of MGM-16 was completely blocked by β-FNA and naltrindole in the chronic pain model. The selective κ-opioid antagonist norBNI was ineffective against MGM-16-mediated antinociception.
Fig. 7.
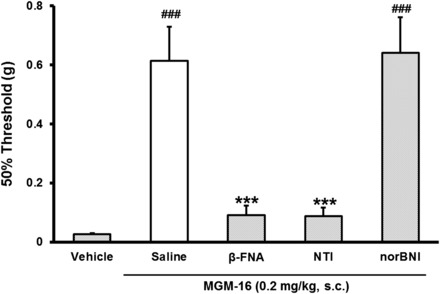
Effects of opioid receptor antagonists on the antiallodynic effect of subcutaneous administration of MGM-16 in partial sciatic nerve–ligated mice. The effects of MGM-16 were determined after subcutaneous administration of the following antagonists: β-FNA (40 mg/kg), NTI (3 mg/kg), and norBNI (20 mg/kg). Measurements were performed 30 minutes after subcutaneous administration of MGM-16 (0.2 mg/kg) in partial sciatic nerve–ligated mice. Each value represents mean ± S.E.M. of six mice. ###P < 0.001 vs. vehicle by Student’s t test. ***P < 0.001 vs. saline and MGM-16 by Bonferroni correction.
Discussion
In the present study, we synthesized the dual-acting μ- and δ-opioid agonists MGM-15 and MGM-16 from 7-hydroxymitragynine for the treatment of acute and chronic pain. MGM-16 showed higher opioid agonist potency than that of 7-hydroxymitragynine and MGM-15 in vitro and in vivo. When administered orally, MGM-16 showed 100-fold-higher analgesic potency than that of gabapentin in sciatic nerve–ligated mice. Therefore, MGM-16 could be useful for the treatment of chronic pain such as mechanical allodynia. To our knowledge, this is the first study to investigate the antiallodynic effect of mitragynine-related indole alkaloids using a neuropathic mouse pain model.
The alkaloid 7-hydroxymitragynine was found to possess the most potent opioid agonistic effects among the constituents isolated from the Thai herbal medicine M. speciosa (Horie et al., 2005). We have previously synthesized opioid analgesics from the indole alkaloid mitragynine (Takayama et al., 2002, 2006; Matsumoto et al., 2004, 2008; Takayama, 2004). We have surveyed these compounds for their opioid agonistic activities in vitro to elucidate the specific structure necessary for its pharmacophore to bind to opioid receptors. A nitrogen atom, a benzene residue, and an oxygen function play a significant role in producing opioid agonistic activity (Matsumoto et al., 2005a). The conversion of an indolenine moiety in 7-hydroxymitragynine (MGM-15) and 10-fluoro-7-hydroxymitragynine into an indoline derivative (MGM-16) led to an increase in agonistic potency. This indicates that the sp3 carbon at the C2 position (Fig. 1), which spatially configured the benzene ring in the ligands, was more efficient at exerting the opioid activity.
Takayama et al. (2006) reported that the dimension or electronegativity of the functional group at the C10 position of mitragynine derivatives is important in eliciting opioid agonistic effects. Among those derivatives, the C10-fluorinated derivative, MGM-9, showed the highest potency (Takayama et al., 2006). MGM-16, the C10-fluorinated derivative of MGM-15, showed the most potent opioid agonistic effects among the derivatives tested previously (Takayama et al., 2002, 2006; Matsumoto et al., 2008). MGM-16 showed full agonistic properties on μ- and δ-opioid receptors. MGM-16 also showed higher affinities and intrinsic efficacies than MGM-15 on μ- and δ-opioid receptors in vitro. A benzene residue of 7-hydroxymitragynine-related indole alkaloids plays an essential role in producing opioid agonistic activity for the specific structure necessary for its pharmacophore to bind to opioid receptors (Matsumoto et al., 2005a). It is speculated that the fluorine group at the C10 position on the benzene residue strengthens pharmacophore binding to opioid receptors because the lone fluorine pair easily forms hydrogen bonds with the receptor molecule (Takayama et al., 2006). Based on receptor-binding studies and tail-flick tests using selective antagonists, MGM-15 and MGM-16 showed similar opioid receptor type selectivity. Taken together, the findings show that fluorination at the C10 position increased the affinity to μ- and δ-opioid receptors but did not change the selectivity to the opioid receptor subtypes.
Subcutaneously and orally administered MGM-16 showed dose-dependent and antiallodynic effects in sciatic nerve–ligated mice. The antinociceptive effect of MGM-16 was completely and partially blocked by the μ-selective antagonist β-FNA and by the δ-selective antagonist naltrindole, respectively, in an acute pain model. The antiallodynic effect of MGM-16 was completely blocked by β-FNA and naltrindole in a chronic pain model. The contribution of δ-opioid receptors to the antiallodynic effect of MGM-16 in the neuropathic pain model was higher than in the acute pain test. As previously reported, upregulation of δ-opioid receptors is induced in a rat model of peripheral nerve injury to the dorsal root ganglion (Kabli and Cahill, 2007). δ-Opioid receptor knockout mice showed an increase in mechanical allodynia under neuropathic pain induced by partial sciatic nerve ligation (Nadal et al., 2006; Gaveriaux-Ruff et al., 2011). However, δ-opioid receptor knockout mice showed normal pain responses to acute pain. Furthermore, δ-opioid selective agonists induce clear antiallodynic effects in various animal models of neuropathic pain but do not show clear antinociceptive effects on acute nociception (Gaveriaux-Ruff et al., 2011). Thus, δ-opioid receptor activation leads to controlled mechanical allodynia. This indicates that the δ-opioid receptor agonistic properties of MGM-16 are involved in its antiallodynic effects in sciatic nerve–ligated mice.
Several studies showed the effectiveness of μ-opioid receptor agonists and demonstrated that μ-opioid receptors contribute to the control of mechanical allodynia in neuropathic pain (Mansikka et al., 2004; Finnerup et al., 2010). Indeed, μ-opioid receptor knockout mice showed an increase in mechanical allodynia under neuropathic pain induced by partial sciatic nerve ligation (Nadal et al., 2006; Gaveriaux-Ruff et al., 2011). Furthermore, μ-opioid receptor agonists attenuate mechanical allodynia in sciatic nerve–ligated mice (Mansikka et al., 2004). This suggests that the μ-opioid receptor contributes to alleviating mechanical allodynia. In this study, we found that the antiallodynic effect of MGM-16 was significantly blocked by a μ-opioid receptor antagonist. Therefore, the μ-opioid receptor agonistic activities of MGM-16 also contribute to its antiallodynic effect in partial sciatic nerve–ligated mice.
The traditional Thai herbal medicine M. speciosa has long been used in Thailand for its opioid-like effects (Burkill, 1935) and as a replacement for opium (Suwanlert, 1975). We have studied the pharmacological activities of mitragynine, a major alkaloid of this herb (Watanabe et al., 1997; Matsumoto et al., 2005b), and 7-hydroxymitragynine, a main active alkaloid of this herb (Matsumoto et al., 2004, 2005a, 2006a). Recently, additional research has been performed on the opium-like effects of M. speciosa and mitragynine and the unique chemical and pharmacological properties of mitragynine-related indole alkaloids (Adkins et al., 2011; Raffa et al., 2013). We synthesized indole alkaloids such as the partial opioid agonist 9-hydroxycorynantheidine, the opioid antagonist corynantheidine, and the μ/κ-opioid dual agonist MGM-9. These compounds showed less rewarding effects, based on the structure-activity relationship study (Takayama, 2004; Matsumoto et al., 2006b, 2008). In this study, we synthesized a novel μ/δ-opioid dual agonist, MGM-16. MGM-16 showed opioid agonistic effects as potent as those of DAMGO and DPDPE in a functional assay using isolated guinea pig ileum and mouse vas deferens, respectively. Previously, there were no reports on G-protein action following the binding of mitragynine-related indole alkaloids to opioid receptors (Raffa et al., 2013). In this study, we show that MGM-16 dose-dependently increases GTPγS binding to CHO-K1 cells expressing recombinant μ- and δ-opioid receptors. We investigated the antinociceptive effects of MGM-16 ingested orally, based on the traditional usage of M. speciosa and the relevance of this route for clinical administration. Following oral administration, the antinociceptive effect of MGM-16 was about 240 times more potent than that of morphine in the mouse tail-flick test. Gabapentin, a synthetic analog of γ-aminobutyric acid, was initially developed as an anticonvulsant and later licensed for the management of postherpetic neuralgia (Gilron, 2007). In our results, gabapentin is effective for mechanical allodynia but very high doses (100 mg/kg p.o.) were needed to alleviate it, as reported previously (Kiso et al., 2008). Under this experimental condition, the antiallodynic effect of MGM-16 is approximately 100 times more potent than that of gabapentin in partial sciatic nerve–ligated mice. MGM-16 exerts a potential ability to treat chronic pain more effectively than existing drugs.
In conclusion, the present study demonstrated that MGM-16 derived from Mitragyna alkaloid 7-hydroxymitragynine has potent μ/δ-opioid dual agonistic effects in vitro and in vivo. Because of its dual mechanism, MGM-16 has efficacy in a broad spectrum of acute and chronic pain models. This is the first study to report the antiallodynic effects of the alkaloids derived from mitragynine. MGM-16 may become a new class of seed compound with potential therapeutic utility for treating neuropathic pain.
Acknowledgments
The authors thank Shionogi & Co., Ltd. for the [35S]GTPγS binding assay and Hiroyuki Kanazawa and Maki Makiyama, Josai International University, for technical assistance with the electrically stimulated twitch contraction in guninea pig ileum and mouse vas deferens.
Abbreviations
- CHO
Chinese hamster ovary
- DAMGO
[d-Ala2, N-MePhe4, Gly-ol5]-enkephalin
- DPDPE
[d-Pen2, d-Pen5]-enkephalin
- β-FNA
β-funaltrexamine hydrochloride
- GIT
gastrointestinal transit
- 7-hydroxymitragynine
(E)-methyl 2-((2S,3S,7aS,12bS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate
- IGIT
inhibitory effects on gastrointestinal transit
- MGM-9
(E)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxyindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate
- MGM-15
(E)-methyl 2-((2S,3S,7aS,12aR,12bS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12,12a,12b-decahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate
- MGM-16
(E)-methyl 2-((2S,3S,7aS,12aR,12bS)-3-ethyl-9-fluoro-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12,12a,12b-decahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate
- norBNI
nor-binaltorphimine dihydrochloride
- NTI
naltrindole
- U69593
(5α,7α,8β)-(+)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
Authorship Contributions
Participated in research design: Matsumoto, Narita, Horie, Suzuki, Takayama.
Conducted experiments: Matsumoto, Muramatsu, Nakayama.
Contributed new reagents or analytic tools: Takayama, Kitajima, Misawa, Nakayama.
Performed data analysis: Matsumoto.
Wrote or contributed to the writing of the manuscript: Matsumoto, Horie, Devi, Tashima.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Culture, Sports, Science, and Technology of Japan; a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; Special Funds for Education and Research (Development of SPECT Probes for Pharmaceutical Innovation) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; and Iodine Research Project at Chiba University.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Adkins JE, Boyer EW, McCurdy CR. (2011) Mitragyna speciosa, a psychoactive tree from Southeast Asia with opioid activity. Curr Top Med Chem 11:1165–1175. [DOI] [PubMed] [Google Scholar]
- Burkill IH. (1935) A Dictionary of the Economic Products of the Malay Peninsula, vol II, pp 1480–1483, Crown Agents for the Colonies, London. [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- Corbett AD, Henderson G, McKnight AT, Paterson SJ. (2006) 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol 147 (Suppl 1):S153–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremeans CM, Gruley E, Kyle DJ, Ko MC. (2012) Roles of μ-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J Pharmacol Exp Ther 343:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Sindrup SH, Jensen TS. (2010) The evidence for pharmacological treatment of neuropathic pain. Pain 150:573–581. [DOI] [PubMed] [Google Scholar]
- Horie S, Koyama F, Takayama H, Ishikawa H, Aimi N, Ponglux D, Matsumoto K, Murayama T. (2005) Indole alkaloids of a Thai medicinal herb, Mitragyna speciosa, that has opioid agonistic effect in guinea-pig ileum. Planta Med 71:231–236. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Nozaki C, Nadal X, Hever XC, Weibel R, Matifas A, Reiss D, Filliol D, Nassar MA, Wood JN, et al. (2011) Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain 152:1238–1248. [DOI] [PubMed] [Google Scholar]
- Gilron I. (2007) Gabapentin and pregabalin for chronic neuropathic and early postsurgical pain: current evidence and future directions. Curr Opin Anaesthesiol 20:456–472. [DOI] [PubMed] [Google Scholar]
- Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. (2005) Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 352:1324–1334. [DOI] [PubMed] [Google Scholar]
- Kabli N, Cahill CM. (2007) Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 127:84–93. [DOI] [PubMed] [Google Scholar]
- Kiso T, Watabiki T, Tsukamoto M, Okabe M, Kagami M, Nishimura K, Aoki T, Matsuoka N. (2008) Pharmacological characterization and gene expression profiling of an L5/L6 spinal nerve ligation model for neuropathic pain in mice. Neuroscience 153:492–500. [DOI] [PubMed] [Google Scholar]
- Litchfield JT, Jr, Wilcoxon F. (1949) A simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther 96:99–113. [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI. (1998) Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain 76:215–222. [DOI] [PubMed] [Google Scholar]
- Mansikka H, Zhao C, Sheth RN, Sora I, Uhl G, Raja SN. (2004) Nerve injury induces a tonic bilateral mu-opioid receptor-mediated inhibitory effect on mechanical allodynia in mice. Anesthesiology 100:912–921. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Hatori Y, Murayama T, Tashima K, Wongseripipatana S, Misawa K, Kitajima M, Takayama H, Horie S. (2006a) Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol 549:63–70. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Horie S, Ishikawa H, Takayama H, Aimi N, Ponglux D, Watanabe K. (2004) Antinociceptive effect of 7-hydroxymitragynine in mice: discovery of an orally active opioid analgesic from the Thai medicinal herb Mitragyna speciosa. Life Sci 74:2143–2155. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Horie S, Takayama H, Ishikawa H, Aimi N, Ponglux D, Murayama T, Watanabe K. (2005a) Antinociception, tolerance and withdrawal symptoms induced by 7-hydroxymitragynine, an alkaloid from the Thai medicinal herb Mitragyna speciosa. Life Sci 78:2–7. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Takayama H, Ishikawa H, Aimi N, Ponglux D, Watanabe K, Horie S. (2006b) Partial agonistic effect of 9-hydroxycorynantheidine on mu-opioid receptor in the guinea-pig ileum. Life Sci 78:2265–2271. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Takayama H, Narita M, Nakamura A, Suzuki M, Suzuki T, Murayama T, Wongseripipatana S, Misawa K, Kitajima M, et al. (2008) MGM-9 [(E)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxyindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate], a derivative of the indole alkaloid mitragynine: a novel dual-acting mu- and kappa-opioid agonist with potent antinociceptive and weak rewarding effects in mice. Neuropharmacology 55:154–165. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Yamamoto LT, Watanabe K, Yano S, Shan J, Pang PK, Ponglux D, Takayama H, Horie S. (2005b) Inhibitory effect of mitragynine, an analgesic alkaloid from Thai herbal medicine, on neurogenic contraction of the vas deferens. Life Sci 78:187–194. [DOI] [PubMed] [Google Scholar]
- Nadal X, Baños JE, Kieffer BL, Maldonado R. (2006) Neuropathic pain is enhanced in delta-opioid receptor knockout mice. Eur J Neurosci 23:830–834. [DOI] [PubMed] [Google Scholar]
- Narita M, Nakamura A, Ozaki M, Imai S, Miyoshi K, Suzuki M, Suzuki T. (2008) Comparative pharmacological profiles of morphine and oxycodone under a neuropathic pain-like state in mice: evidence for less sensitivity to morphine. Neuropsychopharmacology 33:1097–1112. [DOI] [PubMed] [Google Scholar]
- Nozaki C, Le Bourdonnec B, Reiss D, Windh RT, Little PJ, Dolle RE, Kieffer BL, Gavériaux-Ruff C. (2012) δ-Opioid mechanisms for ADL5747 and ADL5859 effects in mice: analgesia, locomotion, and receptor internalization. J Pharmacol Exp Ther 342:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffa RB, Beckett JR, Brahmbhatt VN, Ebinger TM, Fabian CA, Nixon JR, Orlando ST, Rana CA, Tejani AH, Tomazic RJ. (2013) Orally active opioid compounds from a non-poppy source. J Med Chem 56:4840–4848. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. (1990) A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43:205–218. [DOI] [PubMed] [Google Scholar]
- Suwanlert S. (1975) A study of kratom eaters in Thailand. Bull Narc 27:21–27. [PubMed] [Google Scholar]
- Takayama H. (2004) Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pharm Bull (Tokyo) 52:916–928. [DOI] [PubMed] [Google Scholar]
- Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, et al. (2002) Studies on the synthesis and opioid agonistic activities of mitragynine-related indole alkaloids: discovery of opioid agonists structurally different from other opioid ligands. J Med Chem 45:1949–1956. [DOI] [PubMed] [Google Scholar]
- Takayama H, Misawa K, Okada N, Ishikawa H, Kitajima M, Hatori Y, Murayama T, Wongseripipatana S, Tashima K, Matsumoto K, et al. (2006) New procedure to mask the 2,3-π bond of the indole nucleus and its application to the preparation of potent opioid receptor agonists with a Corynanthe skeleton. Org Lett 8:5705–5708. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yano S, Horie S, Yamamoto LT. (1997) Inhibitory effect of mitragynine, an alkaloid with analgesic effect from Thai medicinal plant Mitragyna speciosa, on electrically stimulated contraction of isolated guinea-pig ileum through the opioid receptor. Life Sci 60:933–942. [DOI] [PubMed] [Google Scholar]