

Abstract
Adiponectin, an adipokine predominantly secreted from adipocytes, has been shown to play protective roles against chronic alcohol consumption. Although excessive reactive oxygen species (ROS) production in macrophages is considered one of the critical events for ethanol-induced damage in various target tissues, the effect of adiponectin on ethanol-induced ROS production is not clearly understood. In the present study, we investigated the effect of globular adiponectin (gAcrp) on ethanol-induced ROS production and the potential mechanisms underlying these effects of gAcrp in macrophages. Here we demonstrated that gAcrp prevented ethanol-induced ROS production in both RAW 264.7 macrophages and primary murine peritoneal macrophages. Globular adiponectin also inhibited ethanol-induced activation of NADPH oxidase. In addition, gAcrp suppressed ethanol-induced increase in the expression of NADPH oxidase subunits, including Nox2 and p22phox, via modulation of nuclear factor-κB pathway. Furthermore, pretreatment with compound C, a selective inhibitor of AMPK, or knockdown of AMPK by small interfering RNA restored suppression of ethanol-induced ROS production and Nox2 expression by gAcrp. Finally, we found that gAcrp treatment induced phosphorylation of liver kinase B1 (LKB1), an upstream signaling molecule mediating AMPK activation. Knockdown of LKB1 restored gAcrp-suppressed Nox2 expression, suggesting that LKB1/AMPK pathway plays a critical role in the suppression of ethanol-induced ROS production and activation of NADPH oxidase by gAcrp. Taken together, these results demonstrate that globular adiponectin prevents ethanol-induced ROS production, at least in part, via modulation of NADPH oxidase in macrophages. Further, LKB1/AMPK axis plays an important role in the suppression of ethanol-induced NADPH oxidase activation by gAcrp in macrophages.
Introduction
Reactive oxygen species (ROS), highly reactive oxygen-containing molecules, can react with various cellular molecules. Excessive ROS production results in oxidative stress and causes damage in many organs. Ethanol consumption promotes ROS generation, which causes alterations of various signaling pathways and contributes to the development of damages in the target organs. Therefore, ROS production has long been considered a critical event in the pathogenesis caused by ethanol exposure.
Ethanol-induced ROS generation is mediated via a variety of cellular systems. For example, CYP2E1 plays a critical role in ROS production and oxidative stress in hepatocytes (Gao and Bataller, 2011), whereas NADPH oxidase, composed of multicomponent enzyme complex, including membrane-bound proteins (Nox2 and p22phox) and regulatory cytosolic components (p40phox, p47phox, p67phox, and Rac1/2), is an important source for ROS production in macrophages (Lam et al., 2010). Activation of macrophages and subsequent excessive inflammatory response is one of the key events leading to the pathogenic impact of ethanol on various target organs, including alcoholic liver disease (Gao and Bataller, 2011) and impaired lung function (D’Souza El-Guindy et al., 2007; Brown et al., 2009). Previous studies have shown that p47phox knockout mice exhibit reduced ROS production, lower production of inflammatory cytokines from Kupffer cells, the resident macrophages in liver, and are protected from chronic ethanol-induced liver injury (Kono et al., 2000). In addition, recent studies have reported that NADPH oxidase is a main source of ethanol-induced ROS production in alveolar macrophages, which is closely associated with dysfunction of alveolar macrophages (Yeligar et al., 2012) and is a critical mediator for lipopolysaccharide (LPS)-stimulated ROS production in Kupffer cells after chronic ethanol feeding (Thakur et al., 2006b). We have also shown that ethanol treatment increases ROS production in murine peritoneal macrophages via activation of NADPH oxidase (Kim et al., 2013). Collectively, these results indicate that NADPH oxidase plays a critical role in the activation of macrophages and development of various pathologic conditions by ethanol consumption.
Adiponectin predominantly secreted from adipocytes plays a role in the control of various physiologic and pathophysiologic processes. For example, adiponectin plays a crucial role in glucose and lipid metabolism (Mao et al., 2006) and insulin-sensitizing actions (Kadowaki et al., 2006) and also possesses anti-inflammatory properties (Ouchi and Walsh, 2007). Adiponectin has been also shown to prevent pathologic conditions from chronic ethanol administration, particularly in the liver (Rogers et al., 2008) and stomach (Yamamoto et al., 2012). Growing evidence suggests that adiponectin generates various biologic responses through modulation of oxidative stress (Ouedraogo et al., 2006; Tao et al., 2007). Although emerging evidence suggests that adiponectin negatively regulates oxidative stress, the effect of adiponectin on ethanol-induced ROS production and its potential mechanisms have not been explored.
AMP-dependent protein kinase (AMPK), acting as a molecular sensor in the cellular energy homeostasis, has been considered a key molecule mediating the various beneficial effects induced by adiponectin (Lim et al., 2010). In addition to the critical role in the metabolism of glucose and lipids, recent studies have highlighted that AMPK activation alleviates ROS production in macrophages and endothelial cells (Ouedraogo et al., 2006; Jeong et al., 2009), which, in turn, leads to the negative regulation of nuclear factor-κB (NF-κB) signaling and inflammatory responses (Yang et al., 2010; Salminen et al., 2011). Although it is well recognized that AMPK signaling plays a critical role in the modulation of ROS production and inflammatory response, its role in macrophages treated with ethanol and/or adiponectin has not been reported.
Liver kinase B1 (LKB1, also known as STK11), originally identified as a tumor suppressor gene, exhibits an important role in energy metabolism, mainly via association with AMPK signaling. For example, LKB1 deficiency leads to inactivation of AMPK, and LKB1-induced tumor suppression and modulation of cellular energy homeostasis are mediated through activation of AMPK (Shaw et al., 2004; Alessi et al., 2006). Furthermore, LKB1/AMPK axis is used for adiponectin-induced various biologic responses (Miller et al., 2011; Vu et al., 2013). However, the effect of adiponectin on LKB1 signaling and its role in the modulation of ROS production in macrophages have not been reported yet.
Adiponectin plays a protective role in various organs from chronic ethanol treatment. However, the underlying molecular mechanisms are not clearly understood. Herein, we investigated the effect of globular adiponectin on ethanol-induced ROS production and the potential mechanisms of gAcrp action in macrophages. In the present study, we demonstrated for the first time that adiponectin prevents ethanol-induced ROS generation through modulation of NADPH oxidase and, furthermore, that LKB1/AMPK signaling plays a pivotal role in the suppression of ROS production and NADPH oxidase activation.
Materials and Methods
All the cell culture reagents were obtained from Hyclone Laboratories (South Logan, UT). Recombinant human globular adiponectin (gAcrp) was purchased from Peprotech Inc. (Rocky Hill, NJ). Absolute ethanol was purchased from Merck Chemical (Whitehouse Station, NJ). 5-Chloromethyl-2, 7-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was purchased from Molecular Probes (Eugene, OR). Lucigenin, NADPH, and diphenyleneiodonium chloride were obtained from Enzo Life Sciences (Farmingdale, NY). Bay11-7082, a pharmacologic inhibitor of NF-κB, was obtained from Sigma-Aldrich (St. Louis, MO), and 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside (AICAR), an activator of AMPK, was obtained from Calbiochem (San Diego, CA). Luciferase assay kit and all related products were purchased from Promega (Madison, WI). Polyclonal antibodies against phosphorylated and total forms of AMPKα and LKB1 were obtained from Cell Signaling Technology Inc. (Beverly, MA). Antibody against Nox2 was purchased from Santa Cruz (Delaware, CA).
Isolation and Culture of Murine Peritoneal Macrophages.
Murine peritoneal macrophages were isolated essentially as described previously (Kim et al., 2013). Briefly, 8- to 10-week-old mice were i.p. injected with 1 ml of 4% Brewer thioglycollate medium. After 3 days of injection, peritoneal cells were washed with 10 ml of ice-cold Hanks’ balanced salt solution (HBSS) (calcium and magnesium free). Media prepared from peritoneum were centrifuged at 12,000 rpm for 5 minutes. The pellet was resuspended in RBC lysis buffer (BioLegend, San Diego, CA) and then centrifuged at 12,000 rpm for 5 minutes. After washing with 20 ml of HBSS, cells were seeded on four-well culture slides (5 × 105 cells/well) or 96-well plates (1 × 105 cells/well). These cells were allowed to adhere for 2 hours and then were washed out with 1× phosphate-buffered saline to remove nonadherent cells. After 24 hours of culture, adherent cells were used in the experiments.
Cell Culture.
The RAW 264.7 macrophage cell line was purchased from the Korean cell line bank (Seoul, Korea) and routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin at 37°C in an incubator with a humidified atmosphere of 5% CO2.
Total RNA Isolation, Reverse Transcription, and Quantitative Polymerase Chain Reaction.
To measure the mRNA levels of the target genes, total RNAs were extracted using Qiagen lysis solution (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and reverse-transcribed into cDNA using the GoScript reverse transcription system (Promega). Quantitative real-time polymerase chain reaction (PCR) was then performed with LightCycler 1.5 (Roche Diagnostics, Mannheim, Germany) using QPCR SYBR Green Capillary Mix (ABgene, Surrey, UK) at 95°C for 15 minutes, followed by 40 cycles of 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. The primer sequences used for amplification of the target genes are listed in Table 1. The amount of target mRNA was assessed via the comparative threshold (Ct) method after normalizing target mRNA Ct values to those for glyceraldehyde-3-phosphate dehydrogenase (GAPDH, ΔCt) as a housekeeping gene.
TABLE 1.
Sequences of the primers used for quantitative RT-PCR
| Target Gene | Primer | Sequence |
|---|---|---|
| Nox2 | Forward | 5′- TTGGGTCAGCACTGGCTCTG -3′ |
| Reverse | 5′- TGGCGGTGTGCAGTGCTATC -3′ | |
| p22phox | Forward | 5′- GTCCACCATGGAGCGATGTG -3′ |
| Reverse | 5′- CAATGGCCAAGCAGACGGTC -3′ | |
| GAPDH | Forward | 5′-ACCACAGTCCATGCCATCAC-3′ |
| Reverse | 5′-TCCACCACCCTGTTGCTGTA-3′ |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Measurement of Total ROS Generation.
Intracellular ROS production was assessed by measurement of the changes in the fluorescence of CM-H2DCFDA. In brief, RAW 264.7 macrophages were seeded at a density of 2 × 105 cells/well in four-well culture slides. Cells were pretreated with 0.1 μg/ml of globular adiponectin (gAcrp) for 18 hours, followed by treatment with 100 mM ethanol for an additional 24 hours. Cells were treated with 10 μM CM-H2DCFDA in HBSS in the dark for 30 minutes and then washed with HBSS to remove excess dye. For measurement of ROS production, the change in fluorescence intensity was immediately observed by fluorescence microscopy (Nikon, Tokyo, Japan).The fluorescence intensities were analyzed by quantitative digital analysis using Image Inside from FOCUS (Seoul, South Korea).
Determination of NADPH Oxidase Activity.
NADPH oxidase–dependent ROS formation was determined by measuring lucigenin-derived chemiluminescence using FLUOstar OPTIMA luminometer (BMG Labtech, Ortenberg, Germany). Briefly, peritoneal macrophages and RAW 264.7 macrophages were seeded onto 96-well white culture plates at a density of 0.5–1 × 105 cells/well. After overnight culture, cells were treated with gAcrp and ethanol as indicated in figure legends and then lysed with HBSS containing 0.1% Triton X-100 and 0.1 N NaOH. Cellular lysates were incubated with NADPH (200 µM) and lucigenin (100 µM) in HBSS for 30 minutes at 37°C in the dark. Chemiluminescence was measured in relative light units every 5 minutes over a period of 60 minutes.
Transient Transfection and Luciferase Assay.
Transcriptional activity of NF-κB was determined using luciferase reporter assay kit (Promega) according to the manufacturer’s instructions. Briefly, cells were initially seeded in 24-well plates at a density of 5 × 105 cells/well. After overnight culture, the cells were cotransfected with control (pRL-TK) and expression vectors (pGL4/NF-κB) using Fugene HD (Promega). After 6 hours’ incubation, media were replaced with DMEM containing 10% FBS and cells were cultured for additional 18 hours. Thereafter, the cells were pretreated with 0.1 μg/ml gAcrp followed by 100 mM ethanol for additional 24 hours and extracted with 0.1 ml of passive lysis buffer (Promega). Firefly and Renilla luciferase activities were measured by the Dual Luciferase Reporter Assay System (Promega). Statistical analyses for luciferase expression were carried out on the ratios of relative luciferase activity to Renilla luciferase.
Transient Transfection with Small Interfering RNAs.
RAW 264.7 macrophages were initially seeded on 35-mm dishes at a density of 7 × 105 cells/well. After overnight incubation, cells were transfected with scrambled control small interfering (siRNA) or siRNA targeting AMPKα1 (25 nM) or LKB1 (50 nM) using Hiperfect transfection reagent (Qiagen) according to the manufacturer’s instructions. Gene-silencing efficiency was assessed by qRT-PCR after 24 or 48 hours of transfection. The ON-TARGET plus SMART of murine-specific siRNA targeting AMPKα1 and control siRNA were purchased from Dharmacon (Lafayette, CO). Targeting siRNA of LKB1 was obtained from Bioneer (Daejeon, South Korea). The sequences of the siRNA used are listed in Table 2.
TABLE 2.
Sequences of small interfering RNA used in transfection
| Target Gene | Primer | Nucleotide Sequence |
|---|---|---|
| LKB1 | Forward | 5′- CCACCGAGGUAAUCUACCA -3′ |
| Reverse | 5′- UGGUAGAUUACCUCGGUGG -3′ | |
| AMPKα1 | Forward | 5′- GCAGAAGUUUGUAGAGCAA -3′ |
| 5′- UCUUAUAGUUCAACCAUGA -3′ | ||
| 5′- ACCAGGAAGUCAUACAAUA -3′ | ||
| 5′- CGAGUUGACCGGACAUAAA -3′ | ||
| Scrambled | Forward | 5′-UGGUUUACAUGUCGACUAA-3′ |
| 5′-UGGUUUACAUGUUGUGUGA-3′ | ||
| 5′-UGGUUUACAUGUUUUCUGA-3′ | ||
| 5′-UGGUUUACAUGUUGUCUUA-3′ |
AMPK, AMP-activated protein kinase; LKB1, liver kinase B1.
Preparation of Cellular Extracts and Western Blot Analysis.
RAW 264.7 macrophages were seeded in 35-mm dishes at a density of 1 × 106 cells per well. After overnight incubation, cells were treated with gAcrp and/or ethanol as indicated in figure legends. Total proteins were then isolated using RIPA lysis buffer containing Halt protease and phosphatase inhibitor single-use cocktail (Thermo Scientific, Waltham, MA). For immunoblot analysis, 15 μg of protein was loaded by 10% SDS-PAGE. The proteins were then transferred to polyvinylidene difluoride membranes, blocked with 5% skim milk in phosphate-buffered saline/Tween20 for 1 hour, and incubated with the designated primary antibodies overnight at 4°C. Subsequently, the membrane was washed and incubated with secondary horseradish peroxidase–labeled antibody. The images of the blots were captured using Fujifilm LAS-4000 mini (Fujifilm, Tokyo, Japan). The membranes were then stripped and reprobed with β-actin or total form of AMPKα or LKB1 antibody as the loading control.
Statistical Analysis.
Values were presented as mean ± S.E.M. derived from at least three separate experiments. Data were assessed by one-way analysis of variance and Tukey’s multiple comparison tests using GraphPad Prism software version 5.01 (La Jolla, CA). Differences between groups were considered to be significant at P < 0.05.
Results
Globular Adiponectin Inhibits Ethanol-Induced ROS Production via Modulation of NADPH Oxidase in RAW 264.7 Macrophages and Peritoneal Macrophages Isolated from Mice.
Excessive ROS production in macrophages is a critical event for the development of pathologic conditions by chronic ethanol treatment. We investigated whether adiponectin inhibits ethanol-induced ROS production in macrophages. As shown in Fig. 1A, ethanol treatment enhanced ROS production consistent with previous results, but pretreatment with gAcrp significantly suppressed ethanol-induced ROS production in murine macrophages (RAW 264.7 macrophages). Similar effects were also observed in the primary peritoneal macrophages isolated from mice (Fig. 1B), indicating that adiponectin prevents ethanol-induced ROS production in different types of macrophages. Our results and other previous reports have shown that NADPH oxidase system is the major source for ethanol-induced ROS production in macrophages (Yeligar et al., 2012; Kim et al., 2013).
Fig. 1.
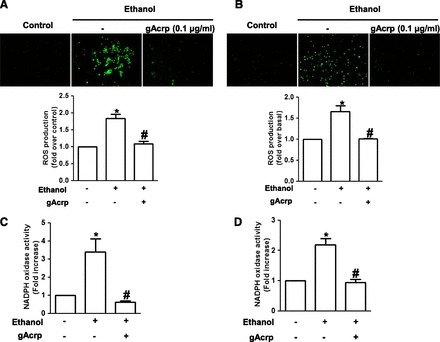
Effects of globular adiponectin on ethanol-induced ROS production and NADPH oxidase activation in macrophages. (A) RAW 264.7 macrophages cultured in four-well culture slides were pretreated with gAcrp (0.1 μg/ml) for 18 hours, followed by stimulation with 100 mM ethanol for an additional 24 hours. Cells were then further stimulated with HBSS containing 10 μM of CM-H2DCF-DA; ROS production was assessed by conversion of CM-H2DCF-DA to the oxidized fluorescent DCF, which was monitored by fluorescence microscopy as described in Materials and Methods. Representative images from three independent experiments are shown along with quantification with green signal intensity (lower panel). Values are expressed as mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (B) Peritoneal macrophages isolated from mice were incubated with 0.1 μg/ml of gAcrp for 18 hours, followed by treatment with 100 mM ethanol for an additional 24 hours. ROS production was assessed as described previously, and representative images from three independent experiments are shown along with the quantitative analysis of green fluorescent intensity (lower panel). Values shown are presented as mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (C) RAW 264.7 macrophages were treated with 0.1 μg/ml of gAcrp for 18 hours, followed by incubation with 100 mM ethanol for 24 hours. NADPH oxidase activity was determined by lucigenin-based assay as described in Materials and Methods. Briefly, cell lysates were incubated with HBSS containing 100 μM lucigenin and 200 μM NADPH for 30 minutes. NADPH oxidase activity was then assessed by chemiluminescence and expressed in relative light units. Values represent fold increase in comparison with cells not treated with ethanol and are expressed as mean ± S.E.M. (n = 6). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (D) Primary murine peritoneal macrophages were pretreated with gAcrp (0.1 μg/ml) for 18 hours followed by incubation with ethanol (100 mM) for an additional 24 hours. NADPH oxidase activity was measured as described previously. Values are presented as fold increase compared with the control cells and are expressed as mean ± S.E.M. (n = 4). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol.
To elucidate the potential mechanisms implicated, we next examined the effect of gAcrp on ethanol-induced activation of NADPH oxidase using lucigenin-based assay. As shown in Fig. 1, C and D, ethanol-induced increase in ROS production by NADPH oxidase was restored by pretreatment with gAcrp in both RAW 264.7 macrophages (Fig. 1C) and primary murine peritoneal macrophages (Fig. 1D). All these data indicate that gAcrp inhibits ethanol-induced ROS production in macrophages via modulation of NADPH oxidase–dependent ROS production.
Globular Adiponectin Suppresses Ethanol-Induced Nox2 and p22phox Expression in RAW 264.7 Macrophages.
NADPH oxidase is composed of multiple components localized in cytosol and plasma membrane. To identify the subunits responsible for regulation of NADPH oxidase by gAcrp, we examined the effect of gAcrp on the expression of Nox2 and p22phox since Nox2 and p22phox are critical subunits for ethanol-induced NADPH oxidase activation and subsequent increase in ROS production (Kim et al., 2013). As shown in Fig. 2A, gAcrp treatment decreased the expression of Nox2 mRNA level. Nox2 protein expression was also significantly decreased by treatment with gAcrp, which showed a pattern similar to the regulation of mRNA expression (Fig. 2B). Globular adiponectin also significantly decreased p22phox mRNA expression in a time-dependent manner (Fig. 2C). We also examined the effect of adiponectin on the expression of other NADPH oxidase subunits, such as p40phox, p47phox, and p67phox; but treatment with gAcrp did not significantly affect the expression of other subunits of NADPH oxidase (data not shown), suggesting that gAcrp affects expression of NADPH oxidase subunits in a selective manner. In addition, ethanol-induced increase in Nox2 expression was restored by pretreatment with gAcrp at both mRNA (Fig. 2D) and protein (Fig. 2E) level, as expected. Pretreatment with gAcrp also significantly suppressed ethanol-induced increase in p22phox expression (Fig. 2F), similar to the pattern for the modulation of Nox2 expression. Taken together, these data suggest that gAcrp modulates ethanol-induced activation of NADPH oxidase probably via down-regulation of Nox2 and p22phox expression.
Fig. 2.
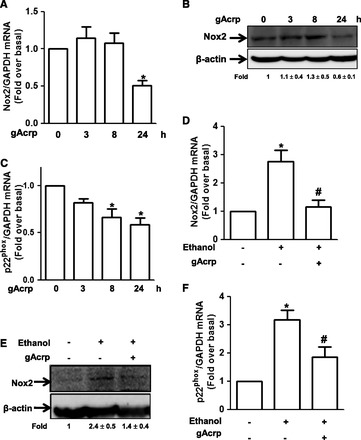
Effects of globular adiponectin on ethanol-induced Nox2 and p22phox expression in RAW 264.7 macrophages. (A) Cells were incubated with gAcrp (0.1 μg/ml) for the indicated time periods. Nox2 mRNA expression was analyzed by qRT-PCR as described in Materials and Methods. Values are represented as fold increase relative to controls and are expressed as mean ± S.E.M. (n = 4). *P < 0.05 compared with cells not treated with gAcrp. (B) Cells were treated with gAcrp (0.1 μg/ml) for the indicated time periods. Nox2 protein expression levels were analyzed by Western blot analysis as described in Materials and Methods. Representative image from four independent experiments is shown along with β-actin for internal loading control. Band intensities were quantified by densitometric analysis and normalized by β-actin. Values are represented as fold changes relative to controls and are expressed as mean ± S.E.M. (C) Cells were treated with gAcrp (0.1 μg/ml) for the indicated time periods. p22phox mRNA expression was analyzed by qRT-PCR as described previously. Values shown are represented as mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol. (D) Cells were pretreated with gAcrp (0.1 μg/ml) for 18 hours, followed by 100 mM of ethanol for an additional 24 hours. Nox2 mRNA expression level was assessed by qRT-PCR as described previously. Values are presented as mean ± S.E.M. (n = 4). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (E) Cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours, followed by ethanol for additional 24 hours. Nox2 protein was measured by Western blot analysis as described previously. Images are representative of three separate experiments that showed similar results. Band intensities were quantified by densitometric analysis and normalized by β-actin. Values are represented as fold changes relative to control and are expressed as mean ± S.E.M. (F) Cells were treated with gAcrp (0.1 μg/ml) for 18 hours, followed by incubation with 100 mM ethanol (100 mM) for 24 hours. p22phox mRNA expression was measured as described previously. Values are presented as mean ± S.E.M. (n = 4). *P < 0.05 compared with controls; #P < 0.05 compared with cells treated with ethanol.
Globular Adiponectin Suppresses Ethanol-Induced Nox2 and p22phox Expression through Modulation of NF-κB Signaling Pathway in RAW 264.7 Macrophages.
To elucidate further the signaling mechanisms underlying suppression of ethanol-induced Nox2 and p22phox by gAcrp, we examined whether gAcrp affects ethanol-induced activation of p38 mitogen-activated protein kinase (MAPK) and NF-κB pathway since p38 MAPK/NF-κB signaling pathway plays a critical role in ethanol-induced Nox2 and p22phox expression and further ROS production in macrophages (Kim et al., 2013). For this, we first confirmed involvement of p38 MAPK and NF-κB signaling in ethanol-induced Nox2 and p22phox expression in our experimental condition. As indicated in Fig. 3, A and B, pretreatment with Bay 11-7082, a pharmacologic inhibitor of IKK, significantly inhibited ethanol-induced mRNA expression of Nox2 and p22phox. In addition, pretreatment with p38 MAPK inhibitor (SB203580) significantly suppressed ethanol-induced Nox2 and p22phox expression (Fig. 3, C and D), consistent with previous observations, confirming the critical role of p38 MAPK/NF-κB signaling in ethanol-induced Nox2 and p22phox expression in macrophages. We next examined the effect of gAcrp on ethanol-induced activation of NF-κB signaling. As indicated in Fig. 3E, pretreatment with gAcrp significantly suppressed ethanol-induced NF-κB activation assessed by luciferase reporter gene assay. All these results imply that gAcrp suppresses ethanol-induced Nox2 and p22phox expression via modulation of NF-κB signaling pathway.
Fig. 3.
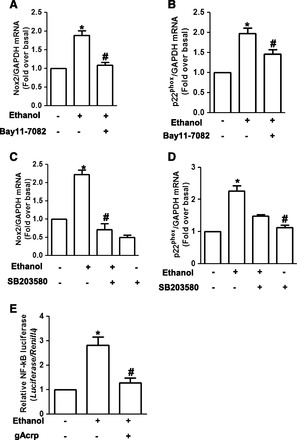
Involvement of p38 MAPK/NF-κB pathway in the suppression of ethanol-induced Nox2 and p22phox expression by gAcrp in RAW 264.7 macrophages. (A, B) Cells were pretreated with Bay 11-7082 (5 μM), a selective inhibitor of IKK, for 1 hour, followed by stimulation with 100 mM ethanol for 24 hours. Messenger RNA levels of Nox2 (A) and p22phox (B) were assessed by qRT-PCR as described previously. Values are expressed as mean ± S.E.M. (n = 8). *P < 0.05 compared with control; #P < 0.05 compared with cells treated with ethanol. (C, D) Cells were pretreated with SB203580 (10 μM), a selective inhibitor of p38 MAPK, for 1 hour followed by treatment with 100 mM ethanol for additional 24 hours. Nox2 (C) and p22phox (D) mRNA levels were measured by qRT-PCR as described previously. Values represent fold change relative to control cells and are expressed as mean ± S.E.M. (n = 4). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (E) Cells were transiently cotransfected with pNFκB-luc and Renilla reporter gene as described in Materials and Methods. After 24 hours of transfection, cells were then pretreated with gAcrp (0.1 μg/ml) for 18 hours, followed by treatment with 100 mM ethanol for an additional 24 hours. Transcriptional activity of NF-κB was measured by luciferase reporter assay. Values are presented as fold increase compared with the control samples and are expressed as mean ± S.E.M. (n = 5). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol.
AMPK Signaling Is Implicated in the Suppression of Ethanol-Induced Nox2 Expression by gAcrp in RAW 264.7 Macrophages.
AMPK is well known as a mediator of adiponectin-induced various biologic responses and modulator of NF-κB signaling (Salminen et al., 2011). To investigate the underlying molecular mechanisms, we studied the role of AMPK signaling in the suppression of ethanol-induced Nox2 expression by gAcrp. For this purpose, we first examined the effect of ethanol on AMPK activity and its role in Nox2 expression in RAW 264.7 macrophages. As shown in Fig. 4A, ethanol treatment decreased the phosphorylation of AMPKα in a time-dependent manner. Furthermore, restoration of AMPK activity by pretreatment with AICAR, a pharmacologic activator of AMPK, attenuated ethanol-induced Nox2 mRNA expression (Fig. 4B), and AICAR treatment also significantly decreased ethanol-induced NF-κB activation (Fig. 4C), which plays a critical role in ethanol-induced Nox2 expression. In these experiments, we confirmed an increased phosphorylation level of AMPK on treatment with AICAR in RAW 264.7 macrophages, as expected (Supplemental Fig. 2). All these results suggest that ethanol treatment causes downregulation of AMPK signaling, which in turn leads to the activation of NF-κB and Nox2 expression. We next examined the potential role of AMPK signaling in prevention of ethanol-induced Nox2 expression by gAcrp. Treatment with gAcrp induces phosphorylation of AMPK up to 24 hours in our experimental condition (Fig. 4D), consistent with previous reports. Furthermore, pretreatment with gAcrp restored ethanol-suppressed AMPK phosphorylation in RAW 264.7 macrophages (Fig. 4E). Finally, we dissected the role of AMPK signaling in the suppression of ethanol-induced Nox2 expression by gAcrp. Pretreatment of the cells with compound C, an inhibitor of AMPK, abolished suppression of Nox2 expression by gAcrp in ethanol-treated cells (Fig. 4F). Moreover, it also abrogated the suppression of ethanol-induced NF-κB activation by gAcrp (Fig. 4G). The role of AMPK activation by gAcrp in suppression of ethanol-induced NF-κB activation was confirmed by gene silencing of AMPKα1. As depicted in Fig. 4H, knocking down AMPKα1 by siRNA reverted inhibition of ethanol-induced NF-κB activation by gAcrp, similar to the results induced by treatment with compound C. Taken together, these data suggest that gAcrp-induced AMPK activation plays an important role in suppression of ethanol-induced expression of Nox2 via modulation of NF-κB signaling in RAW 264.7 macrophages.
Fig. 4.
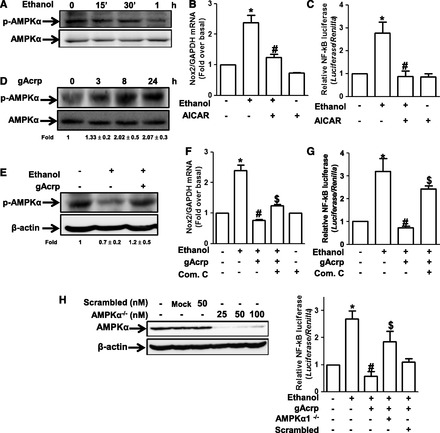
Role of AMPK signaling in the suppression of ethanol-induced Nox2 expression by gAcrp in RAW 264.7 macrophages. (A) Cells were incubated with 100 mM ethanol for the indicated time periods. The level of phosphorylated AMPKα was measured by Western blot analysis as described previously. Images are representative of three independent experiments that showed similar results. Band intensities were quantified by densitometric analysis and normalized by total AMPK. Values are represented as fold changes relative to controls and are expressed as mean ± S.E.M. (n = 3). (B) Cells were pretreated with AICAR (1 mM), an activator of AMPK, for 1 hour, followed by stimulation with 100 mM ethanol for 18 hours. Nox2 mRNA levels were measured by qRT-PCR. Values represent fold change relative to the control cells and are expressed as mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (C) Cells were cotransfected with pNFκB-Luc plasmid and Renilla control reporter gene as described previously. After 24 hours of transfection, cells were preincubated with AICAR (1 mM) for 1 hour, followed by treatment with 100 mM ethanol for additional 24 hours. Transcriptional activity of NF-κB was assessed by reporter assay. Values are the results of six separate experiments and are expressed as mean ± S.E.M. (n = 6). *P < 0.05 compared with the cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (D) Cells were treated with gAcrp (0.1 μg/ml) for the indicated periods. The level of phosphorylation of AMPKα was analyzed by Western blot analysis. Band intensities were quantified by densitometric analysis and normalized by total AMPKα. Values are represented as fold changes relative to control and are expressed as mean ± S.E.M. (E) Cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours, followed by stimulation with 100 mM ethanol for 1 hour. The level of AMPKα phosphorylation was measured by Western blot analysis. Representative images from three independent experiments are shown along with β-actin for loading control. Band intensities were quantified by densitometric analysis and normalized by β-actin. Values are represented as fold changes relative to control and are expressed as mean ± S.E.M. (F) Cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours in the absence or presence of compound C (1 μM), an AMPK inhibitor, followed by 100 mM ethanol treatment. Nox2 mRNA was analyzed by qRT-PCR as described previously. Values are the results of four separate experiments and presented as mean ± S.E.M. (n = 4). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol. (G) Cells were transiently cotransfected with pNFκB-Luc plasmid and Renilla reporter gene as described previously. After 24 hours of culture, cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours in the absence or presence of compound C (1 μM), followed by incubation with 100 mM ethanol for additional 24 hours. Transcriptional activity of NF-κB was assessed by luciferase reporter assay as described previously. Values are expressed as fold increase relative to control cells, mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol; $P < 0.05 compared with cells treated with gAcrp and ethanol. (H) Upper panel: Cells were transfected with different concentrations of siRNA targeting AMPKα1 or scrambled siRNA. After overnight incubation, expression level of AMPKα was determined by Western blot analysis. Lower panel: Cells were transfected with siRNA targeting AMPKα (25 nM) or scrambled control siRNA. After 6 hours’ incubation, cells were then cotransfected with pNFκB-Luc plasmid and Renilla reporter gene. After overnight incubation, cells were pretreated with gAcrp (0.1 μg/ml) for 18 hours, followed by 100 mM ethanol stimulation for additional 24 hours. Transcriptional activity of NF-κB was determined by luciferase reporter assay. Values are presented as fold change relative to control cells and expressed as mean ± S.E.M. (n = 3). *P < 0.05 compared with cells not treated with ethanol; #P < 0.05 compared with cells treated with ethanol.
AMPK Signaling Is Involved in the Suppression of Ethanol-Induced ROS Production by gAcrp in RAW 264.7 Macrophages.
NADPH oxidase is a main source for ethanol-induced ROS production in macrophages, and we found that gAcrp inhibited ethanol-induced NADPH oxidase activation via activation of AMPK signaling (Fig. 4). Next, we finally examined whether AMPK signaling is implicated in modulation of ROS production by gAcrp. As shown in Fig. 5A, although gAcrp treatment significantly suppressed ethanol-induced intracellular ROS production, pretreatment with compound C reversed suppression of ROS production by gAcrp, as assessed by conversion of CM-H2DCF-DA to the fluorescent DCF. Moreover, gene silencing of AMPKα1 reversed inhibitory effect of gAcrp on ethanol-induced ROS production, whereas no significant effect was observed by scrambled siRNA (Fig. 5B). Taken together, these data confirm that the AMPK signaling also plays a critical role in the prevention of ethanol-induced ROS production by gAcrp.
Fig. 5.
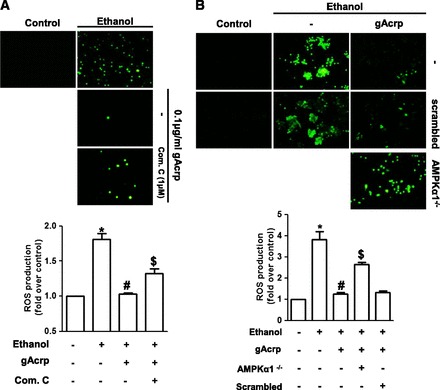
Role of AMPK signaling in suppression of ethanol-induced ROS production by gAcrp in RAW264.7 macrophages. (A) Cells were pretreated with gAcrp in the absence or presence of compound C (1 μM) for 18 hours, followed by incubation with 100 mM ethanol. After 24 hour incubation, the media were replaced with HBSS containing 10 μM CM-H2DCF-DA, and cells were further incubated for 30 minutes in the dark. ROS generation was assessed as described previously, and representative images from three independent experiments are shown along with quantitation of green signal intensity. Values are presented as mean ± S.E.M. (n = 3). * P < 0.05 compared with control; #P < 0.05 compared with cells treated with ethanol; $P < 0.05 compared with cells treated with gAcrp and ethanol. (B) Cells were transfected with siRNA targeting AMPKα (25 nM) or scrambled control siRNA. After 6 hours’ incubation, media were replaced with DMEM containing 10% FBS and then further incubated for additional 22 hours. Cells were treated with 0.1 μg/ml of gAcrp for 18 hours, followed by stimulation with 100 mM ethanol for an additional 24 hours. ROS production was determined as described previously, and representative images from three independent experiments are shown along with quantitation of fluorescence intensity (lower panel). Values are expressed as fold change relative to control cells. *P < 0.05 compared with control group; #P < 0.05 compared with cells treated with only ethanol; $P < 0.05 compared with the cells treated with gAcrp and ethanol.
LKB1 Signaling Is Implicated in the Suppression of Ethanol-Induced Nox2 Expression by gAcrp in RAW 264.7 Macrophages.
Liver kinase B1 (LKB1) is widely known as an upstream signaling molecule of AMPK. To elucidate more fully the upstream signaling mechanisms leading to the AMPK activation and suppression of ROS production by gAcrp in ethanol-treated macrophages, we examined whether LKB1 signaling plays a role in the suppression of Nox2 expression by gAcrp. For this, we first investigated the effect of gAcrp on LKB1. As indicated in Fig. 6A, gAcrp treatment rapidly increased phosphorylation level of LKB1, showing a maximal level at 30 minutes. In addition, suppression of LKB1 phosphorylation by ethanol was restored by pretreatment with gAcrp in RAW 264.7 macrophages (Fig. 6B), which is similar to the pattern for the modulation of AMPK phosphorylation. Furthermore, gene silencing of LKB1 by transfection with siRNA targeting LKB1 significantly restored suppression of Nox2 mRNA expression by gAcrp (Fig. 6C), and it also reverted gAcrp-suppressed NF-κB activity in ethanol-treated cells (Fig. 6D) without significant effect by transfection with scrambled siRNA. We also observed that depletion of LKB1 gene prevented gAcrp-induced AMPK phosphorylation (data not shown) consistent with previous reports. All these data suggest that LKB1 signaling plays an important role in the suppression of ethanol-induced Nox2 expression and NF-κB signaling via acting as an upstream molecule leading to the AMPK activation. LKB1 signaling is regulated by a number of upstream signaling pathways. Particularly, extracellular signal regulated kinase 1/2 (ERK1/2) MAPK has been shown to activate LKB1 signaling in various experimental models (Sapkota et al., 2001). Thus, to identify signaling molecules responsible for gAcrp-induced LKB1 activation, we examined the involvement of MAPKs. As depicted in Fig. 6E, pretreatment of cells with U0126, a selective inhibitor of ERK1/2, suppressed gAcrp-induced LKB1 phosphorylation, whereas inhibitors of JNK (SP600125) and p38 MAPK (SB203580) did not significantly affect gAcrp-induced LKB1 activation, indicating a critical role of ERK1/2 signaling in gAcrp-induced LKB1 activation.
Fig. 6.
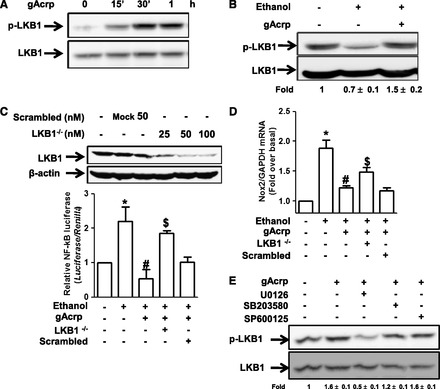
Role of LKB1 signaling in the suppression of ethanol-induced ROS production by gAcrp in RAW 264.7 macrophages. (A) Cells were incubated with gAcrp (0.1 μg/ml) for the indicated periods. The level of LKB1 phosphorylation was analyzed by Western blot analysis as described previously. Images are representative of three independent experiments along with total LKB1 for internal loading control. Band intensities were quantified by densitometric analysis and normalized by total LKB1. Values are represented as fold changes relative to control and are expressed as mean ± S.E.M. (B) Cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours, followed by incubation with 100 mM ethanol for 1 hour. The level of LKB1 phosphorylation was measured by Western blot analysis. Representative images from three independent experiments are shown along with total LKB1 for loading control. (C) Upper panel: Cells were transfected with different concentrations of siRNA targeting LKB1 or scrambled siRNA. After overnight incubation, expression level of LKB1 was determined by Western blot analysis. Lower panel: Cells were transfected with siRNA targeting LKB1 (50 nM) or scrambled control siRNA. After 6 hours’ incubation, media were changed with DMEM containing 10% FBS, and cells were further cultured for an additional 22 hours. Cells were treated with 0.1 μg/ml gAcrp for 18 hours, followed by stimulation with 100 mM ethanol for an additional 24 hours. Nox2 mRNA expression levels were assessed by qRT-PCR as described previously. Values are presented as fold increase relative to control group and expressed as mean ± S.E.M. (n = 5). *P < 0.05 compared with control group; #P < 0.05 compared with cells treated with only ethanol; $P < 0.05 compared with the cells treated with gAcrp and ethanol. (D) Cells were transfected with siRNA targeting LKB1 (50 nM) or scrambled control siRNA. After 6 hours of incubation, cells were then cotransfected with pNFκB-Luc plasmid and Renilla reporter gene. After 6 hours of transfection, media were replaced with DMEM containing 10% FBS and further incubated for 18 hours. Finally, cells were pretreated with 0.1 μg/ml of gAcrp for 18 hours, followed by stimulation with ethanol for an additional 24 hours, and the transcriptional activity of NF-κB was measured as described previously. Values are expressed as fold change relative to control cells, mean ± S.E.M. (n = 3). *P < 0.05 compared with control group; #P < 0.05 compared with cells treated with ethanol; $P < 0.05 compared with cells treated with gAcrp and ethanol. (E) Cells were pretreated with U0126 (5 μM), a selective inhibitor of ERK1/2; SP600125 (10 μM), a selective inhibitor of JNK; or SB203580 (10 μM), a selective inhibitor of p38 MAPK for 1 hour, followed by stimulation with gAcrp (0.1 μg/ml) for an additional 30 minutes. Phosphorylated LKB1 levels were analyzed by Western blot analysis along with total LKB1 for internal control. Band intensities were quantified by densitometric analysis and normalized by total LKB1. Values are represented as fold changes relative to control and are expressed as mean ± S.E.M.
Discussion
ROS has been shown to induce diverse pathologic events such as DNA mutation, carcinogenesis, aging, and such (Cederbaum et al., 2009), although it is required for normal physiologic processes as well. Ethanol treatment caused a significant increase in ROS production, which is a critical event for the development of pathophysiologic condition in various target tissues. Macrophages are the main cellular source responsible for ethanol-induced ROS production, and excessive ROS production by macrophages leads to the damage in target tissues, such as alcoholic liver injury and impairment of lung function. Adiponectin, the most abundant adipokine in plasma, is implicated in a number of beneficial processes. It plays a potent protective role against chronic alcohol consumption (Rogers et al., 2008; Nepal et al., 2012) and prevents ROS production in response to various stimuli (Ouedraogo et al., 2006; Thakur et al., 2006a). Although it is widely accepted that ROS production in macrophages is a critical step leading to the various pathologic conditions by ethanol exposure and adiponectin plays a protective role against chronic ethanol consumption, the effects of adiponectin on ethanol-induced ROS production in macrophages are not clearly understood. In the present study, we examined the effect of adiponectin on ethanol-induced ROS production in macrophages and its potential mechanisms. Herein, we provided the first evidence that globular adiponectin (gAcrp) inhibited ethanol-induced ROS production. Additional mechanistic studies have demonstrated that suppression of ethanol-induced ROS production by gAcrp is mediated via modulation of NADPH oxidase; further, LKB1/AMPK pathway plays an important role in the suppression of NADPH oxidase and ROS production by gAcrp (Fig. 7).
Fig. 7.
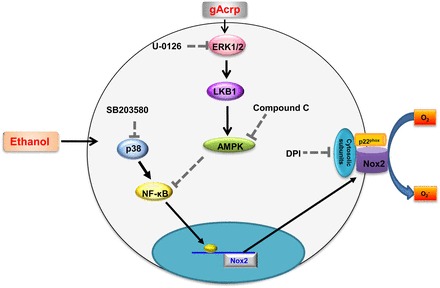
Proposed model for the modulation of ethanol-induced Nox2 expression by globular adiponectin and its potential role in the suppression of ethanol-induced ROS production in macrophages. Ethanol treatment induces increase in ROS production in murine macrophages via NADPH oxidase activation, which is composed of various cytosolic and membrane subunits. Ethanol-induced NADPH oxidase activation is accompanied with Nox2 expression through the p38 MAPK/NF-κB pathway. Globular adiponectin treatment results in prevention of ROS production induced by ethanol treatment. The inhibitory effect of gAcrp on ethanol-induced ROS production is mediated by modulation of NADPH oxidase, particularly via inhibition of Nox2 expression. Abrogation of ethanol-induced Nox2 expression by globular adiponectin is mediated through LKB1/AMPK axis signaling mechanisms. LKB1 and AMPK signaling pathway reverted ethanol-induced Nox2 expression, probably via negative regulation of transcriptional activity of NF-κB. Detailed mechanisms underlying inhibition of NF-κB signaling by LKB1/AMPK axis remain to be determined.
Ethanol consumption promotes the production of ROS via multiple mechanisms in a cell type–specific manner. An accumulated line of evidence demonstrated a critical role of NADPH oxidase in ethanol-induced ROS production in macrophages. For example, ethanol treatment increases ROS production via enhancing expression of Nox2 and p22phox in RAW 264.7 macrophages (Kim et al., 2013), induces oxidative stress through upregulation of NADPH oxidases in alveolar macrophages (Yeligar et al., 2012), and enhances LPS-stimulated TNF-α production via modulation of NADPH oxidase-dependent ROS production (Thakur et al., 2006b), collectively indicating that NADPH oxidase would be a main source for ethanol-induced ROS production in macrophages, and therefore it would be a promising target for modulation of ethanol-induced pathogenic conditions.
Adiponectin has been shown to possess antioxidative and anti-inflammatory properties in various experimental conditions, compatible with the results of the present study. Herein, we demonstrated that gAcrp prevented ethanol-induced ROS production via modulation of NADPH oxidase in both RAW 264.7 macrophages and murine peritoneal macrophages (Fig. 1), providing additional evidence for the antioxidative effect of adiponectin. Although a number of studies have supported the notion that adiponectin acts as an inhibitor of antioxidative stress, other studies have also suggested that adiponectin induces ROS production. Akifusa and colleagues reported that adiponectin treatment increased ROS/RNS production, which causes Bcl-2 modulation and induction of apoptosis in RAW 264 macrophages (Akifusa et al., 2008, 2009). It seems differential effects of adiponectin on ROS production would be derived from experimental conditions. For example, in the current study, pretreatment with 0.1 μg/ml of gAcrp for 24 hours reduced ethanol-stimulated ROS production. However, in previous studies, treatment with higher concentration of gAcrp alone (10 μg/ml) leads to ROS formation. To investigate further the effect of adiponectin on ROS production in macrophages, we examined the effect of gAcrp on ROS production at various dose and time points. In these experiments, relatively higher concentrations (1 and 10 μg/ml) of gAcrp increased ROS production in a time-dependent manner consistent with previous reports, but no significant effect was observed by treatment with 0.1 μg/ml of gAcrp (Supplemental Fig. 1). These results suggest that treatment with a lower concentration of gAcrp does not significantly affect ROS production but has preventive effect on ethanol-induced ROS production in RAW 264.7 macrophages. We could not investigate the effect of higher concentration of gAcrp on ethanol-induced ROS production since treatment of RAW 264.7 macrophages with ethanol and higher concentration of gAcrp (1 or 10 μg/ml) caused severe cytotoxicity. All these results indicate that a lower concentration of gAcrp prevents ethanol-induced ROS production in macrophages, whereas higher concentrations of gAcrp accelerate ethanol-induced cell death in macrophages. Although at this stage we cannot identify detailed mechanisms underlying opposite responses by adiponectin, it is possible that diverse responses to treatment condition of adiponectin are attributable to the different responses by adiponectin.
AMP-dependent protein kinase (AMPK) is well known as a key mediator of adiponectin-induced various biologic responses (Zhou et al., 2009; Nepal and Park, 2013). In the present study, we observed that pretreatment with AMPK inhibitor or targeted knockdown of AMPKα1 efficiently reversed inhibitory effect of gAcrp on ethanol-induced Nox2 expression and ROS production (Fig. 4), providing another evidence that AMPK mediates adiponectin-induced beneficial effects. With regard to the regulation of AMPK signaling, ethanol consumption has been shown to dysregulate AMPK activity in cultured hepatic cell lines, as well as in vivo model (You et al., 2004; Garcia-Villafranca et al., 2008; Shen et al., 2010). Based on previous reports regarding the regulation of AMPK by ethanol and adiponectin, we hypothesized that ethanol-induced ROS production may be mediated via dysregulation of AMPK signaling and that gAcrp prevents ROS production through restoration of AMPK activity. In the current study, we demonstrated that treatment with AMPK activator inhibited ethanol-induced Nox2 expression and NF-κB activation (Fig. 4B and C). Furthermore, inhibition of AMPK signaling restored suppression of ethanol-induced activation of NF-κB signaling, Nox2 expression (Fig. 4F-H), and finally ROS production (Fig. 5, A and B) by gAcrp, clearly demonstrating that AMPK plays a critical role in regulation of ROS production by ethanol and adiponectin in macrophages.
Liver kinase B1 (LKB1) plays important roles in various biologic responses through regulation of multiple downstream kinases. Growing evidence has demonstrated that LKB1 efficiently phosphorylates the catalytic α-subunit of AMPK at Thr172 on binding with Ste20-related adaptor protein (STRAD) and mouse protein 25 (MO25), regulatory proteins of LKB1, and that a number of LKB1-induced biologic responses are mediated through an AMPK-dependent manner (Shaw et al., 2004; Alessi et al., 2006; Shackelford and Shaw, 2009). Recent studies have also shown that the LKB1/AMPK pathway is implicated in adiponectin-induced various biologic responses. For example, adiponectin-induced glucose uptake and suppression of breast cancer cells migration are mediated via LKB1/AMPK-dependent manner (Vu et al., 2013). Although it is well known that LKB1 is implicated in adiponectin-induced biologic responses, the role of LKB1 signaling in antioxidative effect induced by adiponectin has not been demonstrated. To our knowledge, the present study demonstrates for the first time that LKB1 signaling plays a critical role in the inhibition of ROS production and modulation of NADPH oxidase by adiponectin. In the present study, gAcrp treatment induced increase in phosphorylation of LKB1 (Fig. 6A) and further pretreatment with gAcrp ameliorated suppression of LKB1 phosphorylation by ethanol (Fig. 6B), which exactly correlates to the modulation of AMPK signaling by ethanol and adiponectin. Furthermore, we observed that depletion of LKB1 gene prevented gAcrp-induced AMPK phosphorylation (data not shown) and blocked the suppressive effects of gAcrp on Nox2 expression (Fig. 6D). Based on previous reports and data presented here, it seems highly likely that LKB1 acts as upstream of AMPK for the prevention of ethanol-induced ROS production, and further LKB1 would be a promising target for protective effects by gAcrp against chronic alcohol consumption.
LKB1 is phosphorylated at different residues (Ser31, Ser325, Thr366, and Ser428) via various kinases, including PKA, ribosomal S6 protein kinase (RSK), and PKC-ζ. Previous studies have shown that PKC-ζ is responsible for phosphorylation of LKB1 at Ser428. In addition, ethanol has been shown to inhibit phosphorylation of PKC-ζ (Guizzetti and Costa, 2002). Furthermore, in the present study, we found that ethanol decreased and gAcrp reversed the phosphorylation of LKB1 at Ser428 (Fig. 6B), in which phosphorylation is modulated by PKC-ζ. All these previous reports and results from this study suggest ethanol and gAcrp may modulate LKB1 phosphorylation via regulation of PKC-ζ. In the present study, we did not identify the detailed mechanisms underlying LKB1 phosphorylation modulated by ethanol and adiponectin since this project would be beyond the scope of the current study. It would be interesting to investigate the involvement of PKC-ζ signaling in suppression and restoration of LKB1 activation by ethanol and adiponectin for future study.
In conclusion, the data presented in this study demonstrated that globular adiponectin prevents ethanol-induced ROS production through modulation of NADPH oxidase in macrophages and this inhibitory effect is mediated by LKB1/AMPK axis (Fig. 7). Based on these findings, we suggest that inhibition of NADPH oxidase would be a mechanism underlying the hepatoprotective effect and other beneficial effects effected by adiponectin. The present study used RAW 264.7 macrophages and primary peritoneal macrophages to understand the mechanisms underlying modulation of ROS production by gAcrp. Further studies are now required to validate our findings in murine peritoneal macrophages and macrophage cell lines to the appropriate in vitro and in vivo models.
Abbreviations
- AICAR
5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside
- AMPK
AMP-activated protein kinase
- CM-H2DCFDA
5-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK1/2
extracellular signal regulated kinase 1/2
- FBS
fetal bovine serum
- gAcrp
globular adiponectin
- HBSS
Hanks’ balanced salt solution
- LKB1
liver kinase B1
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NADPH oxidase
nicotinamide adenine dinucleotide phosphate-oxidase
- NF-κB
nuclear factor-κB
- ROS
reactive oxygen species
Authorship Contributions
Participated in research design: Kim, Nagy, Park.
Conducted experiments: Kim, Park.
Performed data analysis: Kim, Park.
Wrote or contributed to the writing of the manuscript: Kim, Nagy, Park.
Footnotes
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education 2013R1A1A4A01011110.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Akifusa S, Kamio N, Shimazaki Y, Yamaguchi N, Nishihara T, Yamashita Y. (2009) Globular adiponectin-induced RAW 264 apoptosis is regulated by a reactive oxygen species-dependent pathway involving Bcl-2. Free Radic Biol Med 46:1308–1316. [DOI] [PubMed] [Google Scholar]
- Akifusa S, Kamio N, Shimazaki Y, Yamaguchi N, Yamashita Y. (2008) Regulation of globular adiponectin-induced apoptosis by reactive oxygen/nitrogen species in RAW264 macrophages. Free Radic Biol Med 45:1326–1339. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Sakamoto K, Bayascas JR. (2006) LKB1-dependent signaling pathways. Annu Rev Biochem 75:137–163. [DOI] [PubMed] [Google Scholar]
- Brown SD, Gauthier TW, Brown LA. (2009) Impaired terminal differentiation of pulmonary macrophages in a Guinea pig model of chronic ethanol ingestion. Alcohol Clin Exp Res 33:1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederbaum AI, Lu Y, Wu D. (2009) Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83:519–548. [DOI] [PubMed] [Google Scholar]
- D’Souza El-Guindy NB, de Villiers WJ, Doherty DE. (2007) Acute alcohol intake impairs lung inflammation by changing pro- and anti-inflammatory mediator balance. Alcohol 41:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Bataller R. (2011) Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Villafranca J, Guillén A, Castro J. (2008) Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90:460–466. [DOI] [PubMed] [Google Scholar]
- Guizzetti M, Costa LG. (2002) Effect of ethanol on protein kinase Czeta and p70S6 kinase activation by carbachol: a possible mechanism for ethanol-induced inhibition of glial cell proliferation. J Neurochem 82:38–46. [DOI] [PubMed] [Google Scholar]
- Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, Kim WS, Kim JB. (2009) Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 296:E955–E964. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116:1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Nepal S, Lee ES, Jeong TC, Kim SH, Park PH. (2013) Ethanol increases matrix metalloproteinase-12 expression via NADPH oxidase-dependent ROS production in macrophages. Toxicol Appl Pharmacol 273:77–89. [DOI] [PubMed] [Google Scholar]
- Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, et al. (2000) NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 106:867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam GY, Huang J, Brumell JH. (2010) The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol 32:415–430. [DOI] [PubMed] [Google Scholar]
- Lim CT, Kola B, Korbonits M. (2010) AMPK as a mediator of hormonal signalling. J Mol Endocrinol 44:87–97. [DOI] [PubMed] [Google Scholar]
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. (2006) APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523. [DOI] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Le Lay J, Scherer PE, Ahima RS, Kaestner KH, Foretz M, Viollet B, Birnbaum MJ. (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest 121:2518–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal S, Kim MJ, Subedi A, Lee ES, Yong CS, Kim JA, Kang W, Kwak MK, Arya DS, Park PH. (2012) Globular adiponectin inhibits ethanol-induced apoptosis in HepG2 cells through heme oxygenase-1 induction. Biochem Pharmacol 84:974–983. [DOI] [PubMed] [Google Scholar]
- Nepal S, Park PH. (2013) Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochim Biophys Acta 1833:2111–2125. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Walsh K. (2007) Adiponectin as an anti-inflammatory factor. Clin Chim Acta 380:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, Hough K, Scalia R, Goldstein BJ. (2006) Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes 55:1840–1846. [DOI] [PubMed] [Google Scholar]
- Rogers CQ, Ajmo JM, You M. (2008) Adiponectin and alcoholic fatty liver disease. IUBMB Life 60:790–797. [DOI] [PubMed] [Google Scholar]
- Salminen A, Hyttinen JM, Kaarniranta K. (2011) AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 89:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. (2001) Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell vrowth. J Biol Chem 276:19469–19482. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101:3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Liang X, Rogers CQ, Rideout D, You M. (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 298:G364–G374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, et al. (2007) Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115:1408–1416. [DOI] [PubMed] [Google Scholar]
- Thakur V, Pritchard MT, McMullen MR, Nagy LE. (2006a) Adiponectin normalizes LPS-stimulated TNF-alpha production by rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol 290:G998–G1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. (2006b) Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol 79:1348–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu V, Bui P, Eguchi M, Xu A, Sweeney G. (2013) Globular adiponectin induces LKB1/AMPK-dependent glucose uptake via actin cytoskeleton remodeling. J Mol Endocrinol 51:155–165. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Watabe K, Araki H, Kamada Y, Kato M, Kizu T, Kiso S, Tsutsui S, Tsujii M, Kihara S, et al. (2012) Protective role of adiponectin against ethanol-induced gastric injury in mice. Am J Physiol Gastrointest Liver Physiol 302:G773–G780. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kahn BB, Shi H, Xue BZ. (2010) Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 285:19051–19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Harris FL, Hart CM, Brown LA. (2012) Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 188:3648–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127:1798–1808. [DOI] [PubMed] [Google Scholar]
- Zhou L, Deepa SS, Etzler JC, Ryu J, Mao X, Fang Q, Liu DD, Torres JM, Jia W, Lechleiter JD, et al. (2009) Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J Biol Chem 284:22426–22435. [DOI] [PMC free article] [PubMed] [Google Scholar]