

Abstract
The function of many ion channels is under dynamic control by coincident activation of G-protein-coupled receptors (GPCRs), particularly those coupled to the Gαs and Gαq family members. Such regulation is typically dependent on the subunit composition of the ionotropic receptor or channel as well as the GPCR subtype and the cell-specific panoply of signaling pathways available. Because GPCRs and ion channels are so highly represented among targets of U.S. Food and Drug Administration-approved drugs, functional cross-talk between these drug target classes is likely to underlie many therapeutic and adverse effects of marketed drugs. GPCRs engage a myriad of signaling pathways that involve protein kinases A and C (PKC) and, through PKC and interaction with β-arrestin, Src kinase, and hence the mitogen-activated–protein-kinase cascades. We focus here on the control of ionotropic glutamate receptor function by GPCR signaling because this form of regulation can influence the strength of synaptic plasticity. The amino acid residues phosphorylated by specific kinases have been securely identified in many ionotropic glutamate (iGlu) receptor subunits, but which of these sites are GPCR targets is less well known even when the kinase has been identified. N-methyl-d-aspartate, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, and heteromeric kainate receptors are all downstream targets of GPCR signaling pathways. The details of GPCR–iGlu receptor cross-talk should inform a better understanding of how synaptic transmission is regulated and lead to new therapeutic strategies for neuropsychiatric disorders.
Introduction
Over 40% of U.S. Food and Drug Administration-approved drugs target either G-protein-coupled receptors (GCPR) or ion channels (Overington et al., 2006). Interestingly, these two classes of drug target often regulate one another by direct physical interaction or through activation of signaling pathways. Signaling pathways underlying regulation take many forms, including activation of Ser/Thr or tyrosine kinase cascades by G-protein subunits or β-arrestin, with effector mechanisms involving changes in protein-protein interactions that anchor receptors to synaptic-scaffolding complexes and changes in channel gating or surface trafficking. Adding further nuance to this form of receptor cross-talk, the agonist affinity for metabotropic glutamate receptor 1 (mGlu1) is voltage-dependent due to a gating charge-driven conformational change in the receptor (Ben-Chaim et al., 2006). Thus the ability of mGlu1 to modulate voltage-dependent channels such as N-methyl-d-aspartate (NMDA) receptors will itself be voltage-dependent. The combination of phospho specific antibodies, site-directed mutagenesis, and functional analysis has allowed the secure identification of dynamically regulated phosphorylation sites on many of the ionotropic glutamate receptor subunits. However, convincing linkage of a specific G-protein-coupled receptor (GPCR) to one or more specific phosphorylated amino acids that regulate ionotropic receptor function is uncommon. Several key questions remain for control of glutamate receptor function by GPCRs: Which ionotropic glutamate (iGlu) receptors are regulated by GPCRs, and in what cell types? Does the regulation occur directly (i.e., physical interaction of the GCPR with regulatory/accessory and scaffolding proteins) or indirectly via intracellular signaling molecules? If the latter, which pathways are involved and which of the potential consensus phosphorylation sites on iGlu receptors are actually used? How does cross-talk alter receptor and synaptic function? Here we review the regulation of ionotropic glutamate receptors [α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), NMDA, and kainate] by GPCRs with special attention on Gαq-coupled mGlu and muscarinic acetylcholine (mACh) receptor, as these GPCRs are critically involved in synaptic transmission and plasticity and their altered regulation has significant implications in neuropathies.
Ionotropic Glutamate Receptors
Ionotropic glutamate receptors are classified functionally and by molecular homology into three receptor classes: NMDA (consisting of the GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A, and GluN3B subunits), AMPA (GluA1, GluA2, GluA3, and GluA4), and kainate (GluK1, GluK2, GluK3, GluK4, and GluK5). Ionotropic glutamate receptors are tetrameric assemblies of multiple subunits. Subunit assembly appears exclusive within a receptor class; for example, none of the four AMPA receptor subunits has been shown to coassemble with NMDA receptor subunits to form a functional receptor. We will consider cross-talk regulation of each ionotropic receptor class in turn.
NMDA Receptor Modulation
The regulation of NMDA receptor (NMDAR) currents can occur through phosphorylation of regulatory sites located within the GluN1 or GluN2 subunits or via protein-protein interactions, in which changes in the binding of proteins like postsynaptic density (PSD)-95 or calmodulin help to cluster NMDARs at postsynaptic sites. The distal C terminus of GluN2 subunits contains postsynaptic density 95/disc-large/ZO-1 (PDZ) motifs that noncovalently link NMDA receptors to scaffolding proteins like those in the PSD-95 family and its associated proteins—membrane-associated guanylate kinase and A-kinase anchoring protein. All four GluN2 subunits appear to interact with all four PSD-95 proteins (PSD-95, PSD-93, SAP102, SAP97) typically resulting in a change in surface targeting of NMDA receptors (Bard and Groc, 2011). Over the past two decades attention has also focused on the indirect regulation of NMDARs mediated by the activation of various GPCRs and their downstream signaling molecules. Fig. 1A illustrates several prominent signaling pathways by which GPCRs regulate the channel properties or, more often, surface trafficking, of iGlu receptors.
Fig. 1.
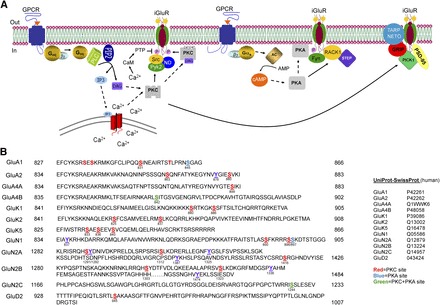
Schematic of the regulation of ionotropic glutamate receptors by GPCR activation. (A) Mechanisms, signaling pathways, and molecules underlying the regulation of ionotropic glutamate receptor by Gαq-coupled and Gαs-coupled GPCR receptor activation. Dashed arrows represent translocation of a signaling molecule, whereas solid arrows represent enzymatic action. Additional pathways mediated by association of GPCR with β-arrestin and subsequent engagement of AKT (protein kinase B) and ERK (extracellular signal-regulated kinase) pathways are omitted for simplicity. (B) Phosphorylation of iGlu receptor C-terminal domains. These phosphorylation sites modulate receptor trafficking, channel activity, and protein-protein interactions. The left column contains the receptor subunit name. UniProt-SwissProt human accession numbers were used for each receptor (Traynelis et al., 2010). The beginning and ending amino acid numbers are on the left and right of the C-terminal domain, respectively. Only a portion of the C-terminal domain is shown for NMDA receptors. Modified residues are colored-coded according to the legend.
In addition to receptor cross-talk mediated via diffusible signaling molecules (described below), the C terminus of the GluN1 subunit has been shown to physically interact with, or reside within the same tight protein complex as, mu opioid receptors (MOR) (Rodriguez-Munoz et al., 2012), D1 dopamine receptors (Lee et al., 2002), and mGlu5a receptors (Perroy et al., 2008). The physical interaction between GluN1 and mGlu5a results in constitutive, agonist-insensitive inhibition of NMDAR, whereas a D1 dopamine receptor agonist both reduces NMDAR currents and weakens the physical interaction between D1 and NMDAR through a protein kinase A (PKA)/protein kinase C (PKC)-independent mechanism. By contrast, the MOR-GluN1 interaction could be disrupted by morphine in a PKC-dependent manner. Interestingly, tolerance to morphine can be prevented by PKC inhibitors (Hull et al., 2010), raising the possibility that the MOR-GluN1 physical interaction is somehow required to maintain chronic sensitivity to morphine.
Regulation of NMDARs by mGlu and mACh Receptors.
Metabotropic glutamate receptors modulate neuronal excitability via regulation of voltage-sensitive calcium channels, G-protein-regulated inward rectifier K+ channels, GABAA receptors, AMPA receptors, and NMDA receptors (Conn and Pin, 1997). Eight mGlu receptors are classified into three groups according to their Gα protein. Group I mGlu receptors (mGlu1 and mGlu5) are coupled to Gαq family members and activate the phospholipase C pathway, which leads to an increase in diacylglycerol and inositol trisphosphate levels, intracellular calcium, and stimulation of PKC (Fig. 1A). An important consequence of group I mGlu activation is modulation of glutamatergic transmission, for example, potentiated NMDAR responses in the hippocampus, cerebellum, cortex, and spinal cord (Bordi and Ugolini, 1999). Activation of group I mGlu receptors in striatal neurons increases GluN1 phosphorylation at serines 896 and 897 in vivo. The GluN1 phosphorylation was attenuated in the presence of an mGlu5 antagonist suggesting that the phosphorylation is mGlu5-mediated (Choe et al., 2006). The kinase(s) responsible for the phosphorylation was not identified in this study. However, functional cross-talk was observed in human embryonic kidney 293 cells expressing NMDARs containing the GluN2A and GluN2B subunits as well as the mGlu1 receptor, where activation of mGlu1 resulted in increased phosphorylation of GluN2A and GluN2B by a Src family tyrosine kinase (Heidinger et al., 2002), suggestive of a β-arrestin or PKC-pathway involvement. Although mGlu1 and mGlu5 are both coupled to Gαq and they both can potentiate NMDA receptor currents in CA3 pyramidal cells, they appear to do so via two distinct signaling pathways, mGlu1 via a G-protein-independent Src kinase pathway possibly involving β-arrestin, and mGlu5 via a Gαq-PKC-PYK2 (proline-rich tyrosine kinase 2)-Src pathway (Benquet et al., 2002).
Neuronal excitability and synaptic transmission are also altered by interactions between the cholinergic and glutamatergic neurotransmitter systems. One mechanism involves potentiation of NMDA receptor currents by M1 muscarinic receptors, which colocalize with the GluN1 subunit in hippocampal CA1 pyramidal cells (Marino et al., 1998). Potentiation of NMDA receptors by M1 mACh receptor activation is mediated by Src kinase and postulated to involve a kinase cascade in which PKC phosphorylates PYK2, which in turn phosphorylates and activates Src kinase (Lu et al., 1999). The molecular target of Src might be three tyrosines on the C terminus of GluN2A (Zheng et al., 1998). Thus M1 mACh and mGlu5 receptors appear to share an identical signaling pathway.
The examples discussed above involve potentiation of NMDA receptor activity; however, there is also evidence for inhibition of NMDA receptor currents following activation of several GPCRs. For example, activation of group I mGlu receptors potentiated NMDA receptor currents in CA1 pyramidal cells, but inhibited NMDA receptor currents in nearby hippocampal CA3 pyramidal cells through a Ca2+-dependent mechanism (Grishin et al., 2004), pointing to cell-specific regulation of NMDA receptors by mGlu receptors. Activation of mGlu7 is reported to inhibit NMDAR currents in neurons of the prefrontal cortex by engaging a β-arrestin/extracellular signal-regulated kinase pathway, which regulates NMDAR surface trafficking by controlling the polymerization state of actin (Gu et al., 2012). A similar pattern of regulation of NMDA receptors was observed following M1 mACh receptor activation, in that NMDA receptor responses were potentiated in hippocampal CA1 pyramidal cells but inhibited in hippocampal CA3 pyramidal cells by a mechanism proposed to involve a calmodulin-activated tyrosine phosphatase (Grishin et al., 2005). More complicated signaling pathways also exist. For example, activation of the Gi/Go-coupled 5HT1A receptor on cortical pyramidal neurons suppresses NMDAR currents by destabilizing microtubules leading to reduced NMDAR transport to dendritic membranes. This effect of 5HT1A is countermanded by activation of Gq-coupled 5HT2A, which through a β-arrestin-Src-extracellular signal-regulated kinase pathway stabilizes microtubules (Yuen et al., 2005). The overall outcome on NMDA receptor activity, whether potentiated or inhibited, thus depends on the cell population that expresses the receptors, the GPCR subtypes expressed and the many scaffolding and intracellular signaling molecules brought into play in multiple signaling cascades.
Signaling Pathways and NMDAR Phosphorylation.
NMDA receptors are known to form stable complexes with intracellular cell signaling proteins such as Src family kinases, calmodulin, and G proteins, which can alter the function and/or distribution of NMDAR subunits. A sequence scan of the intracellular domains of the NMDA receptor subunits reveals consensus phosphorylation sites for both PKA and PKC (Fig. 1B), and the phosphorylation of NMDA receptors by these kinases leads to three functional outcomes depending on NMDAR subunit and phosphorylated residues, namely facilitation of NMDA receptor currents via increased channel opening, increased NMDAR surface expression, and suppression of NMDA-mediated currents (Chen and Roche, 2007; Traynelis et al., 2010). In addition to PKA and PKC, the calcium/calmodulin-dependent protein kinase (CaMKII), and the tyrosine kinases Src and Fyn also phosphorylate NMDA receptors (Chen and Roche, 2007; Traynelis et al., 2010) . The activation of phosphatases (serine/threonine and tyrosine) leads to dephosphorylation of NMDARs and reduced NMDAR activity (e.g., Grishin et al., 2005), suggesting that the set point of NMDAR responsiveness relies on the balance of intracellular kinase and phosphatase activities.
A prominent mechanism by which GPCR activation alters NMDA receptor activity is by altering the surface expression of NMDA receptor subunits. For example, stimulation of mGlu5 in neurons increases the surface expression of NMDA receptors in a tyrosine kinase-dependent manner (Lan et al., 2001). It is now well established that NMDA receptors are involved in elaborate protein-protein interactions with regulatory and accessory proteins at the postsynaptic density. Nonreceptor protein tyrosine kinases, such as Src family kinases, are critically involved in these interactions. Tyrosine kinases like PYK2 and Fyn are intertwined in scaffolding complexes that bind to NMDA receptors and alter NMDA function (MacDonald et al., 2007). Models have been developed describing how this regulation may occur. For example, Pyk2 provides an Src homology 2 docking site for Src that is normally anchored to NMDA receptors by the protein ND2. Following activation of a Gαq-coupled GPCR like the M1 or mGlu5 receptors, PKC is activated leading to phosphorylation of PYK2 that in turn activates Src kinase, which targets a tyrosine near the GluN2B C terminus and enhances NMDA receptor activity by increasing trafficking to the surface membrane (MacDonald et al., 2007). NMDAR-dependent long-term potentiation at CA1 synapses requires both PYK2 and Src (Huang et al., 2001), which emphasizes the importance of this pathway.
GPCR regulation of NMDA receptors is not limited to Gαq-coupled receptors as Gαs-coupled GPCRs also play a role, although some key players are different. For example, the GluN2B subunit interacts with an inhibitory scaffolding protein named RACK1 (receptor for activated C kinase 1). Activation of a Gαs-coupled GPCR by the pituitary adenylate cyclase-activating peptide (PACAP)-enhanced NMDA receptor function via RACK1 in CA1 hippocampal slices (Yaka et al., 2003). The proposed mechanism involves PKA activation that leads to the dissociation of RACK1 from the NMDA receptor, resulting in translocation of RACK1 to the nucleus and exposure of the C terminus of GluN2B to Fyn kinase, which phosphorylates GluN2B on Y1474 thereby enhancing NMDA receptor activity (Yaka et al., 2003). MacDonald et al. (2005) reported that NMDA receptor modulation in CA1 neurons by PACAP is dominantly mediated by activation of PKC and Src, rather than the PKA pathway. The multiple pathways by which PACAP regulates NMDA receptors in hippocampal CA1 neurons deserve further study. In striatal neurons activation of the Gαs-coupled dopamine receptor D1 leads to the rapid increase in synaptic surface expression of NMDA receptor subunits in a tyrosine phosphorylation-dependent manner (Hallett et al., 2006). A model was proposed recently in which dopamine receptor (D1)-mediated potentiation of NMDA receptors occurs via phosphorylation of GluN2B subunits by Fyn kinase (Yang et al., 2012). These examples demonstrate that both PKA and PKC downstream of Gαs and Gαq activation, respectively, regulate NMDA receptors through Src family kinases.
GluN2C subunits in the cerebellum can be phosphorylated by protein kinase B on Ser1096, which controls their interaction with 14-3-3ε, in turn regulating their surface expression (Chen and Roche, 2009). Protein kinase B is itself activated by insulin-like growth factor 1 via β-arrestin and G-protein receptor kinases (Zheng et al., 2012), although the native G-protein-coupled receptor has not yet been identified.
The GluN1 subunit, present in all functional NMDA receptors, is phosphorylated by PKC at serines 890 and 896 with phosphorylation of Ser890 resulting in the disruption of GluN1 clustering (Traynelis et al., 2010). The phosphorylation of Ser896 by PKC does not appear to be important for GluN1 clustering. Interestingly, the dual phosphorylation of Ser896 and Ser897 by PKC and PKA, respectively, promotes exit of this subunit from the estrogen receptor resulting in increased surface expression of GluN1 (Chen and Roche, 2007). Both GluN2A and GluN2B are also phosphorylated by PKC. The phosphorylation of GluN2A at Ser1416 by PKC weakens the binding of CaMKII to the GluN2A subunit, which creates a hierarchy between the PKC- and CaMKII-signaling pathways (Gardoni et al., 2001). Mutagenesis studies suggest that phosphorylation of Ser1303 and Ser1323 in GluN2B is one route to potentiation by PKC of NMDA receptors containing GluN2B (Chen and Roche, 2007). Both the synaptic GluN2A and the extrasynaptic GluN2B subunits are regulated by the PKC, PKA, and the tyrosine kinase Src (Chen and Roche, 2007). The GluN2C receptor contains a common phosphorylation site for both PKA and PKC (Ser1244) that is located in the PDZ-binding motif of the subunit (Fig. 1B) and may play a physiologic role in cerebellar granule cells where the majority of GluN2C receptors are expressed.
AMPA Receptors
A second subgroup of ionotropic glutamate receptors comprises the α-amino-3-hydro-5-methyl-4-isoxazole propionate (AMPA) receptors. These receptors activate and desensitize rapidly in response to glutamate thus mediating rapid excitatory synaptic transmission. An important consequence of AMPA receptor regulation by GPCRs is control of the strength and direction of synaptic plasticity. AMPA receptors are involved in protein-protein interactions with scaffolding proteins such as protein interacting with C kinase 1 (PICK1), the glutamate receptor interacting protein 1 (GRIP1), and the more recently discovered transmembrane AMPA receptor regulatory accessory proteins such as stargazin, which aid in the trafficking of AMPA receptors and present additional targets for regulation.
mGlu and mACh Receptor Regulation of AMPA Receptors.
Muscarinic receptors (mainly M1) and mGlu receptors induce long-term depression (LTD) in the hippocampus and cerebellum mainly through changes in AMPA receptor trafficking (Dickinson et al., 2009; Nomura et al., 2012). Nomura et al. (2012) proposed that cerebellar LTD is mediated by mGlu1 activation and subsequent PKC activation leading to phosphorylation of GluA2, which causes it to dissociate from GRIP1, diffuse laterally to the endocytic zone, and undergo transmembrane AMPA receptor regulatory protein-mediated endocytosis. The mACh receptor-induced LTD in the hippocampus involves the same signaling mechanism (Dickinson et al., 2009). By contrast, mGlu1-mediated LTD on glutamatergic afferents to dopamine neurons in the ventral tegmental area proceeds by PICK1-dependent selective insertion of perisynaptic GluA2 subunits into synaptic receptors (Bellone and Lüscher, 2006), which should reduce channel conductance. Thus cell-specific differences in the signaling pathways and interacting proteins result in specificity of the GPCR-mediated AMPA receptor regulation.
AMPA Receptor Phosphorylation.
Accumulating evidence indicates that AMPA receptor activity is regulated by direct phosphorylation of AMPA receptor subunits by PKA and PKC. Although the C termini of AMPA receptor subunits varies in homology, they all contain consensus phosphorylation sites for serine/threonine kinases (Fig. 1A). GluA1, the most well characterized AMPA receptor subunit, is phosphorylated at serine 831 by both PKC and CaMKII whereas a nearby serine (Ser845) is phosphorylated by PKA. The phosphorylation of Ser845 by PKA increases the open probability of GluA1 channels (Banke et al., 2000) suggesting enhanced AMPA receptor function. However, the phosphorylation of Ser845 is also a critical step in LTD that involves delocalization of AMPA receptor from synaptic regions into perisynaptic sites (Lee et al., 2010), suggesting that trafficking overrides the effect on channel properties. This perisynaptic pool of GluA1 is then available for rapid reuptake into synaptic receptors during long-term potentiation (LTP). One mechanism for synaptic insertion involves phosphorylation of the GluA1 accessory protein stargazin by CAMKII, which facilitates induction of LTP in hippocampal slices by allowing AMPA receptors to bind PSD-95 (Tomita et al., 2005) and become sequestered into the synapse.
A large majority of AMPA receptors in the brain contains the GluA2 subunit, which can also be phosphorylated at multiple sites (Fig. 1B). PKA phosphorylates GluA2 at Ser880, which is in the PDZ-binding domain of the C terminus and regulates the interaction of GluA2 with membrane-scaffolding proteins such as GRIP1 (Matsuda et al., 1999; Chung et al., 2000). Interestingly, GluA4 is phosphorylated by PKA, PKC, and CaMKII at the same C-terminal regulatory site (i.e., Ser842); phosphorylation of this serine is necessary to remove the endoplasmic reticulum retention signal and drive trafficking of GluA4-containing receptors to the synapse (Carvalho et al., 1999). An important issue in GluA4 regulation is whether phosphorylation of this site by one kinase alters the sensitivity of the receptor for the binding of other kinases.
Kainate Receptors
Of the three ionotropic glutamate receptor subgroups the synaptic functions of kainate receptors (KARs) are the least understood. The GluK1-GluK3 subunits have low glutamate affinity and are capable of forming functional homomeric channels. GluK4 and GluK5 are high-affinity KAR subunits that bind glutamate but require coassembly with one or more GluK1-GluK3 subunits to form functional channels. The heteromultimeric assembly of KARs like many other ion channels leads to the formation of receptors with unique pharmacological and functional properties. KAR subunits are distributed throughout the nervous system, including the hippocampus, cortex, amygdala, striatum, hypothalamus, cerebellum, spinal cord, and basal ganglia, where they are involved in synaptic transmission and plasticity (Jin and Smith, 2011). KAR are regulated by several GPCR pathways previously thought to affect only NMDA and AMPA receptors.
mGlu Receptor Regulation of KARs.
One of the earliest demonstrations of regulation of kainite receptors by mGlu receptors was carried out by Cho et al. (2003), who showed that mGlu5 receptor activation potentiates (R,S)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid-activated kainate currents on neurons in the perirhinal cortex. In this study KAR potentiation occurred in the presence of cyclopiazonic acid, which depletes calcium stores and inhibits the mGlu5-dependent increase in internal calcium. The potentiation was blocked by pharmacological inhibitors of PKC; however, it remained unclear whether PKC activation was necessary and sufficient to potentiate KAR responses. It was also not clear which subunits of KAR were involved in the modulation. Rojas et al. (2013) recently demonstrated the regulation of heteromeric KARs by group I mGlu receptors in cultured cortical and hippocampal neurons and Xenopus oocytes. Pronounced potentiation of heteromeric KAR (consisting of high- and low-affinity subunits) currents was observed following activation of group I mGlu receptors (Fig. 2, A and B). On the other hand, homomeric KARs expressed in Xenopus oocytes failed to be potentiated following group I mGlu receptor activation. The potentiation pathway involved phospholipase C, Ca2+ mobilization, and PKC, as potentiation was blocked by inhibitors of all three signaling molecules. There was an interesting difference between the findings by Rojas et al. (2013) and Cho et al. (2003), namely the involvement of Ca2+ mobilization in the regulation of KARs by group I mGlu receptors. In the Cho et al. (2003) study the mGlu5-mediated potentiation of (R,S)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid-activated KAR currents was calcium-independent; however, in Rojas et al. (2013), the potentiation of heteromeric KARs by activation of group I mGlu receptors was clearly calcium-dependent, as the potentiation was lost in the presence of the calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid. This difference in the contribution of a critical signaling molecule suggests that there may be multiple pathways regulating KARs following mGlu receptor activation.
Fig. 2.
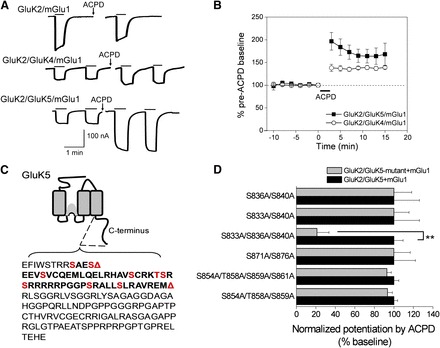
Potentiation of heteromeric KARs by group I mGlu activation. (A) Whole-cell currents were recorded from oocytes expressing GluK2/mGlu1, GluK2/GluK4/mGlu1, or GluK2/GluK5/mGlu1. Following application of 100 μM ACPD (a nonspecific mGlu activator), steady-state activation of the heteromeric receptors by AMPA (30 μM) was potentiated, but not steady-state activation of the homomeric KAR by domoic acid (10 μM). (B) Time-dependent potentiation of GluK2/GluK4/mGlu1 and GluK2/GluK5/mGlu1 AMPA currents by mGlu1 activation with ACPD (n = 5 and n = 16, respectively). (C) Schematic showing the GluK5 C-terminal domain. The first red triangle indicates the location of an inserted stop codon (Δ837) and the second red triangle indicates the position of a more distal stop codon (Δ884). The red residues indicate consensus PKC phosphorylation sites. (D) Alanine mutagenesis of the C-terminal serines and threonines between 833 and 883 revealed that only the triple GluK5 mutant (GluK5-S833A/S836A/S840A) resulted in a significantly reduced mGlu1-mediated potentiation (n ≥ 4; each mutant combination was evaluated against a separate set of oocytes expressing wild-type receptors; **P < 0.001; t test). From Rojas et al. (2012).
mACh Receptor Regulation of KARs.
Benveniste et al. (2010) demonstrated that activation of GluK2 containing heteromeric kainate receptors is potentiated following mACh receptor activation. Similar to mGlu receptor modulation of KARs, muscarinic receptor (M1 and M3) activation by pilocarpine potentiated GluK2 containing heteromeric, but not homomeric, KARs, suggesting that potentiation of KARs by mACh receptors also requires the high-affinity KAR subunits. The regulation was also observed in mossy fibers using the hippocampal slice preparation, where mACh-mediated potentiation seen in native neurons displayed characteristics similar to those seen with recombinant receptors, such as the degree of potentiation, the time-dependence, and the specificity for mACh receptor agonists. The mechanism responsible for mACh receptor potentiation was not investigated.
KAR Phosphorylation.
A sequence scan of the C terminus of KAR subunits reveals a number of consensus phosphorylation sites for the protein kinases PKC, PKA, CaMKII, and Src. PKC phosphorylates the C terminus of both GluK1 and GluK2 in vitro (Hirbec et al., 2003; Nasu-Nishimura et al., 2010; Konopacki et al., 2011; Chamberlain et al., 2012). The phosphorylation of GluK2 by PKC at Ser868 is a critical step in the process of internalization of GluK2 that occurs during KAR-mediated LTP in hippocampal mossy fibers (Chamberlain et al., 2012). On the other hand phosphorylation of C-terminal residues (i.e., Ser880 and Ser886) in GluK1 appears to promote the stability of this subunit in the synapse via an interaction with the PDZ domain containing protein GRIP1 (Hirbec et al., 2003). The C terminus of GluK2 is also phosphorylated by PKA at Ser825 and Ser837 (Kornreich et al., 2007) leading to potentiation of receptor currents, likely via an increase in the receptor open-probability. Site-directed mutagenesis led to the identification of three serines in the GluK5 subunit that are responsible for PKC-mediated potentiation of KAR by group I mGlu receptors (Fig. 2, C and D; Rojas et al., 2013). Selak et al. (2009) reported reduced GluK5 expression via PKC-mediated disruption of the interaction of GluK5 with the synaptic proteins SNAP-25 and PICK1. The authors suggested that PKC interacts with PICK1 resulting in the disruption of the SNAP-25-PICK1-GluK5 complex. These examples show that, as for NMDA receptors and AMPA receptors, KAR functions can be both potentiated and suppressed by PKC.
Conclusion and Outlook
The mechanisms responsible for regulation of iGlu receptors by GPCRs involve many intracellular signaling molecules and regulatory proteins that vary from one cell type to another. The downstream signaling molecules include protein kinases that phosphorylate regulatory sites located within the C terminus of these ionotropic glutamate receptors affecting their interaction with regulatory or interacting proteins that alter iGlu receptor trafficking (Fig. 1A). In some cases synaptic regulation of ionotropic glutamate receptors by the activation of diverse GPCRs proceeds by partially convergent mechanisms shared by the various iGlu receptors, exemplified by the convergence of muscarinic and metabotropic glutamate receptors onto PKC pathways that regulate all three iGlu receptors, albeit via different mechanisms downstream of PKC. Knowing that ionotropic glutamate receptors are subject to many of the same modulatory influences has implications for understanding the physiologic roles of these receptors in concert. One important issue is to understand whether cross-talk is restricted to cellular compartments that coexpress ionotropic glutamate receptors and GPCRs. Second, knowledge of the molecular basis of kinase specificity of cross-talk mediated by multiple GPCRs and whether there is a dominance hierarchy of kinases and ion channel modulation will result in better understanding of how synaptic strength is regulated. It will also be important to identify the conditions under which changes in channel conductance play a role in synaptic strength modification by GPCRs, given the dominant role of receptor trafficking in this process. Finally, the therapeutic implications of cross-talk between NMDAR and dopamine D1 receptors (e.g., for schizophrenia), mu opioid receptors (for chronic pain) and mGlu5a (for cognitive disorders) are considerable.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CaMKII
calcium/calmodulin-dependent protein kinase
- GluA
AMPA receptor subunit
- GluK
kainate receptor subunit
- GluN
NMDA receptor subunit
- GPCR
G-protein-coupled receptor
- GRIP1
glutamate receptor interacting protein 1
- iGlu
ionotropic glutamate
- KARs
kainate receptors
- LTD
long-term depression
- LTP
long-term potentiation
- mACh
muscarinic acetylcholine
- mGlu
metabotropic glutamate receptor
- MOR
mu opioid receptors
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- PACAP
pituitary adenylate cyclase-activating peptide
- PDZ
postsynaptic density 95/disc-large/ZO-1
- PICK1
protein interacting with C kinase 1
- PKA
protein kinase A
- PKC
protein kinase C
- PSD
postsynaptic density
- Pyk2/PYK2
proline-rich tyrosine kinase 2
- RACK1
receptor for activated C kinase 1
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Rojas, Dingledine.
Footnotes
References
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. (2000) Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard L, Groc L. (2011) Glutamate receptor dynamics and protein interaction: lessons from the NMDA receptor. Mol Cell Neurosci 48:298–307. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641. [DOI] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. (2006) Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature 444:106–109. [DOI] [PubMed] [Google Scholar]
- Benquet P, Gee CE, Gerber U. (2002) Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J Neurosci 22:9679–9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste M, Wilhelm J, Dingledine RJ, Mott DD. (2010) Subunit-dependent modulation of kainate receptors by muscarinic acetylcholine receptors. Brain Res 1352:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi F and Ugolini A (1999) Group I metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol 59:55–79. [DOI] [PubMed] [Google Scholar]
- Carvalho AL, Kameyama K, Huganir RL. (1999) Characterization of phosphorylation sites on the glutamate receptor 4 subunit of the AMPA receptors. J Neurosci 19:4748–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SE, González-González IM, Wilkinson KA, Konopacki FA, Kantamneni S, Henley JM, Mellor JR. (2012) SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat Neurosci 15:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53:362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BS, Roche KW. (2009) Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 62:471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Francis JC, Hirbec H, Dev K, Brown MW, Henley JM, Bashir ZI. (2003) Regulation of kainate receptors by protein kinase C and metabotropic glutamate receptors. J Physiol 548:723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe ES, Shin EH, Wang JQ. (2006) Regulation of phosphorylation of NMDA receptor NR1 subunits in the rat neostriatum by group I metabotropic glutamate receptors in vivo. Neurosci Lett 394:246–251. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. (2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci 20:7258–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237. [DOI] [PubMed] [Google Scholar]
- Dickinson BA, Jo J, Seok H, Son GH, Whitcomb DJ, Davies CH, Sheng M, Collingridge GL, Cho K. (2009) A novel mechanism of hippocampal LTD involving muscarinic receptor-triggered interactions between AMPARs, GRIP and liprin-alpha. Mol Brain 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Cattabeni F, Di Luca M. (2001) Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-D-aspartate receptor complex. J Biol Chem 276:7609–7613. [DOI] [PubMed] [Google Scholar]
- Grishin AA, Gee CE, Gerber U, Benquet P. (2004) Differential calcium-dependent modulation of NMDA currents in CA1 and CA3 hippocampal pyramidal cells. J Neurosci 24:350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AA, Benquet P, Gerber U. (2005) Muscarinic receptor stimulation reduces NMDA responses in CA3 hippocampal pyramidal cells via Ca2+-dependent activation of tyrosine phosphatase. Neuropharmacology 49:328–337. [DOI] [PubMed] [Google Scholar]
- Gu Z, Liu W, Wei J, Yan Z. (2012) Regulation of N-methyl-D-aspartic acid (NMDA) receptors by metabotropic glutamate receptor 7. J Biol Chem 287:10265–10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. (2006) Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci 26:4690–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, Behrens MM. (2002) Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci 22:5452–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Coutinho V, Meyer G, Isaac JT, et al. (2003) Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron 37:625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, et al. (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29:485–496. [DOI] [PubMed] [Google Scholar]
- Hull LC, Llorente J, Gabra BH, Smith FL, Kelly E, Bailey C, Henderson G, Dewey WL. (2010) The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by mu-opioid agonists of different efficacy. J Pharmacol Exp Ther 332:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XT, Smith Y. (2011) Localization and functions of kainate receptors in the basal ganglia. Adv Exp Med Biol 717:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopacki FA, Jaafari N, Rocca DL, Wilkinson KA, Chamberlain S, Rubin P, Kantamneni S, Mellor JR, Henley JM. (2011) Agonist-induced PKC phosphorylation regulates GluK2 SUMOylation and kainate receptor endocytosis. Proc Natl Acad Sci USA 108:19772–19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornreich BG, Niu L, Roberson MS, Oswald RE. (2007) Identification of C-terminal domain residues involved in protein kinase A-mediated potentiation of kainate receptor subtype 6. Neuroscience 146:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Zheng X, Bennett MV, Zukin RS. (2001) Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci 21:6058–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chéry N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. (2002) Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 111:219–230. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. (1999) G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci 2:331–338. [DOI] [PubMed] [Google Scholar]
- Macdonald DS, Weerapura M, Beazely MA, Martin L, Czerwinski W, Roder JC, Orser BA, MacDonald JF. (2005) Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires G alpha q, protein kinase C, and activation of Src. J Neurosci 25:11374–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. (2007) G protein-coupled receptors control NMDARs and metaplasticity in the hippocampus. Biochim Biophys Acta 1768:941–951. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. (1998) Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci USA 95:11465–11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H. (1999) Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem 73:1765–1768. [DOI] [PubMed] [Google Scholar]
- Nasu-Nishimura Y, Jaffe H, Isaac JT, Roche KW. (2010) Differential regulation of kainate receptor trafficking by phosphorylation of distinct sites on GluR6. J Biol Chem 285:2847–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Kakegawa W, Matsuda S, Kohda K, Nishiyama J, Takahashi T, Yuzaki M. (2012) Cerebellar long-term depression requires dephosphorylation of TARP in Purkinje cells. Eur J Neurosci 35:402–410. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996. [DOI] [PubMed] [Google Scholar]
- Perroy J, Raynaud F, Homburger V, Rousset MC, Telley L, Bockaert J, Fagni L. (2008) Direct interaction enables cross-talk between ionotropic and group I metabotropic glutamate receptors. J Biol Chem 283:6799–6805. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Berrocoso E, Garzón J. (2012) The mu-opioid receptor and the NMDA receptor associate in PAG neurons: implications in pain control. Neuropsychopharmacology 37:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Wetherington J, Shaw R, Serrano G, Swanger S, and Dingledine R (2013) Activation of group I metabotropic glutamate receptors potentiates heteromeric kainate receptors. Mol Pharmacol 83:106–121. [DOI] [PMC free article] [PubMed]
- Selak S, Paternain AV, Aller MI, Picó E, Rivera R, Lerma J. (2009) A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron 63:357–371. [DOI] [PubMed] [Google Scholar]
- Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. (2005) Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron 45:269–277. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, He DY, Phamluong K, Ron D. (2003) Pituitary adenylate cyclase-activating polypeptide (PACAP(1-38)) enhances N-methyl-D-aspartate receptor function and brain-derived neurotrophic factor expression via RACK1. J Biol Chem 278:9630–9638. [DOI] [PubMed] [Google Scholar]
- Yang K, Trepanier C, Sidhu B, Xie YF, Li H, Lei G, Salter MW, Orser BA, Nakazawa T, Yamamoto T, et al. (2012) Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J 31:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Jiang Q, Chen P, Gu Z, Feng J, Yan Z. (2005) Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J Neurosci 25:5488–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ. (1998) Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1:185–191. [DOI] [PubMed] [Google Scholar]
- Zheng H, Worrall C, Shen H, Issad T, Seregard S, Girnita A, Girnita L. (2012) Selective recruitment of G protein-coupled receptor kinases (GRKs) controls signaling of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA 109:7055–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]