

ABSTRACT
Naringin, a natural occurring flavonoid compound, enriches in citrus fruits. We aimed to evaluate the inhibitory effect of naringin on colitis and chronic inflammation-driven carcinogenesis. Male C57BL/6 mice were exposed to AOM/DSS to induce colorectal inflammation and carcinogenesis. Naringin by oral administration prevented AOM/DSS-induced ulcerative colitis and carcinogenesis without significant side effects. Naringin attenuated the severity of colitis and colorectal adenomas through inhibiting myeloid-derived suppressor cells (MDSCs), pro-inflammatory mediators GM-CSF/M-CSF, IL-6 and TNF-α and the NF-κB/IL-6/STAT3 cascades in colorectal tissues. Naringin-treated mice exhibited normalized structures of colorectal tissues. Electron microscopy analysis showed the suppression of robust endoplasmic reticulum (ER) stress-induced autophagy. Naringin inhibited the secretion of the ER-spanning transmembrane proteins, such as GRP78 ATF6, IRE1α and activated PERK phosphorylated eIF-2α and complex of autophagosomes ATG3, ATG5, ATG7, ATG12, ATG16 and ATG16L1 in the colorectal mucosal cells. Conclusion: Naringin prevented colitis and colorectal carcinogenesis through suppressing robust ER stress-induced autophagy in colorectal mucosal cells. Naringin could develop a promising therapeutic agent for the prevention of ulcerative colitis and colorectal tumor.
KEYWORDS: Colitis, colorectal carcinogenesis, Endoplasmic reticulum (ER) stress, autophagy, MDSCs, Naringin, Aspirin
Introduction
Inflammatory bowel disease (IBD) has been considered a major risk of developing colorectal cancer (CRC). IBD, including ulcerative colitis and Crohn's disease, is a chronic, relapsing, and remitting inflammatory condition in colorectal tract. CRCs are known to develop through a series of histological events, called the ‘‘adenoma-carcinoma’’ sequence.1,2 Clinical studies showed that patients with IBD exhibiting a 2- to 8-fold higher risk of CRC.3,4 Many factors, such as the environment, imbalance of bacterial flora, and diet, such as low fruit and vegetable consumption, as well as high intake of red meat – could increase the risk of IBD.5 Although the exact mechanism of IBD remains unknown, the pathogenic paradigms have highlighted that the interactions between various constituents of the innate and adaptive immune systems play key roles in the pathogenesis of IBD. It is accepted that during IBD, there are various activated myeloid-derived cells in which there is a vicious cycle of tissue destruction and repair due to either irremovable injurious stimuli or a dysfunction. Myeloid-derived cells include monocytes, macrophages, myeloid-derived suppressor cells (MDSCs), neutrophils, and dendritic cells (DCs). In particular, MDSCs which possessed immunosuppressive function was widely studied in IBD and colorectal tumor.6 Once activated, MDSCs mediate suppression of T-cell functions through upregulation of immune suppressive factors such as arginase and inducible nitric oxide synthase (iNOS) and increase production of ROS.6 MDSCs produce various pro-inflammatory mediators such as GM-CSF and M-CSF, IL-6, Il-10 and TNF-α to further exacerbate their immune suppressive activity. MDSCs also amplify the NF-κB/IL-6/STAT3 signaling pathway in the pathogenesis of IBD and colorectal tumor.7 Regardless of IBD pathogenesis, chronic inflammation are known to account for many malignancies, as they can induce genomic instability, inhibition of apoptosis or tumor suppressors, modulation of cell proliferation, and angiogenesis.6,7 These cancer-related inflammations represent the targets for designing innovative therapeutic strategies to prevent chronic inflammation and inflammation-driven carcinogenesis.
There is no cure for IBD. Current therapeutics e.g., corticosteroids, immunomodulators, antibiotics, aminosalicylates, and biologic therapies are aimed at inducing and maintaining the remission of diseases by modifying chronic inflammatory processes.8 Moreover, the results of these drugs are highly variable, as response to the treatment often diminishes over time, resulting in disease complications. Although aspirin produces regression of existing colorectal adenomas and prevents formation of new polyps, the lowest effective doses, treatment duration, target populations, and the effects on patient survival are not entirely clear. Moreover, prolonged aspirin treatment could induce haemorrhage, especially in the gastrointestinal tract.9 Therefore, attempts have been made to find drugs targeting the IBD for reducing the risk of colorectal cancer.
A promising source of chemoprevention agents is traditional medicine derived from natural compounds. It is well known that polyphenolic flavonoids possess diverse biological activities including anti-inflammatory and anti-carcinogenesis effects owing to their specific structural features. Of note, naringin, 4′,5,7-trihydroxy avanone-7-rhamnoglucoside, a major flavanone glycoside in citrus species, has drawn growing attention in for its various biological properties.10 In mammals, naringin is broken down by intestinal microflora into its aglycone naringenin, which is rapidly metabolized in the liver into the glucuronide intermediates. Both naringin and naringenin have been shown to possess strong antioxidant, anti-inflammatory, anti-carcinogenesis potential.11 Naringin could protect neural cells on 3-nitropropionic acid-induced mitochondrial dysfunction through modulating the Nrf2 signaling pathway.12 Naringin inhibited autophagy-mediated gastric adenocarcinoma cell growth by downregulating the PI3K/Akt/mTOR cascade via activation of the MAPK pathway.13 Naringin also prevented cisplatin-induced oxidative stress, inflammatory response and apoptosis in rat striatum via suppressing the ROS-mediated NF-κB and p53 signaling pathways.14 Naringin inhibited the growth of hepatocellular carcinoma HepG2 cells and DU145 prostate cancer cells through inducting apoptosis.15,16 In our group, we found that naringin could prevent intestinal adenomas through modulating the GSK-3β and APC/β-catenin pathways in APCMin/+ mice.17 Our continuous study aimed to investigate the inhibitory effects of naringin on AOM/DSS-induced colorectal inflammation and carcinogenesis in C57BL/6 mice. Naringin by oral administration exhibited a strong chemopreventative effect on ulcerative colitis and colorectal adenomas without significant side effects. We suggest that naringin could be developed a potential chemopreventive agent for reducing the risk of colonic cancer.
Results
Naringin prevented AOM/DSS-induced ulcerative colitis and colorectal carcinogenesis
Mice received AOM/DSS exhibited the symptoms of ulcerative colitis such as diarrhea, hair bristling, anal prolapses due to colorectal ulcerative inflammation. In model mice, body weight loss started from d 6 (Fig. 1A). Bloody stools were also seen from d 6. The severity of disease as scored by DAI reached highest level on d 7 after third DSS cycle (Fig. 1B). In contrast, naringin strongly prevented the symptoms of ulcerative colitis (i.e., body weight loss, diarrhea, and rectal bleeding). Naringin by 50 and 100 mg/kg significantly reduced the DAI scores (P < 0.01 vs. model mice), demonstrating the prevention of bloody stools (P < 0.01 vs. model mice) and body weight loss (P < 0.05 vs. model mice). Aspirin also inhibited AOM/DSS-induced DAI score (P < 0.05 vs. model mice) but did not prevent body weight loss and bloody stools (P > 0.05 vs. model mice).
Figure 1.
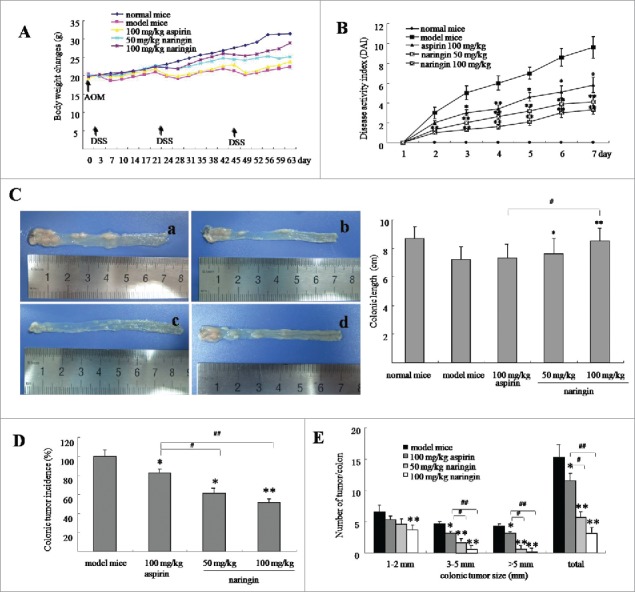
Naringin attenuated colorectal inflammation and adenomas in mice. (A) Body weight changes of mice during AOM/DSS-induced colitis and carcinogenesis. (B) Naringin attenuated colorectal inflammation expressed as reduced DAI after third DSS cycle. (C) Gross view of colorectal tissues and adenomas (left). C-a. model mice. C-b. naringin 50 mg/kg. C-c. naringin 100 mg/kg. C-d. aspirin 100 mg/kg. Naringin prevented shortening of colonic length (right). (D) Naringin prevented the incidence of colorectal adenomas. (E) Tumor size distribution in mice model and drug-treated mice. Each dot represents ten mice (n = 10). *P < 0.05, **P < 0.01 between model mice and drug-treated mice. #P < 0.05, ##P < 0.01 between naringin and aspirin.
Colorectal ulcerative inflammation led to shortening of colorectal length (Fig. 1C). Average colorectal length was 8.8 cm in normal mice. AOM/DSS induced shortening of colorectal length to 7.1 cm in model mice (Fig. 1C–a, P < 0.05 vs. normal mice). Naringin prevented shortening of colorectal length, showing average length was 7.4 cm (Fig. 1C–b, 50 mg/kg, P < 0.05 vs. model mice) and 8.5 cm (Fig. 1C–c, 100 mg/kg, P < 0.01 vs. model mice). Aspirin did not significantly prevent shortening of colorectal length (Fig. 1C–d, P < 0.05 vs. model mice).
AOM/DSS induced colorectal neoplasms by 100% in model mice, whereas by 64.3% and 51.4%, respectively, in naringin-treated mice by 50 mg/kg (Fig. 1D, P < 0.05 vs. model mice) and 100 mg/kg (Fig. 1D, P < 0.01 vs. model mice). Further, model mice developed larger numbers and sizes of colorectal neoplasms as compared to naringin-treated mice (Fig. 1D, P < 0.01 vs. model mice). Although naringin and aspirin both presented the inhibition of colorectal neoplasms, naringin demonstrated higher efficacy than aspirin (Fig. 1D, P < 0.05). Larger sized cancers (>5 mm) were observed in aspirin-treated mice, but not in naringin-treated mice (Fig. 1E, P < 0.05).
Pathology analysis showed that AOM/DSS induced severe inflammatory lesions in the colorectal tissues, showing different degrees of tissues structural changes, such as swelling and degeneration of epithelial cells, extensive denudation and collapse of villi, surface erosion with exuberant inflammatory exudate, patchy reepithelization, lamina propria fibrosis with inflammatory infiltrate and submucosa edema (Fig. 2A–b). In contrast, naringin-treated mice presented less severity of inflammatory lesions, demonstrating the integrity and normal structure of colorectal tissues (Fig. 2A–c,d). Aspirin achieved a mild inhibitory effect on chronic inflammation (Fig. 2A–e). The inhibitory effect of naringin and aspirin on inflammation was recorded by determining the inflammation scores as showed in Fig. 2B (P < 0.01 between naringin and model mice; P < 0.05 between naringin and aspirin).
Figure 2.
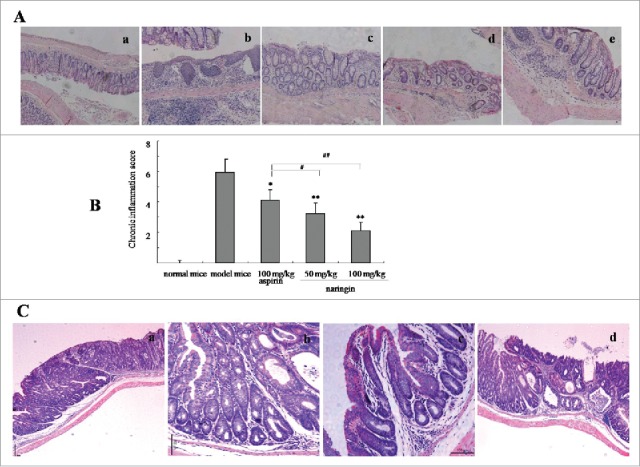
H&E-stained colorectal sections from model mice and drug-treated mice (50× ). (A) Naringin prevented colorectal inflammation. A-a. normal mice; A-b. model mice; A-c. Naringin with 50 mg/kg treated mice; A-d. Naringin with 100 mg/kg treated mice; A-e. Aspirin-treated mice. (B) Naringin reduced colitis evaluated by inflammation score. *P < 0.05, **P < 0.01 between model mice and drug-treated mice. #P < 0.05, ##P < 0.01 between naringin and aspirin. (C) Naringin prevented inflammation-driven colorectal adenomas. C-a. model mice; C-b. Naringin 50 mg/kg; C-c. Naringin 100 mg/kg; B-d. Aspirin 100 mg/kg.
Further analysis revealed that AOM/DSS-induced colorectal neoplasms were histologically determined as adenoma. In model mice, colorectal tissues presented advanced tubular adenoma with focal high grade dysplasia marked nuclear pleomorphism, lack of nuclear polarity, frequent mitoses and architectural distortion. Gross ulceration was frequently seen in large-sized adenomas (Fig. 2C–a). Naringin strongly inhibited colorectal adenomas, showing attenuated dysplasia and mitoses, less architectural distortion and fair restore effects on colonic tissue structures (Fig. 2C–b,c). In addition to attenuated inflammation, aspirin did not show high quality evidence of preventing effect on colonic tissue lesions (Fig. 2C,d).
Naringin prevented AOM/DSS-induced infiltration of MDSCs and inflammatory mediators in the intraepithelial and intraepithelial propria of colon tissues
During colitis, MDSCs was infiltrated, while CD4+ and CD8+ T lymphocytes and macrophages were decreased in the intraepithelial and intraepithelial propria of colon tissues.18 We used flow cytometry assay to analyze the profiles of these immunocytes in AOM/DSS-treated mice. MDSCs are defined as CD11b+Gr-1+ cells that are further divided into 2 subsets, monocytic (CD11b+Ly6C+ defined as M-MDSCs) and granulocytic (CD11b+Ly6Ghigh defined as G-MDSCs) cells.18 Naringin significantly prevented the infiltration of M-MDSCs (Fig. 3A, P < 0.01 vs. model mice) and G-MDSCs (Fig. 3B, P < 0.01 vs. model mice). As expected, an increase of CD4+ (Fig. 3C, P < 0.05 vs. model mice) and CD8+ T lymphocytes (Fig. 3D, P < 0.01 vs. model mice) was seen in intraepithelial and intraepithelial propria. Macrophagys identified by CD11b+F4/80 and CD11b were also increased in intraepithelial and intraepithelial propria of colonic tissues (Fig. 3E, P < 0.05 vs. model mice). Aspirin did not significantly prevent the infiltration of G-MDSCs and M-MDSCs (P > 0.05 between aspirin and model mice). The increases of lymphocytes CD4+ and CD8+ T and macrophages were not significantly in aspirin-treated mice (P > 0.05 between aspirin and model mice).
Figure 3.

Flow cytometry analyzed the profiles of immunocytes in the intraepithelial and intraepithelial propria of colon tissues. (A) Naringin prevented the infiltration of M-MDSCs as determined by anti-CD11b+Ly6C+. A-a. normal mice. A-b. model mice. A-c. Naringin 50 mg/kg; A-d. Naringin 100 mg/kg; A-e. Aspirin 100 mg/kg. (B) Naringin prevented the infiltration of G-MDSCs as determined by anti-CD11b+Ly6G+. B-a. normal mice. B-b. model mice. B-c. Naringin 50 mg/kg; B-d. Naringin 100 mg/kg; B-e. Aspirin 100 mg/kg. (C) Naringin increased CD4+ T lymphocytes as determined by anti-CD3+/CD4+. C-a. normal mice. C-b. model mice. C-c. Naringin 50 mg/kg; C-d. Naringin 100 mg/kg; C-e. Aspirin 100 mg/kg. (D) Naringin increased CD8+ T lymphocytes as determined by anti-CD3+/CD8+. (E) Naringin increased macrophages as determined by anti-CD11b+F4/80+. E-a. normal mice. E-b. model mice. E-c. Naringin 50 mg/kg; E-d. Naringin 100 mg/kg; E-e. Aspirin 100 mg/kg.
ELISA assay showed high level of GM-CSF and M-CSF in the supernatant of colonic tissues in model mice. Naringin significantly reduced the levels of GM-CSF (Fig. 4A, P < 0.01 vs. model mice) and M-CSF (Fig. 4B, P < 0.01 vs. model mice). Since myeloid cells are a major source of IL-6 and TNF-α in immunosuppressive function of MDSCs,18,19 we thus determined high levels of IL-6 and TNF-α in model mice. As expected, inhibition of IL-6 (Fig. 4C, P < 0.05 vs. model mice) and TNF-α (Fig. 4D, P < 0.05 vs. model mice) was observed in naringin-treated mice. Aspirin treatment led to an inhibition of IL-6 (P < 0.05 vs. model mice) and TNF-α (P < 0.05 vs. model mice), but not GM-CSF (P > 0.05 vs. model mice) and M-CSF (P < 0.05 vs. model mice).
Figure 4.
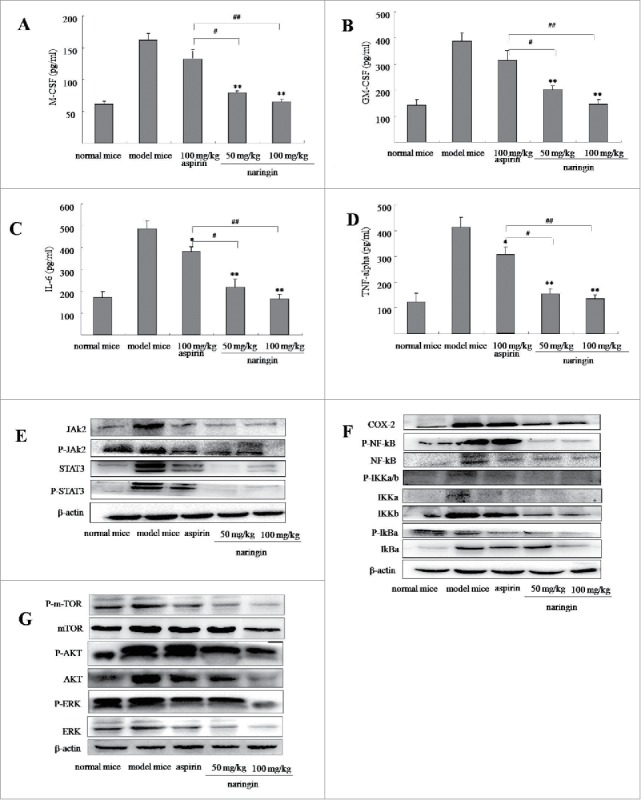
Naringin reduced inflammatory mediator in colorectal inflamed mucosa and colorectal adenomas. (A) ELISA analyzed concentration of M-CSF in colorectal mucosa and adenomas. (B) Naringin reduced GM-CSF in colorectal mucosa and adenomas. (C) Naringin reduced IL-6 in colorectal mucosa and adenomas. (D) Naringin reduced TNF-α in colorectal mucosa and adenomas. Each dot represents eight mice (n = 10). *P < 0.05, **P < 0.01 between model mice and drug-treated mice. #P < 0.05, ##P < 0.01 between naringin and aspirin. (E) and (F) Western blotting analyzed levels of JAK2/STAT3 and COX-2/NF-κB cascades in colorectal mucosa and adenomas.
During colitis and tumor formation, activated UPR pathway augmented STAT3 and activated nuclear factor-κB (NF-κB) through activation of phosphorylated m-TOR pathway.20 Western blotting showed that naringin-treated mice showed suppressed STAT3 (Fig. 4E), NF-κB signaling cascades (Fig. 4F) and phosphorylated m-TOR pathway (Fig. 4G). Aspirin showed a mild effect on the phosphorylated m-TOR-induced activation of STAT3 and NF-κB signaling pathways (Fig. 4E–G).
Naringin prevented AOM/DSS-induced robust ER-stress-mediated autophagy in colonic mucosal cells
In colitis-associated carcinoma model, continuous AOM/DSS induced misfolded proteins beyond correction effects, leading to robust ER stress enhancing cell survival through sustaining autophagy. Autophagy could support cells to adapt the ER stress-induced hostile environment, leading to colitis and adenomas.21 Electron microscopy analyzed the ultrastructure of ER lumen, showing the ER lumen was expanded with numerous swellings and vacuole-like structures. Most of ribosomes attached ER and free ribosomes were disappeared (Fig. 5A). Naringin-treated mice exhibited the integrity of ER structure, showing continuous ER and normal structures with extensive parallel membranes. Ribosomes were frequently seen attached to the ER as well as free in the cytoplasm. These results indicated the restoring effect of naringin on the ER structure. The restoring effect of naringin on the ER was more visible than aspirin (Fig. 5A).
Figure 5.

Naringin inhibited robust ER-stress-mediated autophagy in colorectal mucosal cells. (A) Electron microscopy analyzed the ultrastructures of ER, indicating ER expansion with double-membrane structures. Naringin attenuated ER expansion. (B) Electron microscopy showed double membrane autophagy vesicles in colorectal mucosal epithelial cells. Autophagy vesicles were reduced in naringin-treated mice. (C) AOM/DSS induced robust ER stress as evidenced by high expressions of GRP78, IRE1α, PERK and ATF6α. Naringin inhibited the expression of GRP78, IRE1α, PERK and ATF6α in colorectal mucosal cells. (D) Naringin prevented the formation of robust ER stress induced-ATG16L1 complex in colorectal mucosal cells. (E) Naringin reduced ER stress-induced high level of LC3B and Bcelin-1 in colorectal mucosal cells.
Electron microscopy further analyzed robust ER stress-induced autophagy. Double membrane autophagy vesicles were frequently observed in model mice, whereas few autophagy vesicles were seen in naringin-treated mice (Fig. 5B). Aspirin had mild inhibitory effect on robust ER stress-autophagy vesicles (Fig. 5B).
Western blotting analysis of the ER-resident sensors showed that naringin suppressed the secretion of major ER-spanning transmembrane proteins, including GRP78 ATF6, IRE1α and activated PERK phosphorylated eIF-2α. Naringin strongly prevented robust ER stress-induced GRP78, ATF6α, phosphorylated eIF-2α and IRE1α. Statistical analysis indicated that naringin had higher inhibitory effects on robust ER stress-induced secretion of ER-resident sensors than aspirin (Fig. 5C). Upon phagophore, a conjugate of ATG3, ATG5, ATG7, ATG12, ATG16 may multimerize with ATG16L1 to form a complex of autophagosomes and then lipidates LC3 (LC3-II).22 Naringin significantly inhibited the formation of the autophagosomes (Fig. 5D), thus, leading to the inhibition of autophagy in the inflamed colonic epithelial cells and adenoma cells. Because AOM/DSS induced colorectal adenoma in 100% of mice, we thus suggest that autophagy is mostly occurred in the adenoma cells. Naringin inhibited the ER-stress-mediated autophagy, thus preventing inflammation-driven colorectal adenoma.
Discussion
Chemoprevention is a rational and appealing approach to reduce the risk of colorectal cancer by using pharmacologic means, such as aspirin. Aspirin has long been considered the role in the chemoprevention of colorectal cancer. In APCmin/+ mouse model, aspirin prevented 44% tumor formation at a dosage of 80 mg/kg.23 However, its clinical efficacy in the prevention of human CRC has remained controversial.9 Can we select patients for colorectal cancer prevention with aspirin? Rothwell published a 20 y follow-up of 5 RCTs (Randomised controlled trials) which supported the role of low dose aspirin in reducing 30–40% colorectal cancer incidence and mortality, with most benefit for proximal colon cancers.24 However, a report issued from Physicians Health Study (PHS) and Women's Health Study (WHS) showed that the incidence or mortality of colorectal cancer was not seen a significant reduction on short-term follow-up.24 In many cases, it is prohibited due to upper gastrointestinal bleeding, peptic ulceration and haemorrhagic stroke.25 Aspirin also exacerbated asthma, urticaria and angioedema and hepatic dysfunction.26 It has been understood that the ideal chemopreventative agent, in addition to being efficacious in the prevention of cancers, must be easily administered and well tolerated. In this study, naringin prevented AOM/DSS-induced colorectal inflammation and carcinogenesis in mice without significant side effects. Subchronic toxicity study by oral administration at doses of 50, 250 and 1250 mg/kg for 13 weeks did not find any toxicity in SD rats. During subchronic oral toxicity study, there were no mortality and toxicologically significant changes in clinical signs, food consumption, opthalmoscopic examination, hematology, clinical biochemistry, macroscopic findings, organ weights and histopathological examination.27 Pharmacokinetic analysis revealed that naringin has low bioavailability (11%), implying that the prokinetic effect of naringin was largely due to the local activation in the intestine rather than a systemic effect after absorption. This local mode of action might be advantageous for preventing possible systemic side effects since naringin is not well absorbed into the system circuit.28 We thus suggest that the advantage of naringin being superior to aspirin might be its low side effects. Naringin could be developed as a potential therapeutic agent for prevention colitis and reduction the risk of colonic cancers.
Inflammatory tumor microenvironment (ITME) has recently received increasing attention due to its driving roles in the development of cancers. ITME is a complex structure composed of malignant cancer cells surrounded by a stroma containing various inflammatory cells, endothelial cells, and fibroblasts, etc.7 These stromal cells interact amongst themselves and with cancer epithelial cells, directly through cell-cell contacts, and indirectly through paracrine/exocrine signaling, release of cytokines, and chemokines. Among the inflammatory cells, MDSCs which function as potent immune suppressive activity are the major components. It is widely accepted that during colitis, there are various activated immune cells in which there is a vicious cycle of tissue destruction and repair due to either irremovable injurious stimuli or a dysfunction.29 Persistent infiltration of colonic MDSCs could inhibit CD8+ T cells to inhibit adenoma cells and their immunosuppressive functions and is thus considered to promote colitis and adenomas. MDSCs have potent suppressive effects on various T cell responses and macrophages.18 Thus, activated MDSCs and reduced T cells and macrophages together contribute to down-regulation of immune responses in the colonic epithelial barrier. Meanwhile, activated MDSCs suppress T-cell functions through upregulation of many immune suppressive factors which further promotes inflammatory response. Activated MDSCs largely produce GM-CSF and M-CSF.18 In addition, a reciprocal regulation was found between the accumulation of MDSCs and their productions GM-CSF and M-CSF in colonic inflamed mucosa.30 It is thus suggest a central role for GM-CSF and M-CSF in the pathogenesis of colitis, leading to a complex, chronic, relapsing, and remitting inflammatory condition in colon tissues. MDSCs were also a major source of NF-κB, IL-6 and TNF-α which plays a key role in the immunosuppressive function through activating the NF-κB/IL-6/Stat3 pathway which further exacerbates colitis-associated carcinogenesis.18.30 In this study, naringin-treated mice presented strong inhibition on colitis and colorectal carcinogenesis, showing reduced MDSCs and their consequent inflammatory mediators, while increase of CD8+ T and CD4+ T cells and macrophages. In the same time, level of GM-CSF and M-CSF were strongly reduced, thus resulting less severe inflammation. These results demonstrated that naringin prevented colorectal inflammation and adenomas mainly through inhibiting the infiltration of MDSCs and the consequent inflammatory mediators.
It is widely studied that both robust ER stress and activation of UPR are involved in the pathology of colitis and inflammation-driven carcinogenesis. Normally, ER regulates cell-intrinsic and cell-extrinsic stresses to adaptive ER protein-folding machinery readily to handle secretory pathway.20 During colitis, numerous environmental, physiological and pathological insults could disrupt the steady state of ER protein-folding environment to cause misfolding protein beyond tolerable threshold, thus the ER-resident sensors trigger UPR to improve ER protein-folding capacity. Robust ER stress confers survival and advantages to carcinogenesis. In colitis and inflammation-driven carcinogenesis model, continuous AOM/DSS exposure produced robust ER stress, prolonging UPR activation owing to unresolved misfolding protein, leading intestinal epithelial cells to transform tumorigenesis through UPR activation as a survival strategy through autophagy. Under ER stress, epithelial cells might constantly adjust the sizes and shapes of their organelles according to need. In colitis and inflammation-driven carcinogenesis model, robust ER stress induces ER expansion.31,32 On the other hand, expanded ER further alleviates ER stress. In the same time, ER expansion generates a double-membrane structure called phagophore. Upon phagophore, ATG16L1 forms a complex with an ATG5-ATG12 conjugate, which multimerizes and then lipidates LC3 (LC3-II). Autophagy could counterbalance ER expansion during unfolded protein response, facilitates cell survival and transform tumorigenesis.33 Accumulating reports revealed that ER expansion is mainly regulated by three major ER-spanning transmembrane proteins: inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK) and activating transcription factor 6α (ATF6α).34 These sensors exhibit a similar activation mechanism and regulate many unique and overlapping facets of the ER stress response. Each is bound intraluminally to chaperone GRP78 (BiP) which locks them in monomeric, inactive state.34 If misfold proteins exceed the folding capacity of ER-resident chaperones, such as glycosylases and oxido-reductases, GRP78 would dissociate from IRE1α, PERK and ATF6α. These sensors subsequently drive mutually reinforcing signaling pathways to correct the proteins. If the burden can be reduced quickly, cells successfully adapt to the insult for survival through autophagy, whereas insufficient clearance results in apoptotic cell death. In model mice, continuous AOM/DSS induced robust ER stress, thus activated autophagy and inflammation-driven carcinogenesis. Inhibition of UPR and ER stress responses is thus an important therapeutic rationale for colitis and colonic cancer.32-34 Naringin reduced ER-stress-induced autophagy, therefore preventing colitis and inflammation-driven adenomas.
In conclusion, naringin, the naturally food containing component, prevented AOM/DSS-induced mice colorectal inflammation and carcinogenesis. The mechanisms of chemopreventive effects were associated with its multiple biological activities including anti-inflammation, anti-proliferation and prevention of the ER-stress-induced autophagy in colorectal epithelium. Long term use of naringin did not produce considerable adverse effects to mice. We thus suggest that naringin could be developed a therapeutic option for reducing the risk of CRC.
Materials and methods
Chemicals
Naringin (purity ≥ 98%) was purchased from Sigma Chemical Co. (St. Louis, MO). Aspirin was also purchased from Sigma with ≥ 99.0% purity. Naringin and aspirin were dissolved in 5% sodium carboxymethyl cellulose (CMC-Na) (Sigma) before use.
Animal care and diet
Male C57BL/6 mice (6 weeks old) were purchased from Charles River Laboratories (Beijing, China). Mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Capital Medical University. Mice were housed five per cage under specific pathogen-free conditions and kept in an air-conditioned room with controlled temperature (22 ± 1°C), humidity (50%), and d/night cycle (12 h light, 12 h dark). They were fed with standard mouse pellet diet, which contains 52% carbohydrate, 12% fat, 23% protein, 4% fiber, 6% ash, and 3% moisture.35 After one week of acclimation, mice (6 wk old) were administrated either vehicle (CMC-Na 5%, v/v), naringin (50 and 100 mg/kg) or aspirin (100 mg/kg) by p.o. gavage daily (0.2 ml/10 g body weight).
Animal model of AOM/DSS–induced colitis and inflammation-driven carcinogenesis
Forty male mice were divided four groups were then injected intraperitoneally with AOM (10 mg/kg, Sigma) with regular diet and water. Second d after injection of AOM, mice were received 1.0% dextran sulfate sodium (DSS, MP Biomedicals) in drinking water for one week, followed by two weeks of receiving regular drinking water for recovery and then subjected the mice to another 2 cycles DSS treatment. A group of mice did not expose AOM/DSS were served as normal control. An AOM/DSS only group which developed colitis and adenomas was orally administered CMC-Na (0.2 ml/mouse). Two treatment groups were orally administered naringin (50 and 100 mg/kg/d). Aspirin (100 mg/kg/d) was served as a positive control drug. Administration was started one week after AOM injection using an oral Zonde needle for 8 consecutive weeks. Mice were weighed and observed daily for any signs of illness. DSS induced colorectal ulcerative inflammation was scored as disease activity index (DAI) obtained based on weight loss, stool consistency, and bloody excreta as follows: weight loss score = 0: < 1%, 1: 1–5%, 2: 5–10%, 3:10–15%, 4: >15%; stool consistency score = 0: normal, 2: loose, 4: diarrhea; blood in excreta score = 0: normal; 2: reddish, 4: bloody.36
Tissue processing and colonic cancer scoring
Mice were sacrificed after 8 weeks administration and blood was obtained from the postorbital venous plexus of the mice. Whole colorectal tissues were removed and macroscopic tumors were counted and measured with digital calipers under the dissecting microscope based on size (diameter) into 1–2 mm, 3–5 mm and >5 mm. Portions of colonic tissues were either snap-frozen in liquid nitrogen for Western blot analysis or placed in 4% phosphate-buffered formalin for histopathology/immunohistochemical (IHC) analysis.
Preparation of supernatant of colonic mucosa and determination of protein concentration
Fresh colonic tissues were weighed and homogenised in PBS (pH 7.2) at 4˚C. The supernatant of colorectal mucosa was centrifuged and concentration of proteins was quantitatively determined by using BAC 100 protein determination kit (Sigma).
Isolation of colorectal immunocytes from colon tissues and flow cytometry analysis
Colorectal immunocytes were isolated according to previous report.18 Colonic tissues were separated and cut into small pieces (2–3 mm). Intraepithelial immunocytes were separated by incubating colorectal tissues in 0.015% dithioerythritol (Sigma). For isolating immunocytes of lamina propria, the remaining tissue was incubated with EDTA and digested by digestion buffer (RPMI medium containing 5% FBS and 200 units/ml of collagenase (Gibco). Immunocytes were enriched by using discontinuous (44% and 67%) percoll (GE) separation method. Colorectal immunocytes were stained with anti-CD11b+Ly6C+ (101206, 128008, Biolegend) to determine monocytic myeloid-derived suppressor cells (M-MDSCs), anti-CD11b+Ly6G+ (101206, 1127627, Biolegend) to granulocytic myeloid-derived suppressor cells (G-MDSC). Anti-CD3+/CD4+ (100234, 100424, Biolegend) and anti-CD3+/CD8+ (100234, 100730, Biolegend) were used to determine T lymphocytes, anti-CD11b+F4/80+ to determine macrophages.18 Flow cytometric profiles were analyzed by counting 20,000 events using Kaluza software program (Beckman Coulter).
Histopathology analysis
Histopathology was performed according to regular methods.
Enzyme-linked immunosorbent (ELISA) assay
Levels of colonic tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), granulocyte-macrophage colony stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) were quantified by using sandwich ELISA kits, according to manufacturer's protocol. TNF-α kit and IL-6 kit (583361) were purchased from Cayman Chemical. GM-CSF (CSB-E04569m) kit and M-CSF kit (SB-E04659m) were purchased from CUSABIO. The concentrations of these cytokines were normalized to the total protein.
Western blotting assay
Western blotting assay was performed to determine the levels of following proteins prepared from colonic mucosa. Equal amounts of proteins were resolved by SDS-PAGE and transferred to PVDF membranes. After blocking in 5% non fat milk for 1 h, the membranes were immunoblotted overnight at 4°C with following primary antibodies: GRP78 (ab108615, Abcam), ATF6 (65880), eIF2α (3294), P-eIF2α (3398), IRE1α (3294), mTOR (2983), P-mTOR (5536), AKT (9272), P-AKT (9271), ERK (4695), P-ERK (4370), NF-κB (8242), P-NF-κB (3033), Ikkα (11930), Ikkβ (8943), P-Ikkα/β (2697), IκBα (4814), P-IκBα (2859), STAT3 (12640), P-STAT3 (9145), JAK2 (3230), P-JAK2 (3776), COX-2 (12282), ATG3 (3415), ATG5 (12994), ATG7 (8558), ATG12 (4180), ATG16L1 (8089), Beclin-1 (3738), mTor (2983), p-mTor (5536), Akt (9272), p-Akt (9271), Erk (4695), p-Erk (4370), LC3B (L7543) and β-actin (A1978, Sigma). The bound antibodies were visualized using an enhanced chemiluminescence reagent and quantified by densitometry using FluorChem FC3 image analyzer (Molecular Devices). Densitometric analyses of bands were adjusted with β-actin.
Statistical analysis
Data were described as mean ± S.D. Statistical analysis was done with SPSS/Win19.0 software (SPSS, Chicago, IL). Comparison between two groups of mice was conducted by two-tailed Student's t test. The incidence of colorectal neoplasms in naringin-treated mice and aspirin-treated mice was analyzed by comparison with total neoplasms as 100% in model mice. A P value less than 0.05 was considered statistically significant.
Funding Statement
This project was supported by the Shandong Natural Science Foundation (ZR2009CQ019). This project was supported by the Natural Science Foundation of China (91629303).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge and thank teachers and students in professor Qu's group in Capital Medical University.
References
- 1.Danese S, Fiorino G, Peyrin-Biroulet L. Early intervention in Crohn's disease: towards disease modification trials. Gut. 2017;66(12):2179–87. doi: 10.1136/gutjnl-2017-314519. PMID:28874419. [DOI] [PubMed] [Google Scholar]
- 2.Vancamelbeke M, Vanuytsel T, Farré R, Verstockt S, Ferrante M, Van Assche G, Rutgeerts P, Schuit F, Vermeire S, Arijs I, et al.. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23(10):1718–29. doi: 10.1097/MIB.0000000000001246. PMID:28885228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biancone L, Armuzzi A, Scribano ML, D'Inca R, Castiglione F, Papi C, Angelucci E, Daperno M, Mocciaro F, Riegler G, et al.. Inflammatory bowel disease phenotype as risk factor for cancer in a prospective multicentre nested case-control IG-IBD study. J Crohns Colitis. 2016;10(8):913–24. doi: 10.1093/ecco-jcc/jjw048. PMID:26933032. [DOI] [PubMed] [Google Scholar]
- 4.Burisch J, Munkholm P. The epidemiology of inflammatory bowel disease. Scand J Gastroenterol. 2015;50(8):942–51. doi: 10.3109/00365521.2015.1014407. PMID:25687629. [DOI] [PubMed] [Google Scholar]
- 5.Satia JA, Tseng M, Galanko JA, Martin C, Sandler RS. Dietary patterns and colon cancer risk in Whites and African Americans in the North Carolina Colon Cancer Study. Nutr Cancer. 2009;61(2):179–93 doi: 10.1080/01635580802419806. PMID:19235034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leal MC, Däbritz J. Immunoregulatory role of myeloid-derived cells in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(12):2936–47. doi: 10.1097/MIB.0000000000000511. PMID:26244650. [DOI] [PubMed] [Google Scholar]
- 7.Colangelo T, Polcaro G, Muccillo L, D'Agostino G, Rosato V, Ziccardi P, Lupo A, Mazzoccoli G, Sabatino L, Colantuoni V. The tumour microenvironment dilemma in colorectal cancer. Biochimica et Biophysica Acta. 2017;1867(1):1–18. PMID:27864070. [DOI] [PubMed] [Google Scholar]
- 8.Kraus S, Sion D, Arber N. Can we select patients for colorectal cancer prevention with aspirin? Curr Pharm Des. 2015;21(35):5127–34. doi: 10.2174/1381612821666150915111000. PMID:26369678. [DOI] [PubMed] [Google Scholar]
- 9.Dubinsky MC. Reviewing treatments and outcomes in the evolving landscape of ulcerative colitis. Postgrad Med. 2017;129(5):538–53. doi: 10.1080/00325481.2017.1319730. PMID:28425825. [DOI] [PubMed] [Google Scholar]
- 10.Bharti S, Rani N, Krishnamurthy B, Arya DS. Preclinical evidence for the pharmacological actions of naringin: a review. Planta Med. 2014;80(6):437–51. doi: 10.1055/s-0034-1368351. PMID:24710903. [DOI] [PubMed] [Google Scholar]
- 11.Alam MA, Subhan N, Rahman MM, Uddin SJ, Reza HM, Sarker SD. Effect of citrus flavonoids, naringin and naringenin, on metabolic syndrome and their mechanisms of action. Adv Nutr. 2014;5(4):404–17. doi: 10.3945/an.113.005603. PMID:25022990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulasekaran G, Ganapasam S. Neuroprotective efficacy of naringin on 3-nitropropionic acid-induced mitochondrial dysfunction through the modulation of Nrf2 signaling pathway in PC12 cells. Mol Cell Biochem. 2015;409(1-2):199–211. doi: 10.1007/s11010-015-2525-9. PMID:26280522. [DOI] [PubMed] [Google Scholar]
- 13.Raha S, Yumnam S, Hong GE, Lee HJ, Saralamma VV, Park HS, Heo JD, Lee SJ, Kim EH, Kim JA, et al.. Naringin induces autophagy-mediated growth inhibition by downregulating the PI3K/Akt/mTOR cascade via activation of MAPK pathways in AGS cancer cells. Int J Oncol. 2015;47(3):1061–9. doi: 10.3892/ijo.2015.3095. PMID:26201693. [DOI] [PubMed] [Google Scholar]
- 14.Chtourou Y, Aouey B, Kebieche M, Fetoui H. Protective role of naringin against cisplatin induced oxidative stress, inflammatory response and apoptosis in rat striatum via suppressing ROS-mediated NF-κB and P53 signaling pathways. Chem Biol Interact. 2015;239:76–86. doi: 10.1016/j.cbi.2015.06.036. PMID:26120027. [DOI] [PubMed] [Google Scholar]
- 15.Banjerdpongchai R, Wudtiwai B, Khaw-On P, Rachakhom W, Duangnil N, Kongtawelert P. Hesperidin from Citrus seed induces human hepatocellular carcinoma HepG2 cell apoptosis via both mitochondrial and death receptor pathways. Tumour Biol. 2015;37(1):227–37. doi: 10.1007/s13277-015-3774-7. PMID:26194866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewinska A, Siwak J, Rzeszutek I, Wnuk M. Diosmin induces genotoxicity and apoptosis in DU145 prostate cancer cell line. Toxicol In Vitro. 2015;29(3):417–25. doi: 10.1016/j.tiv.2014.12.005. PMID:25499067. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YS, Li Y, Wang Y, Sun SY, Jiang T, Li C, Cui SX, Qu XJ. Naringin, a natural dietary compound, prevents intestinal tumorigenesis in Apc (Min/+) mouse model. J Cancer Res Clin Oncol. 2016;142(5):913–25. doi: 10.1007/s00432-015-2097-9. PMID:26702935. [DOI] [PubMed] [Google Scholar]
- 18.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24(5):631–44. doi: 10.1016/j.ccr.2013.10.009. PMID:24229710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9(3):162–74. doi: 10.1038/nri2506. PMID:19197294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017;168(4):692–706. doi: 10.1016/j.cell.2016.12.004. PMID:28187289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, Van Assche G, Agostinis P, Rutgeerts P, Vermeire S, et al.. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy. 2013;9(12):2046–55. doi: 10.4161/auto.26337. PMID:24247223. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Chen M, Yang J, Zhang Z. LC3 fluorescent puncta in autophagosomes or in protein aggregates can be distinguished by FRAP analysis in living cells. Autophagy 2013;9(5):756–69. doi: 10.4161/auto.23814. PMID:23482084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JM, Kanaoka Y, Eguchi N, Aritake K, Grujic S, Materi AM, Buslon VS, Tippin BL, Kwong AM, Salido E, et al.. Hematopoietic prostaglandin D synthase suppresses intestinal adenomas in ApcMin/+ mice. Cancer Res. 2007;67(3):881–9. doi: 10.1158/0008-5472.CAN-05-3767. PMID:17283118. [DOI] [PubMed] [Google Scholar]
- 24.Smith T, Hutchison P, Schrör K, Clària J, Lanas A, Patrignani P, Chan AT, Din F, Langley R, Elwood P, et al.. Aspirin in the 21st century-common mechanisms of disease and their modulation by aspirin: a report from the 2015 scientific conference of the international aspirin foundation, 28 August, London, UK. Ecancermedicalscience. 2015;9:581. PMID:26557879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiagarajan P, Jankowski JA. Aspirin and NSAIDs; benefits and harms for the gut. Best Pract Res Clin Gastroenterol. 2012;26(2):197–206. doi: 10.1016/j.bpg.2012.01.007. PMID:22542157. [DOI] [PubMed] [Google Scholar]
- 26.Singh Ranger G. The role of aspirin in colorectal cancer chemoprevention. Crit Rev Oncol Hematol. 2016;104:87–90. doi: 10.1016/j.critrevonc.2016.05.011. PMID:27289249. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Wang S, Guan X, Liu B, Wang Y, Xu K, Peng W, Su W, Zhang K. Acute and 13 weeks subchronic toxicological evaluation of naringin in Sprague-Dawley rats. Food Chem Toxicol 2013;60:1–9. doi: 10.1016/j.fct.2013.07.019. PMID:23871784. [DOI] [PubMed] [Google Scholar]
- 28.Jang Y, Kim TK, Shim WS. Naringin exhibits in vivo prokinetic activity via activation of ghrelin receptor in gastrointestinal motility dysfunction rats. Pharmacology. 2013;92(3-4):191–7. doi: 10.1159/000354579. PMID:24080610. [DOI] [PubMed] [Google Scholar]
- 29.Kontaki E, Boumpas DT, Tzardi M, Mouzas IA, Papadakis KA, Verginis P. Aberrant function of myeloid-derived suppressor cells (MDSCs) in experimental colitis and in inflammatory bowel disease (IBD) immune responses. Autoimmunity. 2017;50(3):170–81. doi: 10.1080/08916934.2017.1283405. PMID:28276713. [DOI] [PubMed] [Google Scholar]
- 30.Ma N, Liu Q, Hou L, Wang Y, Liu Z. MDSCs are involved in the protumorigenic potentials of GM-CSF in colitis-associated cancer. Int J Immunopathol Pharmacol. 2017;30(2):152–62. doi: 10.1177/0394632017711055. PMID:28534709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharifi MN, Mowers EE, Drake LE, Macleod KF. Measuring autophagy in stressed cells. Methods Mol Biol. 2015;1292:129–50. doi: 10.1007/978-1-4939-2522-3_10. PMID:25804753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han F, Zhang H, Xia X, Xiong H, Song D, Zong X, Wang Y. Porcine β-defensin 2 attenuates inflammation and mucosal lesions in dextran sodium sulfate-induced colitis. J Immunol. 2015;194(4):1882–93. doi: 10.4049/jimmunol.1402300. PMID:25601921. [DOI] [PubMed] [Google Scholar]
- 33.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013;110(12):4628–33. doi: 10.1073/pnas.1217611110. PMID:23487760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.George Z, Omosun Y, Azenabor AA, Partin J, Joseph K, Ellerson D, He Q, Eko F, Bandea C, Svoboda P, et al.. The roles of unfolded protein response pathways in chlamydia pathogenesis. J Infect Dis. 2017;215(3):456–65. PMID:27932618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meidenbauer JJ, Ta N, Seyfried TN. Influence of a ketogenic diet, fish-oil, and calorie restriction on plasma metabolites and lipids in C57BL/6J mice. Nutr Metab (Lond). 2014;11:23. doi: 10.1186/1743-7075-11-23. PMID:24910707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69(2):238–49. PMID:8350599. [PubMed] [Google Scholar]