

Abstract
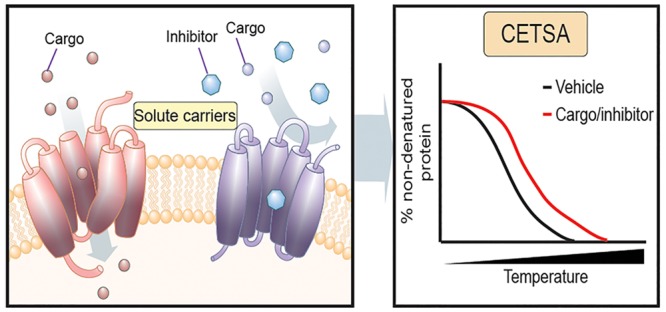
Solute carriers (SLCs) are transmembrane proteins that transport various nutrients, metabolites, and drugs across cellular membranes. Despite the relevance of SLCs to cell homeostasis, metabolism, and disease states, for the majority of SLCs we lack experimental evidence regarding the nature of the cognate ligands, whether endobiotic or xenobiotic. Moreover, even for the roughly 20 SLCs for which inhibitors have been characterized, engagement assays in cells are limited to the accessibility of radiolabeled or fluorescent probes. The cellular thermal shift assay (CETSA) has been introduced as a powerful method to assess target engagement by monitoring ligand-induced changes in the thermal stability of cellular proteins. We addressed the question of whether CETSA could be modified to become routinely applicable to membrane transporters such as SLCs. We used SLC16A1 (MCT1) and SLC1A2 (EAAT2) as targets to establish robust conditions by which chemical engagement of SLCs can be detected. Using immunoblotting, we demonstrate that treatment with the SLC16A1 inhibitors AZD3965 and AR-C155858 stabilized endogenous SLC16A1 in HEK293 cell lysates as well as intact cells. In addition, the high-affinity ligand of SLC16A1, l-lactate, and the low-affinity ligand, formate, resulted in strong and weak stabilization of SLC16A1, respectively. Moreover, we observed stabilization of SLC1A2 upon treatment with the selective inhibitor WAY-213613. We propose that the experimental approach presented here should be generally and easily applicable for monitoring the engagement of chemical ligands by SLCs in cellular settings and thus assisting in their deorphanization.
Solute carriers (SLCs) are integral membrane proteins localized on the cell surface and in organellar membranes, where they mediate transport of a wide variety of small molecules, such as amino acids, metal ions, nucleosides, and vitamins.1 The SLC family comprises >400 distinct genes, which are differentially expressed to orchestrate the supply of essential metabolites and energy resources and regulate cell growth, apoptosis, metabolism, and differentiation.2,3 As several drugs are thought to depend on SLC-mediated transport to enter cells, distinct SLC expression profiles seem to influence drug distribution in tissues and cells.4 Inhibition of specific SLCs can therefore influence multiple processes and be broadly therapeutically relevant. Despite the evident importance of this protein family, our knowledge of SLCs is still limited, as exemplified by the large numbers of SLCs with unknown function and unknown cargoes. Furthermore, only 26 SLCs are currently targeted by drugs, or drugs in development, even though several SLCs have been associated with disease states.2,5 To overcome this shortfall, there is strong demand for novel methods for experimentally matching chemical compounds to SLCs in an easy manner.
The thermal shift assay (TSA) is a method for detecting target engagement by monitoring thermostability of purified proteins.6,7 This method is based on the finding that ligand–target interactions change the thermodynamic parameters of the target, affecting its stability vis-à-vis a temperature increase. The recently developed cellular thermal shift assay (CETSA) showed that target engagement can likewise be assessed in whole cell lysates or intact cells based on altered protein thermostability.8,9 A CETSA thus allows the investigation of target engagement under physiological conditions.10−12 Stabilization can occur directly by ligand engagement of the target, but also by more remote changes induced indirectly through protein interactions or downstream effectors of drug treatment. Furthermore, a CETSA can be aimed at a specific protein using specific antibodies, or in an unbiased way employing mass spectrometry, known as thermal proteome profiling.10−12 Membrane proteins, including SLCs, have been among the species detected in thermal proteome profiles.10−13 Moreover, a CETSA has been shown to work for monitoring thermal stabilization of G-protein-coupled receptors and ATP binding cassette proteins, upon treatment with their ligands.13,14
Given that solute carrier proteins make up one of the target classes for which deorphanization is most needed, we set out to adapt a CETSA to monitor engagement of these transporters by known ligands. To test the feasibility of the CETSA approach for detection of SLC binding events, two physiologically important SLCs (SLC1A2 and SLC16A1) were chosen, which are also currently studied as drug targets for neurodegenerative diseases and cancer, respectively.15−17 We used these two proteins for a proof-of-concept study and have been able to develop and validate an approach that should be feasible for all members of this large class of proteins.
Results and Discussion
To test the applicability of a CETSA for identification of SLC binders, we searched for SLCs of known biological relevance, for which inhibitors and naturally transported cargo molecules have already been established. One such SLC is SLC16A1, also known as MCT1 (monocarboxylate transporter 1). SLC16A1 is a 12-transmembrane domain cotransporter of monocarboxylates and protons, which is expressed on the plasma membrane of various cells and tissues.18 Importantly, SLC16A1 has a primary role in lactate transport, and inhibition of SLC16A1 has been proposed to selectively target highly glycolytic cancer cells.19 AR-C155858 is a specific and potent inhibitor of SLC16A1, with Ki values in the low nanomolar range.20,21 AR-C155858, whose structure is depicted in Figure 1a and does not relate to the structure of endogenous SLC16A1 cargoes (Figure 1c,d), binds to an intracellular site involving transmembrane helices 7–10.20 The recently developed AZD3965 is derived from AR-C155858 (Figure 1a,b), displays a high affinity for SLC16A1 (Ki = 1.6 nM), as well,15 and is currently undergoing clinical trials for application in advanced solid tumors and lymphoma.22,23
Figure 1.

Scheme of CETSA for SLCs. Chemical structures of investigated compounds are shown: (a) AZD3965, (b) AR-C155858, (c) l-lactate, and (d) formate. (e) Workflow of a CETSA for SLCs in cell lysates and intact cells.
To test the binding between SLC16A1 and its inhibitors in HEK293 cell lysates using a CETSA, we optimized the cell lysis method. We used a lysis solution [100 mM ammonium sulfate, 400 mM NaCl, and 10% (v/v) glycerol] containing n-dodecyl β-d-maltoside [DDM, final concentration of 0.5% (w/v)], which was the detergent previously used in a TSA for membrane proteins to increase solubility and stability.6Figure 1e depicts the overall experimental strategy, for a CETSA in both cell lysates and intact cells. We first focused on cell lysates, which were incubated with the inhibitors or vehicle for 30 min on ice and subsequently exposed to different temperatures before immunoblotting. In Figure 2a, the results of the immunoblot showed that the abundance of SLC16A1 in the cell lysate decreased with increasing temperatures (45–85 °C), showing that the thermostability of endogenous SLC16A1 could be monitored by this method. Importantly, in the range from 65 to 85 °C, the lysates treated with AZD3965 (50 nM) or AR-C155858 (50 nM) showed abundances of SLC16A1 protein higher than the abundances of those treated with dimethyl sulfoxide (DMSO), suggesting ligand-dependent stabilization. In contrast, the protein levels of SLCO1A2 (organic anion-transporting polypeptide 1A2) and tubulin, which were chosen as controls, failed to show any differences (Figure 2a,b). In addition, the increase in the thermostability of SLC16A1 caused by AZD3965 treatment was found to be concentration-dependent (5–1000 nM) in the cell lysates heated to 65 and 75 °C (Figure 2c). These results indicated that, using the modified CETSA protocol, we could observe engagement of SLC16A1 by both inhibitors leading to its stabilization in whole cell lysates.
Figure 2.

CETSA for SLC16A1 inhibitors in HEK293 cell lysates and intact cells. The results of immunoblotting show the thermostability of SLC16A1 and SLCO1A2 following heat treatment at the indicated temperatures in the presence (+) or absence (−) of (a) AZD3965 (50 nM) and (b) AR-C155858 (50 nM). (c) For analysis of dose responses, levels of thermostable SLC16A1 and SLCO1A2 were analyzed in lysates heated to 65 or 75 °C, at 0, 5, 10, 20, 50, 100, and 1000 nM AZD3965. (d) For a cell-based CETSA, cells were incubated with AZD3965 (20 nM). The results of the immunoblot show the increased thermostability of SLC16A1 and SLCO1A2 following the indicated heat treatment. The expression of tubulin was analyzed as a loading control. For quantification of thermostable proteins, the signal intensity was normalized to the respective intensity at 45 °C. In the isothermal dose–response fingerprint (c), the blot intensity was normalized to samples treated with 1000 nM AZD3965. Data are means ± the standard deviation of biological triplicate measurements. The immunoblots are representative of three biological replicates. Legend: AR-C, AR-C155858; L.E., long exposure; Temp., temperature.
Next, to test if a CETSA also allows detection of SLC–inhibitor interactions in intact cells, we performed cell-based drug treatment before analysis by temperature shift. To this end, HEK293 cells were treated with 20 nM AZD3965 or vehicle in culture plates and only subsequently lysed in lysis buffer containing 0.5% DDM. Immunoblotting of the lysates revealed the increased thermostability of SLC16A1 in cells treated with AZD3965 as compared to those treated with DMSO at 60, 65, and 70 °C, whereas the protein levels of tubulin and SLCO1A2 remained unchanged (Figure 2d). Furthermore, an isothermal drug-response fingerprinting (ITDRF) assay was performed using AZD3965 and AR-C155858 in intact cells, which exhibited IC50 values of 49.44 ± 1.65 and 47.80 ± 1.26 nM, respectively (Supplementary Figure 1). To examine detergent effects, we also tested a cell-based CETSA on cells that were lysed in buffer containing NP-40 [final concentration of 0.2% (v/v)]. However, this detergent did not allow detection of a proper “melting curve” or thermostabilization of SLC16A1 upon AZD3965 treatment (Supplementary Figure 2), suggesting that the use of DDM as a detergent is critical for monitoring SLC thermostabilization.
Taken together, binding of the inhibitor to SLC16A1 could be detected by using a CETSA in both cell lysates and intact cells. The ITDRF assay also confirmed that the IC50 values of SLC16A1 inhibitors are still in the lower nanomolar range, thus correlating with the Ki values described in previous reports.15,20,21 These results suggested that the method may be applicable also for other SLCs and perhaps also using naturally transported metabolites.
As natural ligands are known to be transported efficiently in intact cells and thus have only a very transient engagement with the transporter, it may be not amenable to the CETSA experimental setup. However, engagement of transported metabolites by SLCs should stabilize specific conformations of the transporter, when lysates are incubated with enough ligand. To test this hypothesis, HEK293 cell lysates were treated with vehicle or l-lactate, the major cargo of SLC16A1 (Km = 4.5 mM in tumor cells), and subjected to the CETSA procedure.24 As shown in Figure 3a, the presence of 10 mM l-lactate in HEK293 cell lysate significantly increased the stability of SLC16A1 at 55, 60, 65, and 70 °C.
Figure 3.

CETSA for SLC16A1 and l-lactate in HEK293 cell lysates. The results of immunoblotting show the thermostability of SLC16A1 following heating in the presence (+) or absence (−) of (a) 10 mM l-lactate or (b) 10 mM formate. Quantification of thermostable SLC16A1 is shown at the left of the immunoblots. The signal intensity was normalized to the intensity of the 45 °C sample. Data are means ± the standard deviation of biological triplicate measurements. The immunoblots are representative of three biological replicates. Legend: L.E., long exposure; Temp., temperature.
To rule out possible pH-dependent effects resulting from l-lactate-induced acidification, we tested the stability of SLC16A1 upon formate treatment. Both lactate and formate are short monocarboxylates showing similar pKa values (3.86 and 3.74, respectively) (Figure 1c,d). In line with these pKa values, the pH values in the cell lysates treated with l-lactate (10 mM) and formate (10 mM) were 3.18 and 3.12, respectively, whereas the pH in vehicle (water)-treated control samples was 6.82. Notably, formate also engages SLC16A1, albeit with an affinity much lower than that of l-lactate (the Km value for formate is >100 mM).24 Indeed, formate treatment increased the stability of SLC16A1 only slightly in the cell lysates heated to 60, 65, and 70 °C (Figure 3b). We concluded that the stabilization of SLC16A1 observed upon l-lactate treatment was a pH-independent effect. Moreover, treatment of l-lactate and formate showed strong and weak thermostabilization of SLC16A1 in CETSA, respectively, implying a correlation between thermostabilization and transport affinity. Taken together, these findings suggest that a CETSA may be useful not only for screening for potent inhibitors but also for validating and perhaps even identifying relationships between SLC transporters and their naturally transported metabolites (or xenobiotics).
The approach described so far depends on the availability of high-affinity antibody reagents to detect endogenous transporter proteins, displaying different levels of natural expression. Notoriously, however, there are not many good antibodies directed against members of this large class of transporters.2 We therefore used a tagged form of SLC1A2 (also known as EAAT2 or glutamate transporter 1) to overcome the limitations resulting from antibody dependence and differential endogenous expression levels. SLC1A2 is a Na+-dependent glutamate transporter, which is expressed in the plasma membrane of astrocytes and glia, where it is responsible for glutamate influx.17 WAY-213613, the structure of which is depicted in Figure 4a, was found to be a potent and selective inhibitor of human SLC1A2 (IC50 = 85 ± 5 nM).25
Figure 4.

Lysate-based CETSA for SLC1A2 transiently expressed in HEK293 cells. (a) Chemical structure of WAY-213613. (b) The results of immunoblotting show the thermostability of FLAG-SLC1A2 following heating in the presence (+) or absence (−) of WAY-213613 (100 μM). For melting curve analysis, the signal intensity of the thermostable protein was normalized to the intensity of the 45 °C sample. Data are means ± the standard deviation of biological triplicate measurements. The immunoblots are representative results of three biological replicates. Tubulin was analyzed as a control. Legend: L.E., long exposure; WAY, WAY-213613.
To validate the WAY-213613–SLC1A2 interaction with a CETSA, we transiently expressed C-terminally FLAG-tagged SLC1A2 (SLC1A2-FLAG) in HEK293 cells. The three identified bands via immunoblotting (around 70, 90, and >180 kDa) could possibly result from both glycosylation and oligomerization of SLC1A2 (Supplementary Figure 3a).26,27 Because the fragments at 90 and >180 kDa disappeared in the cell lysates treated with peptide-N-glycosidase F, we inferred that these two upper bands corresponded to glycosylated monomers and possibly trimers of SLC1A2, respectively (Supplementary Figure 3b). Immunocytochemistry additionally confirmed that transfected SLC1A2-FLAG was localized at the plasma membrane (Supplementary Figure 3c). We then examined the effect of WAY-213613 treatment on the stability of SLC1A2-FLAG using a CETSA. As depicted in Figure 4b, a lysate-based temperature shift showed that SLC1A2-FLAG was destabilized in a temperature-dependent manner. Addition of 100 μM WAY-213613 increased the thermostability of SLC1A2-FLAG in the cell lysates heated to 60, 65, and 70 °C, thus confirming binding of WAY-213613 to SLC1A2. Taken together, these results suggest that the modified CETSA method described here is also applicable in the context of exogenously expressed tagged SLCs, which significantly extends its value for transporter research.
Tedious transport assays using cells and vesicles have so far been the main methods for studying engagement of SLC transporters by their ligands, drugs, or metabolites. However, the practicality of such approaches is seriously limited by the availability of radiolabeled or fluorescent compounds or by the laborious employment of mass spectrometry-based methods to detect ligands. On the other hand, as a biophysical approach, a cell-free TSA using purified proteins has been developed to assess engagement of the target to both soluble and membrane proteins.6,7 While a TSA in principle would provide for a straightforward binding assessment, it is limited by the difficulty of purifying sufficient amounts of SLC protein as well as by the inevitable loss of the physiological environment provided by modification, interacting protein partners, and the context of natural membranes. Compared to these approaches, a CETSA allows one to probe for engagement by ligands in the natural environment of the target protein. Moreover, it is exceedingly simple, inexpensive, and amenable to high-throughput configurations. The evidence provided in our study should motivate the community of scientists working at the deorphanization of the hundreds of still poorly characterized transporters to adopt it and further develop it. However, it should be considered that a cell-based CETSA is likely to be applicable with only relatively high-affinity ligands. The dilution of drug concentration upon cell lysis limits the sensitivity for the effects of low-affinity ligands, for which the lysate-based CETSA is rather the method of choice. Another clear limitation of this approach lies in the fact that stabilization per se does not necessarily reflect transport or binding as it may be an indirect consequence of a biological process caused by ligand addition. However, the rather short chemical treatments would most of the time argue against effects mediated by differential transcription, translation, or relocalization. Indirect effects by a chain of differential protein binding in a protein “mikado” situation are to be considered more likely. However, stabilization by direct engagement of the ligand is probably the most common cause in the majority of cases. The use of several structurally related compounds may allow assessment of structure–activity relationships that should be helpful to distinguish direct from indirect engagement.
In this study, we investigated the properties of thermostability as read-out for target engagement. However, differential sensitivity to proteases may also be used to assess changes in the thermodynamic and conformational status of proteins upon ligand engagement. Indeed, limited proteolysis has recently been successfully used to assess engagement of metabolites at a large-scale, unbiased level.28 We have started to use limited proteolysis to monitor ligand engagement of SLCs in preliminary studies and found it to be applicable both alone and in combination with a CETSA, however requiring further optimization (unpublished data).
One of the attractive features of the approach described here is that it allows assessment of SLC engagement even in the absence of high-affinity antibodies recognizing the endogenous protein. Through the systematic exogenous or endogenous tagging of SLC proteins, multiplexing or multiwell approaches become feasible, enabling parallel testing of specificity across the entire SLC superfamily. Potentially, fusion of SLCs to fluorescent proteins may lead to further simplification and automation of the approach. Finally, we predict that this method will also find use in those drug discovery campaigns in which evidence of SLC–target engagement in intact cells is strategically important.
Methods
Chemicals
AZD3965, WAY-213613, and AR-C155858 were obtained from MedChem Express, Sigma, and Tocris, respectively. These chemicals were dissolved in DMSO. Sodium l-lactate and sodium formate were purchased from Sigma and dissolved in distilled water. The chemical structures of the mentioned compounds are shown in Figures 1a–d and 4a. Tris-buffered saline with Tween (TBS-T) buffer [150 mM NaCl, 0.05% (v/v) Tween 20, 50 mM Tris-HCl buffer (pH 7.6)] was prepared by diluting 10× TBS-T buffer in doubly distilled H2O. The blocking buffer was 5% bovine serum albumin (Sigma) diluted in TBS-T. DDM and complete (EDTA-free) protease inhibitor cocktail were obtained from Cayman and Roche, respectively.
Cell Culture
All cells were maintained in a humidified atmosphere at 37 °C with 5% CO2. HEK293 cells were obtained from ATCC and grown in Dulbecco’s modified Eagle’s medium, supplemented with 10% (v/v) fetal bovine serum, 100 units mL–1 penicillin, and 100 units mL–1 streptomycin. The medium and supplements for cell culture were purchased from Thermo Fisher Scientific.
Cellular Thermal Shift Assay
For a CETSA in HEK293 cell lysates, 1.0 × 106 cells were cultured in 10 cm dishes for 24 h. The cells were washed with PBS and harvested in 3 mL of a lysis solution [100 mM ammonium sulfate, 400 mM NaCl, 10% glycerol, 0.5% DDM, and 0.1% proteinase inhibitor cocktail (Roche)]. After incubation on ice for 20 min, the lysates were centrifuged at 14000g for 20 min at 4 °C, supernatants were transferred to new tubes, and the protein concentration was measured using the Pierce BCA protein assay kit. For a CETSA, the cell lysates (0.8 mg mL–1, 30 μL) were incubated with the respective drugs or substrates at different concentrations for 30 min on ice.
For a CETSA in living HEK293 cells, cells were seeded in 12-well cell culture plates (3.0 × 105 cells/well) and exposed to compounds at the indicated concentrations for 30 min in the incubator. Control cells were incubated with an equal volume of a vehicle. Following incubation, the cells were washed with PBS to remove excess drug and directly lysed in 500 μL of a lysis solution in the culture plate. After the cell lysates were centrifuged at 14000g for 20 min at 4 °C, supernatants were transferred to 1.5 mL Eppendorf tubes.
After preparation of lysates, 30 μL aliquots of the supernatants were heated individually on a Thermomixer compact (Eppendorf) at different temperatures for 6 min and then cooled at room temperature for 3 min. Following centrifugation at 14000g for 40 min at 4 °C, supernatants were transferred to new tubes and stored at −80 °C until immunoblotting was performed.
Immunoblotting
CETSA samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and immunoblotting was performed as described previously29 using a rabbit polyclonal anti-MCT1 antibody (Sigma, HPA003324, 1:1000 dilution), a rabbit polyclonal anti-SLCO1A2 antibody (Sigma, SAB4502814, 1:1000 dilution), a mouse monoclonal anti-FLAG M2 antibody (Sigma, F1804, 1:1000 dilution), and a mouse monoclonal anti-α-tubulin antibody (Abcam, ab7291, 1:5000 dilution).
Data Analysis
The band intensity of immunoblot films was quantified using ImageJ (version 1.51t).30 For analysis of melting shift and isothermal dose–response fingerprint, quantified protein levels were analyzed in GraphPad Prism 7 (version 7.02, GraphPad Software) using the Boltzmann sigmoid equation and the four-parameter logistic curve.9
Information about the vectors, transient expression system, PNGase treatment, and immunocytochemistry is described in the Supplementary methods.
Acknowledgments
The authors thank L. X. Heinz for helpful discussion. This work was supported by an ERC Advanced Investigator Grant (ERC 695214 Game of Gates, M.H. and G.S.F.), the Austrian Science Fund (FWF 29250 Vitra, E.G.), and an EMBO Long Term Fellowship (ALTF245-2017, R.E.).
Glossary
Abbreviations
- CETSA
cellular thermal shift assay
- DMSO
dimethyl sulfoxide
- DDM
n-dodexyl β-d-maltoside
- EAAT2
excitatory amino acid transporter 2
- ITDRF
isothermal drug-response fingerprinting
- MCT1
monocarboxylate transporter 1
- SLC
solute carrier
- TSA
thermal shift assay
- TBS-T
Tris-buffered saline with Tween
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00270.
Supplementary methods and Supplementary Figures 1–3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hediger M. A.; Clémençon B.; Burrier R. E.; Bruford E. A. (2013) The ABCs of membrane transporters in health and disease (SLC series): introduction,. Mol. Aspects Med. 34, 95–107. 10.1016/j.mam.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- César-Razquin A.; Snijder B.; Frappier-Brinton T.; Isserlin R.; Gyimesi G.; Bai X.; Reithmeier R. A.; Hepworth D.; Hediger M. A.; Edwards A. M.; Superti-Furga G. (2015) A Call for Systematic Research on Solute Carriers,. Cell 162, 478–487. 10.1016/j.cell.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Nigam S. K. (2015) What do drug transporters really do?. Nat. Rev. Drug Discovery 14, 29–44. 10.1038/nrd4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K. M.; Huang S. M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; Hoffmaster K. A.; Ishikawa T.; Keppler D.; Kim R. B.; Lee C. A.; Niemi M.; Polli J. W.; Sugiyama Y.; Swaan P. W.; Ware J. A.; Wright S. H.; Yee S. W.; Zamek-Gliszczynski M. J.; Zhang L.; Consortium I. T. (2010) Membrane transporters in drug development. Nat. Rev. Drug Discovery 9, 215–236. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Yee S. W.; Kim R. B.; Giacomini K. M. (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discovery 14, 543–560. 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A. I.; Mileni M.; Chien E. Y.; Hanson M. A.; Stevens R. C. (2008) Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359. 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screening 6, 429–440. 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Martinez Molina D.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundbäck T.; Nordlund P.; Martinez Molina D. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells,. Nat. Protoc. 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Huber K. V.; Olek K. M.; Müller A. C.; Tan C. S.; Bennett K. L.; Colinge J.; Superti-Furga G. (2015) Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 12, 1055–1057. 10.1038/nmeth.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Martinez Molina D.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Franken H.; Mathieson T.; Childs D.; Sweetman G. M.; Werner T.; Tögel I.; Doce C.; Gade S.; Bantscheff M.; Drewes G.; Reinhard F. B.; Huber W.; Savitski M. M. (2015) Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593. 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- Reinhard F. B.; Eberhard D.; Werner T.; Franken H.; Childs D.; Doce C.; Savitski M. F.; Huber W.; Bantscheff M.; Savitski M. M.; Drewes G. (2015) Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods 12, 1129–1131. 10.1038/nmeth.3652. [DOI] [PubMed] [Google Scholar]
- Ashok Y.; Nanekar R.; Jaakola V. P. (2015) Defining thermostability of membrane proteins by western blotting. Protein Eng., Des. Sel. 28, 539–542. 10.1093/protein/gzv049. [DOI] [PubMed] [Google Scholar]
- Curtis N. J.; Mooney L.; Hopcroft L.; Michopoulos F.; Whalley N.; Zhong H.; Murray C.; Logie A.; Revill M.; Byth K. F.; Benjamin A. D.; Firth M. A.; Green S.; Smith P. D.; Critchlow S. E. (2017) Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 8, 69219–69236. 10.18632/oncotarget.18215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow S. E., Hopcroft L., Mooney L., Curtis N., Whalley N., Zhong H., Logie A., Revill M., Xie L., Zhang J., Yu D.-H., Murray C., and Smith P. D. (2012) Pre-clinical targeting of the metabolic phenotype of lymphoma by AZD3965, a selective inhibitor of monocarboxylate transporter 1 (MCT1). In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research, American Association for Cancer Research, Chicago. [Google Scholar]
- Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105. 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Halestrap A. P. (2012) The monocarboxylate transporter family–Structure and functional characterization,. IUBMB Life 64, 1–9. 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- Cheong H.; Lu C.; Lindsten T.; Thompson C. B. (2012) Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 30, 671–678. 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovens M. J.; Davies A. J.; Wilson M. C.; Murray C. M.; Halestrap A. P. (2010) AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem. J. 425, 523–530. 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Påhlman C.; Qi Z.; Murray C. M.; Ferguson D.; Bundick R. V.; Donald D. K.; Ekberg H. (2013) Immunosuppressive properties of a series of novel inhibitors of the monocarboxylate transporter MCT-1. Transplant Int. 26, 22–29. 10.1111/j.1432-2277.2012.01579.x. [DOI] [PubMed] [Google Scholar]
- http://science.cancerresearchuk.org/ (accessed May 4, 2018).
- Polański R.; Hodgkinson C. L.; Fusi A.; Nonaka D.; Priest L.; Kelly P.; Trapani F.; Bishop P. W.; White A.; Critchlow S. E.; Smith P. D.; Blackhall F.; Dive C.; Morrow C. J. (2014) Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 20, 926–937. 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröer S.; Schneider H. P.; Bröer A.; Rahman B.; Hamprecht B.; Deitmer J. W. (1998) Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 333 (Part 1), 167–174. 10.1042/bj3330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J.; McIlvain H. B.; Carrick T. A.; Jow B.; Lu Q.; Kowal D.; Lin S.; Greenfield A.; Grosanu C.; Fan K.; Petroski R.; Williams J.; Foster A.; Butera J. (2005) Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol. Pharmacol. 68, 974–982. 10.1124/mol.105.012005. [DOI] [PubMed] [Google Scholar]
- Ye R.; Rhoderick J. F.; Thompson C. M.; Bridges R. J. (2010) Functional expression, purification and high sequence coverage mass spectrometric characterization of human excitatory amino acid transporter EAAT2. Protein Expression Purif. 74, 49–59. 10.1016/j.pep.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendreau S.; Voswinkel S.; Torres-Salazar D.; Lang N.; Heidtmann H.; Detro-Dassen S.; Schmalzing G.; Hidalgo P.; Fahlke C. (2004) A trimeric quaternary structure is conserved in bacterial and human glutamate transporters,. J. Biol. Chem. 279, 39505–39512. 10.1074/jbc.M408038200. [DOI] [PubMed] [Google Scholar]
- Piazza I.; Kochanowski K.; Cappelletti V.; Fuhrer T.; Noor E.; Sauer U.; Picotti P. (2018) A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication,. Cell 172, 358–372. 10.1016/j.cell.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Rebsamen M.; Pochini L.; Stasyk T.; de Araújo M. E.; Galluccio M.; Kandasamy R. K.; Snijder B.; Fauster A.; Rudashevskaya E. L.; Bruckner M.; Scorzoni S.; Filipek P. A.; Huber K. V.; Bigenzahn J. W.; Heinz L. X.; Kraft C.; Bennett K. L.; Indiveri C.; Huber L. A.; Superti-Furga G. (2015) SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481. 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.