

ABSTRACT
Tumors represent a dynamic system where the genomic plasticity permits to adapt to the perturbation induced by environmental pressures, supporting the importance of longitudinal tumor sampling strategies to deciphering the temporal acquisition of driver event that could impact treatment outcome. We describe the case of a metastatic colorectal cancer (mCRC) patient, RAS wild-type, who responded to anti-EGFR therapy and underwent liver surgery, revealing a KRAS mutations in the metastatic lesion, not detectable prior to initiation of therapy in the colonic biopsy. After liver surgery, the patient received chemotherapy alone, then underwent left colectomy and the final pathological report confirmed the KRAS wild-type status. We can speculate the existence of two distinct populations of KRAS wild-type and mutant CRC cells sharing the same genetic origin. The anti-EGFR treatment represented a selective pressure which allowed the selection of KRAS mutant subclones. The prognostic and /or predictive role of intratumor heterogeneity has not been assessed prospectively. Our case report is of clinical relevance because patients with mCRC who respond to anti-EGFR antibodies often develop resistance within several months of initiating therapy, thus outlining the importance to better ascertain the molecular landscape of tumors to design better therapeutic strategies.
KEYWORDS: Colorectal cancer, Cancer Biology, Heterogeneity, Mutation, Liver metastases, RAS, Target therapy, Treatment resistance, Tumor Metastases
Introduction
Mutations in KRAS or NRAS occur in ∼50% of patients with metastatic colorectal cancer (mCRC) and are negative predictive indicator for treatment with anti-EGFR antibodies (moAbs).1
Chemotherapy doublets in association with anti-EGFR moAbs are commonly used showing a response rate of 55%–65%, thus allowing dramatic tumor downstaging and the possibility to convert the borderline resectable disease to a resectable one.
However, even in the wild-type RAS mCRC setting, 35%–45% of patients exhibit resistance and don't respond to this treatment.2 Mutations in KRAS and NRAS represent the most common molecular mechanisms driving resistance to anti-EGFR therapy.3 Nonetheless, resistance to EGFR blockade is caused by several factors and several molecular predictors have been intensively investigated. Primary, we have to distinguish intrinsic resistance, that is related to constitutive activation of signal pathways downstream of EGFR, from acquired resistance that occurs during the anti-EGFR treatment and could be driven by emerging mutations disrupting the binding of anti-EGFR moAbs to EGFR itself, by so called “pathway-bypass” mutations (for examples in RAS and BRAF genes), or by the activation of parallel pathways (such as MET and HER2 signaling pathways).1,3
According to recent studies, epigenetic factors also play an important role in the development of anti-EGFR moAbs resistance, since they regulate gene expression at the posttranscriptional level and are involved in different signaling pathways.4
These data suggest that tumors represent a dynamic system where the plasticity of their genome permits to adapt to the perturbation induced by environmental pressures, such as the anti-EGFR treatment. The extent of intratumor heterogeneity has only recently been recognized and its prognostic and/or predictive role has not been assessed prospectively.
Here we report a clinical case that describes how intratumor heterogeneity and subclonal diversity might contribute to the limitations of targeted therapies and how it can be leveraged to better understand the evolutionary history of a tumor and to optimize treatment.
Clinical case report
We report the case of a 73-y ear-old Caucasian, well-nourished woman, no smoker, with a history of hypertension and familiar history for cancer: a maternal aunt died for pancreatic cancer and two other maternal aunts had a history of breast cancer. The patient sought our medical attention in September 2015 following a positive fecal occult blood test (132 ng/mL). No contributory medical history was elicited. On physical examination the abdomen was bulging on palpation, with tenderness of the left pelvic fossa. Lab tests, including blood count, liver and renal function, were within normal limits. Tumor marker CEA was 30 ng/ml. Complete colonoscopy revealed an excavated lesion located at about 28 cm from the anal rhyme, with an extension up to 35 cm, ulcerated, occupying about 2/3 of the lumen. Pathological findings were consistent with a diagnosis of adenocarcinoma. We additionally completed the entire mutation status of RAS (KRAS and NRAS in exons 1, 2, 3, and 4) and BRAF (V600E): no mutations in any of the genes analyzed were found. A thoraco-abdominal CT scan confirmed endoscopy findings and revealed multiple and bilateral repetitive lesions varying in size from a maximum of 3.5 cm to a few millimeters. The case was discussed during the weekly multidisciplinary team meeting (MDT) on liver cancer, the patient was considered borderline resectable from our team of surgeons, and preoperative chemotherapy was recommended. From October 2015 to December 2016 the Patient underwent 4 cycle of chemotherapy with FOLFOX regimen + panitumumab. The radiological evaluation performed in December 2016 revealed a partial response, according to RECIST criteria v1.1, with a concomitant important decrease in the value of CEA (8 ng/ml). Indeed, in February 2016 the Patient underwent ultrasound-guided liver resection of multiple liver lesions (n = 10), with uneventful postoperative course. Pathological findings confirmed the diagnosis of adenocarcinoma, CDX2 +, compatible with liver metastases from colorectal cancer (Fig. 1). Whereas the initial genotyping carried out within the primary colon tumor revealed a RAS wild-type status, we then analyze two of these metastases and unexpectedly we found a mutation in KRAS gene, exon 4 [A146V] in one of them. From March to June 2016 the Patient was treated with 5 more cycle of chemotherapy with FOLFOX regimen without panitumumab. In August 2016, the radiological reassessment showed no extracolic disease, and CEA value was 6 ng/ml and the Patient underwent laparoscopic resection of sigmoid colon without significant morbidity. Pathological diagnosis was G2 adenocarcinoma, with necrosis and microcalcifications, with invasion through muscularis propria into the subserosa, with vascular and lymphatic invasion, and metasteses in 0 out of 21 regional lymph nodes. Surgical resection margins were negatives. Degree of tumor regression according to Dworak classification was 1 (minimum regression). Histopathologic stage (TNM v7) was ypT3N1C. No mutations were found in either RAS or BRAF genes.
Figure 1.
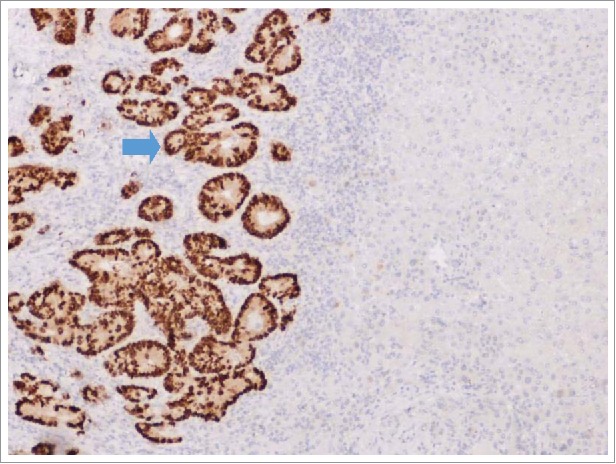
IHC analysis of CDX2 expression performed on formalin-fixed, paraffin-embedded tissue sections of the resected liver metastasis, which revealed a mutation in KRAS gene. IHC: immunohistochemical.
Discussion
In our case, we sequenced the paired primary and metastatic lesions. The metastatic tissue samples best described the advanced disease phenotype, revealing a somatic mutation in KRAS, not detectable prior to initiation of therapy in the colonic biopsy.
We retrospectively compared the radiological response to preoperative therapy and all the lesions (including the one found with KRAS mutation) showed tumor shrinkage (Fig. 2). Though counterintuitive, this is in line with known mechanisms of resistance to targeted agents: even if a resistant mutation is present in a fraction of cells, we might still observe a response to treatment, but we could expect a shorter duration of response, because the resistant clone can act as a reservoir of resistant cells that expand to repopulate the tumor.5 Normanno et al. performed a quantitative estimation of neoplastic cells carrying KRAS, NRAS, BRAF and/or PIK3CA mutations and found that patients with low KRAS mutant cells responded to anti-EGFR therapy, but their progression free survival (PFS) was similar to those with high KRAS mutant cells tumors.5
Figure 2.
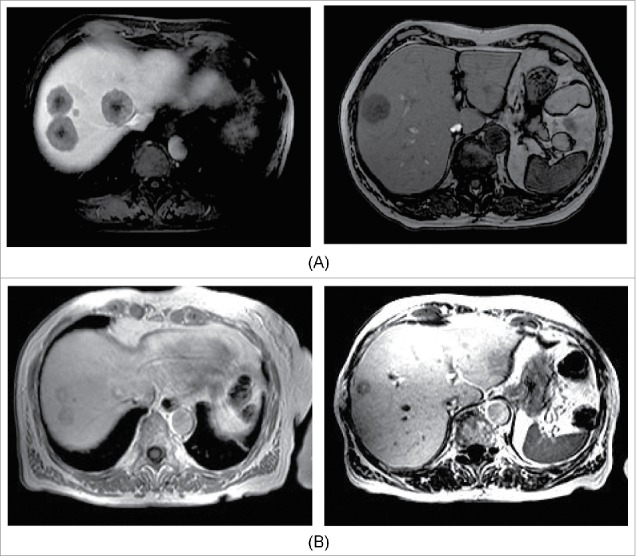
Radiological assessment before (A) and after (B) 4 cycles of chemotherapy with FOLFOX and panitumumab. A: Basal CT scan, October 23, 2015. B: CT scan, December 17, 2015.
Regardless if the emergence of mutant clones derived from selection of a preexisting clones or as result of ‘de novo’ mutation our report has formidable implications for translational research and patient care.
In fact, given that in clinical practice we use single tumor samples taken at one point in the disease course, we likely underestimate the true extent of tumor heterogeneity and the consequences of emerging resistant subclones on disease biology and tumor responsiveness to treatment in the face of continued drug exposure.
Recently, Løes et al. analyzed tumor samples from 94 patients with ≥2 liver metastases from CRC (CRCLM) and found mutation heterogeneity (defined as different mutation status between metastases from the same patient at the same surgical procedure) in 13 of them for at least one of the 4 genes analyzed: KRAS, BRAF, PIK3CA and TP53. A negative prognostic effect for KRAS and BRAF mutation heterogeneity, as compared to homogenous mutation or no mutations at all was also reported.6
Despite the effort to maximize tumor content by macrodissecting, tissue biopsies represent a small fraction of the entire tumor burden and mutated alleles could be present at a prevalence not detectable by commonly used techniques (<1 in 10,000 tumor cells). Recent studies showed that a subset of CRC cellular models, which are KRAS wild-type by conventional Sanger sequencing (LOD ∼ 10%), resulted KRAS mutated with more sensitive techniques.7
It could be questionable if the emergence of mutant clones derives from selection of a preexisting or “de novo” RAS mutant clones under the pressure of anti-EGFR treatment, or as result of random emergence of RAS mutant clones.
Several preclinical findings showed the emergence of anti-EGFR resistant variants during EGFR inhibition.
Previous studies reported that the proportion of mutant KRAS alleles in circulating tumor DNA (ctDNA) may decrease after anti-EGFR treatment discontinuation, supporting the importance of longitudinal tumor sampling strategies to deciphering the temporal acquisition of driver event that could impact treatment outcome.8 Misale et al. generated cetuximab-resistant variants of two CRC cellular models by continuous drug exposure, demonstrating that resistant cells differed by two molecular alterations: reduction of EGFR gene copy number and acquisition of KRAS mutations and amplification. When they performed a deep sequencing in the parental cells, they found a KRAS mutations in approximately 0.2% of them. They also examined tumor biopsies from 10 KRAS wild-type metastatic CRC refractory to either cetuximab or panitumumab, and identified KRAS mutation in 5 of them.9 Concordant data were obtained through the analysis of circulating cell-free tumor DNA from plasma samples.10
In their conclusions, authors emphasized that resistance to anti-EGFR therapy may result from either the selection of pre-existing KRAS mutant clones or from a continuous mutation process.9 Recent clinical works echoed these conclusions.11 From a clinical standpoint, similar findings might explain the regained sensitivity to anti-EGFR treatments observed in patients already exposed to the anti-EGFR moAb cetuximab.12
Unfortunately, current methods are underpowered to detect subclonal drivers of tumor present at low variant allele frequencies or spatially and/or temporally separated within tumors. This caveat could be overcome by using “liquid biopsy”. Some efforts in this field have been launched and investigators have reported excellent correlation of ctDNA and tumor samples in mCRC, including the ability to follow changes in gene mutation at multiple time points throughout therapy. These data underpin that liquid biopsy holds the potential to be a powerful tool for following intratumor heterogeneity and guiding cancer treatment in clinical settings.10,13
We acknowledge that, due to tumor heterogeneity, clonal redistribution is difficult to grasp in tissue samples and liquid biopsies could be more effective, though noninvasive, methods to monitor clonal dynamics and detect mutated clones emerging throughout the treatment. However, liquid biopsies are not yet used in routine clinical practice and validation is still needed to enable the enlargement of the potential applications of liquid biopsies for CRC management, while tumor tissue is still to be considered the gold standard source for clinical molecular analyses. Therefore, we did not look at RAS mutations in ctDNA.
Further to our case, we can speculate the existence of two distinct populations of KRAS wild-type and mutant CRC cells sharing the same genetic origin. The anti-EGFR treatment represented a selective pressure that allowed the selection of KRAS mutant subclones, due to a hypothetical paradoxical stimulatory effect. The panitumumab discontinuation could have prevented a continuous selective pressure in favor of mutant KRAS clones, thus supporting the impossibility to detect them in the primary tumor.
Our observations have relevant implications for patients' care. First, since a considerable fraction of patients eligible for anti-EGFR treatment develop secondary resistance in a short time frame, we underscore the importance of high-sensitivity sequencing technologies for the detection of emergent resistant subclones as a consequence of continued drug exposure. Second, our case further calls into question the use of targeted therapies after liver resection: do we have to reassess the molecular landscape in CRCLM after targeted therapy to make sure no selective pressure effects have been elicited? Do we have enough evidence to support the use of targeted therapy in the postoperative setting after CRCLM resection?
Awareness of heterogeneity between primary and metastatic lesions has existed for a long time but results of different works are controversial in mCRC: some authors have described a high degree of concordance between primary tumors and metastases while discordant mutations were observed in other study reports.15 He et al. demonstrated a significant discordance of PIK3CA mutations status and this was supported by a systematic review that showed highly concordance in the mutational status of KRAS, NRAS and BRAF, but not in PIK3CA mutations.16 The following factors have been associated with discordance in KRAS and PIK3CA mutation: i) metachronous resection, ii) intervening chemotherapy between resection of primary and metastasis iii) number of lines of intervening chemotherapy.17 The evidence of genetic discordance, even between different metastatic lesions at the same time, adds another piece to this complex scenario.
Regarding the second question, considerations about the role of targeted therapy in the postoperative setting are warranted.
In the EORTC trial patients with resectable CRCLM were randomized to FOLFOX before and after surgery versus surgery alone; chemotherapy was associated with improved PFS.18 Subsequently a meta-analysis confirmed the benefit of chemotherapy in this setting. Recently, a population-based study showed that post-operative oxaliplatin-based chemotherapy is associated with an improvement in survival in this population.19 Indeed, oxaliplatin-based regime is widely used even in the absence of randomized clinical trials versus fluoropyrimidines alone.
In contrast, no established role for biological-targeted agents use after CRCLM resection exist, even if many teams advocate the postoperative use of a regimen with anti-EGFR, if effective in the preoperative setting [20]. Evidence of intratumor heterogeneity and the putative association with therapy resistance makes it more difficult to decide to use the anti-EGFR drugs after resection of CRCLM. Further research is warranted to address this question and better stratify patients for therapy.
Our case provides evidence of the potential utility to reassess the molecular landscape of tumors both in primary and in metastatic lesions, and emphasizes the need to better understand the clinical significance of subclonal driver mutations to improve therapeutic strategies in cancer medicine.
Disclosure of potential conflicts of interest
The authors certify that they have no conflict of interest and no affiliations with or involvement in any organization or entity with any financial interest, or non-financial interest in the subject matter discussed in this manuscript.
References
- 1.Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, Schilsky RL. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol. 2016;34:179–85. doi: 10.1200/JCO.2015.63.9674. PMID:26438111. [DOI] [PubMed] [Google Scholar]
- 2.Molinari F, Felcioni L, Buscarino M, De Dosso S, Buttitta F, Malatesta S, Movilia A, Luoni M, Boldorini R, Alabiso O, et al.. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin Cancer Res. 2011;17:4901–14. doi: 10.1158/1078-0432.CCR-10-3137. PMID:21632860. [DOI] [PubMed] [Google Scholar]
- 3.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–61. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 4.Mekenkamp LJ, Tol J, Dijkstra JR, de Krijger I, Vink-Börger ME, van Vliet S, Teerenstra S, Kamping E, Verwiel E, Koopman M, et al.. Beyond KRAS mutation status: influence of KRAS copy number status and microRNAs on clinical outcome to cetuximab in metastatic colorectal cancer patients. BMC Cancer. 2012;12:292. doi: 10.1186/1471-2407-12-292. PMID:22804917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Normanno N, Rachiglio AM, Lambiase M, Martinelli E, Fenizia F, Esposito C, Roma C, Troiani T, Rizzi D, Tatangelo F, et al.. On the behalf of the CAPRI-GOIM investigators. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann Oncol. 2015;26:1710–4. doi: 10.1093/annonc/mdv176. PMID:25851630. [DOI] [PubMed] [Google Scholar]
- 6.Løes IM, Immervoll H, Sorbye H, Angelsen JH, Horn A, Knappskog S, LønningInt PE. Impact of KRAS, BRAF, PIK3CA, TP53 status and intraindividual mutation heterogeneity on outcome after liver resection for colorectal cancer metastases. Int J Cancer. 2016;139:647–56. doi: 10.1002/ijc.30089. PMID:26991344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al.. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. doi: 10.1038/ng.2891. PMID:24487277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, et al.. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. PMID:26030179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al.. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. PMID:22722830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al.. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. PMID:24553385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein-Scory S, Maslova M, Pohl M, Eilert-Micus C, Schroers R, Schmiegel W and Baraniskin A. Significance of Liquid Biopsy for Monitoring and Therapy Decision of Colorectal Cancer. Transl Oncol. 2018;11(2): 213–20. doi: 10.1016/j.tranon.2017.12.010. PMID:29367069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santini D, Vincenzi B, Addeo R, Garufi C, Masi G, Scartozzi M, Mancuso A, Frezza AM, Venditti O, Imperatori M, et al.. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol. 2012;23:2313–8 doi: 10.1093/annonc/mdr623. PMID:22396447. [DOI] [PubMed] [Google Scholar]
- 13.Punt CJ, Koopman M and Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017;14:235–46. doi: 10.1038/nrclinonc.2016.171. PMID:27922044. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz RE, J Berlin JD, Lenz HJ, Nordlinger B, Rubbia-Brandt L, Choti MA. Systemic cytotoxic and biological therapies of colorectal liver metastases: expert consensus statement. HPB (Oxford) . 2013;15:106–15. doi: 10.1111/j.1477-2574.2012.00558.x. PMID:23297721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Q, Xu Q, Wu W, Chen L, Sun W, Ying J. Comparison of KRAS and PIK3CA gene status between primary tumors and paired metastases in colorectal cancer. Onco Targets Ther. 2016;20:2329–35. doi: 10.2147/OTT.S97668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goswami RS, Patel KP, Singh RR, Meric-Bernstam F, Kopetz ES, Subbiah V, Alvarez RH, Davies MA, Jabbar KJ, Roy-Chowdhuri S, et al.. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin Cancer Res. 2015;21:2644–51. doi: 10.1158/1078-0432.CCR-14-2391. PMID:25695693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, Bechstein WO, Primrose JN, Walpole ET, Finch-Jones M, et al.. Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): long-term results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2013;14: 1208–15. doi: 10.1016/S1470-2045(13)70447-9. PMID:24120480. [DOI] [PubMed] [Google Scholar]
- 18.Krishnamurthy A, Kankesan J, Wei X, Nanji S, Biagi JJ, Booth CM. Chemotherapy delivery for resected colorectal cancer liver metastases: Management and outcomes in routine clinical practice. EJSO. 2017;43:364–71. doi: 10.1016/j.ejso.2016.08.022. PMID:27727025. [DOI] [PubMed] [Google Scholar]
- 19.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson A, Bodoky G, et al.. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422. doi: 10.1093/annonc/mdw235. PMID:27380959. [DOI] [PubMed] [Google Scholar]