

ABSTRACT
In recent years there has been great progress with the implementation and utilization of Clustered Regularly Interspaced Palindromic Repeats (CRISPR) and CRISPR-associated protein (Cas) systems in the world of genetic engineering. Many forms of CRISPR-Cas9 have been developed as genome editing tools and techniques and, most recently, several non-genome editing CRISPR-Cas systems have emerged. Most of the CRISPR-Cas systems have been classified as either Class I or Class II and are further divided among several subtypes within each class. Research teams and companies are currently in dispute over patents for these CRISPR-Cas systems as numerous powerful applications are concurrently under development. This mini review summarizes the appearance of CRISPR-Cas systems with a focus on the predominant CRISPR-Cas9 system as well as the classifications and subtypes for CRISPR-Cas. Non-genome editing uses of CRISPR-Cas are also highlighted and a brief overview of the commercialization of CRISPR is provided.
KEYWORDS: CRISPR products, Cas9, Cas13, genome editing, NHEJ, HDR, ZFN, TALEN, CRISPR-Cas figure, Cas classes, Cas types
Introduction
The CRISPR-Cas system (clustered regularly interspaced short palindromic repeats) has become one of the most powerful tools in the arsenal of molecular biologists and geneticists since its discovery by Ishino et al. in 1987 [1]. Mojica et al. [2] performed much of the initial characterization of CRISPR-Cas systems during the 1990s and the term CRISPR was coined for the first time by Jansen et al. in 2002 [3]. Since then, the discoveries and characterisations of the proteins and molecules involved, as well as the processes that generally occur across all types of the CRISPR-Cas system [4]. Using the predominant Class 2, Type II CRISPR-Cas9 [4–6] system as an example, CRISPR-Cas systems effectively consist of a three-stage process: expression, interference and adaptation [4,5]. During expression, the CRISPR array which contains many sequences homologous to specific target sequences (protospacers) are transcribed into what is called pre-CRISPR RNA (pre-crRNA) [4–6] (Figure 1(a)), and these pre-crRNA form homologous bonds with smaller transactivating crRNAs (tracrRNA) [4–6]. Once this complex has formed, it attaches to a Cas9 protein where the long pre-crRNAs are cut and separated by RNase III into individual crRNA/tracrRNA complexes (Figure 1(b)). Interference begins as the crRNA/tracrRNA guides the Cas9 complex to a target sequence and the crRNA binds to the target sequence after the so called protospacer adjacent motif (PAM) (Figure 1(c)). It is this short sequence that allows for self/nonself-discrimination as the sequence is absent from the hosts own CRISPR array [4–6]. The target sequence is unwound at this stage and cut by the Cas9 protein’s nuclease domains (RuvC and HNH) [4–6], leaving a double stranded break in the target DNA sequence, after which the Cas9 complex detaches. The desired DNA repair template is then inserted and attached to the blunt ends of the cleaved target DNA product at the end of the interference by Homology Directed Repair (HDR). The repaired spacer sequence is then transcribed and adapted into the genome (Figure 1(d)) [4–6]. Adaptation in the majority of the known CRISPR-Cas systems is controlled by the Cas1 and Cas2 proteins (and to some extent Cas4) that adapt the desired spacer sequences into the CRISPR array by integrating the RNA and then inducing reverse transcription of the RNA into DNA [4–6]. It is by these processes that some bacteria are able integrate viral genomes into their own (i.e. the CRISPR array) during an infection, thus allowing a more effective immune response during future infections [4–7]. More recently these processes have been modified to function as powerful tools for molecular biology and genetic engineering. The CRIPSR-Cas system classifications and developments are reviewed below, along with of non-genome editing CRIPSR-Cas systems and the current state of CRISPR-Cas commercialization.
Figure 1.
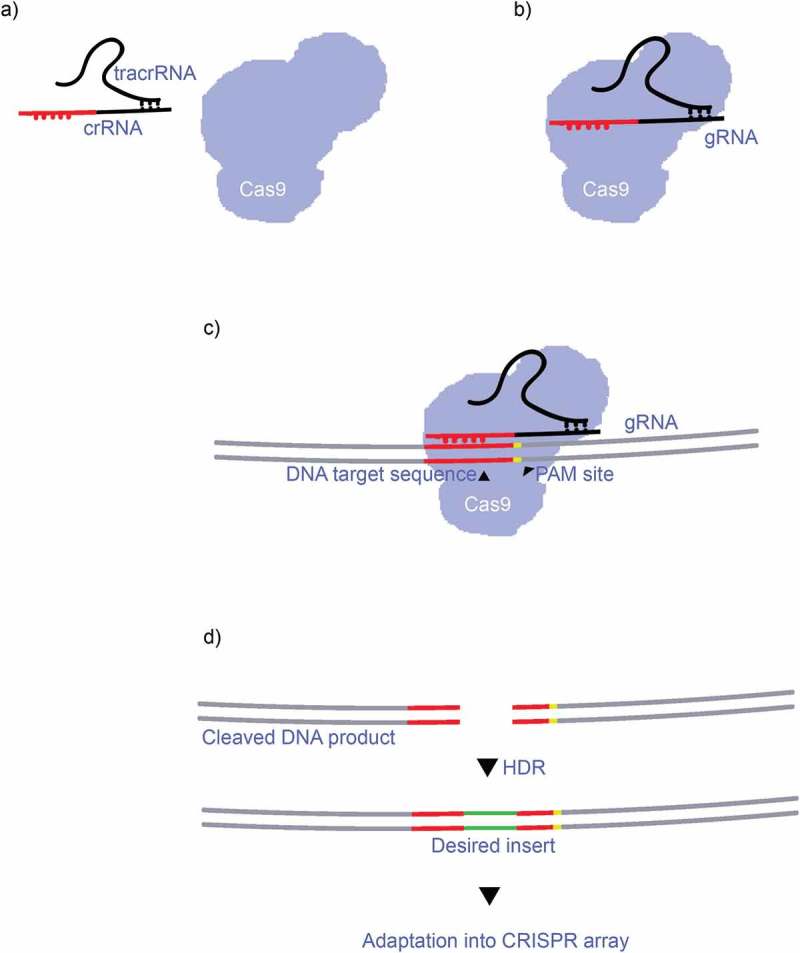
a) The crRNA from the CRISPR array combines with a smaller tracrRNA molecule, becoming a gRNA complex. b) The gRNA binds with a Cas9 protein, forming a gRNA:Cas9 complex. c) The gRNA guides the Cas9 protein, targeting a specific DNA sequence, which it first recognizes by the PAM motif. The RuvC and HNC nuclease sites cuts the target sequence, leaving two homologous blunt ends. d) The desired DNA repair template inserts the desired gene and repairs the strands by HDR, the product DNA then undergoes adaptation into the organism’s genome.
CRISPR-Cas subtypes and classifications
The ever-evolving interaction between prokaryotes and the viruses that infect them has resulted in wide variation among the CRISPR-Cas systems [7]. The general classification divides the known Cas systems into two classes, six types, and 19 subtypes. Currently, they are classified according to the structure shown in Table 1 [4–8]. This system is widely distributed in archaeal (87%) and bacterial (50%) genomes, Class I being the most commonly found (90%) [5–7].
Table 1.
| |
Class I |
Class II |
||||
|---|---|---|---|---|---|---|
| Type I | Type III | Type IV | Type II | Type V | Type VI | |
| Integration module | Cas1/2 [3] | Cas1/2 | Unknown | Cas1/2 | Cas1/2 | Cas1/2 |
| Effector module | 4 – 7 Cas protein Cascade | Cas9 [10] | Cas12a (cpf1)/Cas12b/Cas12c/[17] | Cas13a/Cas13b/Cas13c [20] | ||
| Molecule substrate | DNA | RNA | ||||
| Organism | bacteria and archaea | archaea | bacteria | bacteria and archaea | bacteria | |
| Nuclease domain | HDa fused to Cas3 | HD fused to Cas10 | unknown | RuvC and HNH | RuvC and Nuc | HEPN domains (2) |
| tracrRNA | no | No | no | yes | cpf1-no | no |
| Cleavage motif | subtype dependant (7) | subtype dependant (2) | subtype dependant (2) | CG rich NGG (blunt ends) | AT rich (staggered ends) | non-G PFS (ssRNA) |
aHistidine-aspartate domain
The main difference between the classes are how the effector modules are composed. In Class I, the effector is comprised of a complex formed by several proteins with different functions, whereas in Class II, the effector is associated with a single multi-domain protein [4,7]. Considering the integration module (adaptation step), the proteins that are involved in this process are Cas1 and Cas2, which integrate a viral protospacer into the bacterial/archaea genome, and its function remains conserved throughout classes, apart from Type IV, in which the processes remain unknown [4,5]. By contrast, the effector modules involved in the target recognition and cleavage steps, are generally variable throughout types. The CRISPR-Cas system classifications are further discussed below. Class I is divided into three types (Type I, III and IV) where the effector module is generated by a cascade of different Cas and other accessory proteins [4–7]. Class II systems are used more in research and will be reviewed in further detail below.
Class II CRISPR-Cas classifications
Most researchers have used Class II types to date because they enable them to work with only one multidomain protein [4,7]. Within this Class, the predominantly researched group is Type II, in which we find the well-known Cas9 effector. Cas9 is a protein with two nuclease domains (RuvC and HNH) [6] that requires two combined RNA molecules to produce a double stranded break (DSB) with blunt ends in the target DNA sequence. These RNA molecules are a crRNA, and a tracrRNA, that together guide the interference. Researchers have bioengineered these RNA molecules into one guide RNA molecule (gRNA) which alone contains both functions of the crRNA and tracrRNA, thereby making CRISPR systems even easier to utilize [9]. The Cas9 effector spCas9 from Streptococcus pyogenes is the most widely used effector due to its high efficiency at producing DSB. However, spCas9 has three major limitations. Firstly, the PAM is NGG, which dictates the need for sequences with two consecutive GG to produce the DBS, making its use problematic in AT rich sequences [10]. Secondly, the size of this protein is 1,368 amino acids, which can be a hindrance when introducing these sequences into viral vectors [11]. Thirdly, Cas9 is prone to producing off target effects, which means that DSB may be generated at incorrect locations [12].
Several solutions to these problems have been tested. One possible solution is to modify the spCas9 sequence to obtain better variants with less off target effects. This has been progressed through generation of a highly specific Cas9 that contains mutations that reduce the interactions between the nuclease domain and the non-specific DNA: spCas9 HF (high fidelity) [13]. To overcome the PAM restrictions, the same spCas9 was engineered to obtain different PAM motifs to make this tool even more adaptable to different types of DNA sequences, such as VQR, EQR and VRER [14]. Another alteration to spCas9 is the removal of one of the nuclease domains. As a result, nCas9 (nickase Cas9) was generated, nCas9 can induce a single stranded break, and using two of these enzymes with two gRNA; a deletion or other alterations with a reduced amount of off target effects can be achieved [15]. To solve the problem related to spCas9’s large size, Cas9 homologues from other organisms can be used. One example, saCas9 (Staphylococcus aureus Cas9) is smaller and has a different PAM site [16]. Cas9 proteins are continually being altered to obtain even more variations in size, site recognition and target effect.
The second type of Class II is the Type V. This group has the characteristic of sharing the RuvC nuclease domain with Type II, but not the HNH domain. This type is divided in three subtypes (A, B and C) of which the most cited and used is subtype A, which includes the Cpf1 effector nuclease (also referred interchangeable as Cas12a) [6]. This type of nuclease has four distinctive characteristics that has allowed it to develop as a complementary product for the Cas9 effector. Firstly, this type of nuclease does not need a tracrRNA sequence to be functional, making the design easier and more cost effective. Secondly, the size of this protein is even smaller than Cas9, making insertion into viral vectors easier. Thirdly, when this enzyme generates a cleavage, it leaves staggered ends, improving the chances of a non-homologous end join (NHEJ) knock-in. Fourthly, it recognizes AT rich PAM sites, making this enzyme complementary to Cas9 (CG rich zones). Due to all these features, Cpf1 has become an extremely useful tool for genome editing [12,17]. Cpf1 continues to be modified to improve efficiency and it has been reported to be an excellent tool for plant editing, even better than Cas9 [18].
The high efficiency of CRISPR-Cas systems and their relatively ease of use makes it possible to generate organisms with several mutations in the genome. A rapid method to obtain a multi-mutated organism is the use of lentiviral gRNA libraries with several gRNA that can be integrated into any of the Cas mentioned above. In this way, an organism can be generated with multiple mutations in only one generation [18,19].
The last integrant of Class II is the recently discovered Type VI. Its most unusual characteristic is that it can edit RNA instead of DNA. This is discussed further below, together with other non-genome editing CRISPR-Cas systems.
Delivery methods for CRISPR-Cas systems
An important consideration is the selection of effective methods to deliver the CRISPR-Cas system to organisms that are to be mutated. There are several methods available, and choice largely depends on the characteristics of the organism to be transfected. In plant models, plasmids in Agrobacterium tumefaciens are often used as a vector, whereas in mammalian cells use of the complex gRNA-Cas9 is preferred [9,18].
Non-genome editing methods of the CRISPR-Cas systems
RNA-targeting with CRISPR-Cas13 and rCas9
One of the most recent discoveries in CRISPR-Cas is the Cas13 (Cas13a, Cas13b and Cas13c) Class II, type VI group, described in 2015 by Shmakov et al. [6]. It should be noted that Cas13a was previously referred to as C2c2 (Cas13b = C2c4, Cas13c = C2c7) [6,20,21] and some literature uses these terms interchangeably. What separates Cas13 from the other predominant CRISPR-Cas systems, such as CRISPR-Cas9, is that it targets single-stranded RNA (ssRNA) rather than double-stranded (dsDNA), and it tends to cleave RNA non-specifically (Figure 2(a)) [20,21]. Unlike most of the previously described systems, Cas13 is guided by a lone crRNA molecule rather than a crRNA-tracrRNA complex. Another mechanism that sets Cas13 apart from Cas-types is its twin HEPN nuclease domains (Table 1), which generate blunt ends in the target RNA after cutting [20,21]. As described by Nakade et al. [12], O’Connell et al. and Nelles et al. [22,23] are working to generate variants of the CRISPR-Cas9 (CRISPR-rCas9) system that can target ssRNA similarly to the CRISPR-Cas13 system, by modifying PAM-presenting oligonucleotides (PAMmers). These PAMmers will navigate the Cas9 to bind specifically to target ssRNA sequences [22,23].
Figure 2.
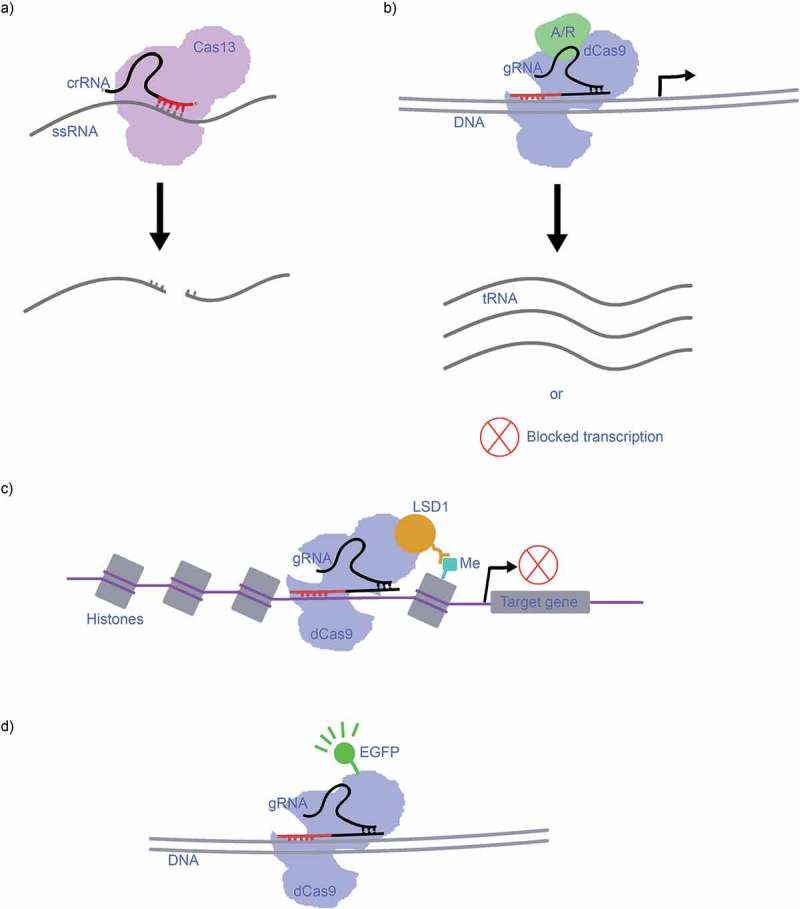
a) CRISPR-Cas13 targets ssRNA with its crRNA, and the twin HEPN nuclease domains cleaves the sequence non-specifically after the first crRNA guided cleavage at the binding site, leaving blunt ends. b) The dCas9 combines with an activator/repressor domain to activate/repress an upstream gene, resulting in transcription of that gene into RNA or blocked transcription. c) dCas9-LSD1 complex targets the genome at the chromatin to repress transcription of the targeted gene by demethylation. d) CRISPR-dCas9-EGFP as a fluorescent marker complex for imaging.
CRISPRa/CRISPRi, epigenetic modifications and markers
Lundh et al. [24] demonstrated that the Cas9 protein can be enzymatically deactivated (dCas9) to lose its ability to cleave while retaining ability to target and bind to specific DNA sequences. This dCas9 protein can then be combined with activator- or repressor domains to systematically activate or repress upstream genes, which is a reversible process as the genome is not directly edited [24–27]. A simple model is presented in Figure 2(b); an activator or repressor domain attaches to the dCas9 complex, resulting in the activation (and thus transcription) or the repression of one or several upstream genes. This system is called CRISPRa when an activator domain is used, and CRISPRi when a repressor domain is used [24–27]. These techniques have been developed into useful genetic screening tools [28,29]. CRISPR-dCas9 also has another use: epigenetic modification. By attaching the dCas9 complex to known epigenetic modifiers such as histone demethylase (LSD1) or human acetyltransferase (p300) dCas9 can target the genome with great proficiency. What differentiates this mechanism from other CRISPR-Cas systems, is that the dCas9-LSD1 complex works on the chromatin, while the genome is still wrapped up in histones. Thus, these modifications are useful tools for heritable gene expression. These complexes can also serve to activate or repress transcription, e.g. ‘LSD1 repress pluripotency maintenance genes (e.g. Oct4 and Tbx3) in mouse embryonic stem cells’, which is visualized in Figure 2(c) [25].
Both the Cas9 and Cas13 systems have been modified by researchers to function as genetic markers; dCas9 for DNA and dCas13 for RNA. For example, Chen et al. [30] demonstrated that a dCas9 complex tagged with an enhanced green fluorescent protein (eGFP) can be guided by a gRNA to target sequences that will then fluoresce during dynamic imaging (Figure 2(d)) [30,31].
CRISPR-Cas commercialization status quo
In contrast to other genome editing techniques such as zinc finger nucleases (ZFNs) [32] and transcription activator-like effector nucleases (TALENs) [33], CRISPR was originally developed inside academic research institutions [34]. In 2012, Jennifer Doudna and Emmanuelle Charpentier from the University of California, Berkeley, published a paper and initiated their patent application which demonstrated the use of CRISPR-Cas9 system to edit DNA [35]. By the end of that year, another group led by Feng Zhang at the Broad Institute of MIT (Massachusetts) and Harvard in Cambridge, initiated another patent which demonstrated the application of CRISPR–Cas9 in mammalian cells [19,36]. Their paper was published in 2013, and it initiated a conflict as to which group would have the rights to CRISPR-Cas9 intellectual property. This issue currently remains unresolved, and four additional researchers have now also claimed rights to this system [37]. Since its development, the number of patents related to CRISPR products has increased at an unprecedented rate compared to other editing technologies; several private commercialisations have been generated in a short period of time [34]. In 2015 there was a 5-fold increment in investment in CRISPR and biotechnology companies received a total of $1.2 billion in venture capital funds [34]. The spread of this technology to the private sector has occurred in two ways. Firstly, the original developers have generated their own companies, for example Caribou Biosciences by Charpentier, CRISPR Therapeutics by Doudna and Editas by Feng Zhang. Secondly, numerous leading biotechnology companies have developed new market opportunities with the technology, such as AstraZeneca, DuPont, Novartis, Thermo Fisher Scientific and Sigma Aldrich and several others, and entered into the market [38].
FDA regulations on the use of CRISPR products remain unclear in relation to oncological trials, and it may take several years to obtain final approvals [34]. However, the situation in agricultural sciences is clearer and some knock-out and mutated crops have been approved as non-GMO products in the USA [39]. While the issue regarding who can claim the CRISPR-Cas9 original patent remains unresolved, CRISPR is well placed to be commercialized by companies, and to be further developed by researchers.
Conclusion
The discovery, characterization and development of CRISPR-Cas systems constitutes a major milestone for molecular biology in the 21st century. The current state of these systems, and furthermore their future potential as ever more easy-to-use variants are developed, promises to open many doors for genetic engineering – both in the areas of genome editing and non-genome editing. Further research is necessary to fully map out all the molecular mechanisms involved in the classes and subtypes (e.g. Class I, Type IV in Table 1). There also remain limitations to some of the existing systems, such as CRISPR-Cas9, but recent discoveries have bypassed several of these limitations and more are under development. The CRISPR-Cas patent disputes will eventually be resolved, which may or may not change the availability and cost of commercially available CRISPR-Cas systems. Regardless of how the patent disputes are resolved, CRISPR-Cas systems will play major roles in a wide range of areas in the near future including genetic engineering and screening, mammalian gene therapy and plant and livestock breeding.
Acknowledgments
We acknowledge the resources provided by the Centre for Plant Genetics and Breeding (PGB) at The University of Western Australia (UWA) for the Master’s studies conducted by Fernando Perez Rojo and Rikard Karl Martin Nyman.
Author contributions
PK, FPR and RKMN conceived and designed the research. FPR and RKMN performed the literature search, prepared the figures and wrote the manuscript with contributions from AATJ, MPN, MHR, WE and PK. All authors read and approved this manuscript.
Disclosure statement
The authors declare that they have no competing interests.
References
- [1].Ishino Y, Shinagawa H, Makino K, et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mojica FJM, Diez-Villaseñor C, García-Martínez J, et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. [DOI] [PubMed] [Google Scholar]
- [3].Jansen R, Embden JDA, Gaastra W, et al. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. [DOI] [PubMed] [Google Scholar]
- [4].Mohanraju P, Makarova KS, Zetsche B, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. [DOI] [PubMed] [Google Scholar]
- [5].Jackson SA, McKenzie RE, Fagerlund RD, et al. CRISPR-Cas: adapting to change. Science. 2017;356:eaal5056. [DOI] [PubMed] [Google Scholar]
- [6].Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR–cas systems. Nat Rev Microbiol. 2017;15:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR–cas systems. Nat Rev Microbiol. 2015;13(11):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hille F, Charpentier E.. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci. 2016;371:1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang J-H, Adikaram P, Pandey M, et al. Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered. 2016;7(3):166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nakade S, Yamamoto T, Sakuma T. Cas9, Cpf1 and C2c1/2/3—What’s next? Bioengineered. 2017;8(3):265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523(7561):481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 155:479–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ran FA, Cong L, Yan WX, et al. In vivo genome editing using staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Scheben A, Wolter F, Batley J, et al. Towards CRISPR/Cas crops – bringing together genomics and genome editing. New Phytol. 2017;216(3):682–698. [DOI] [PubMed] [Google Scholar]
- [19].Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR–cas13. Nature. 2017;550(7675):280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].O’Connell MR, Oakes BL, Sternberg SH, et al. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014;516(7530):263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nelles DA, Fang MY, O’Connell MR, et al. Programmable RNA tracking in live cells with CRISPR/Cas9. Cell. 2016;165(2):488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lundh M, Pluciñska K, Isidor MS, et al. Bidirectional manipulation of gene expression in adipocytes using CRISPRa and siRNA. Mol Metab. 2017;6:1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lo A, Qi L. Genetic and epigenetic control of gene expression by CRISPR–cas systems. F1000. 2017;6:Faculty Rev–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gilbert LA, Horlbeck MA, Adamson B, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jost M, Chen Y, Gilbert LA, et al. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol Cel. 2017;68(1):210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kampmann M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem Biol. 2017; 13:406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen B, Gilbert LA, Cimini BA, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma H, Naseri A, Reyes-Gutierrez P, et al. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci USA. 2015;112:3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010;79:213–231. [DOI] [PubMed] [Google Scholar]
- [33].Li T, Huang S, Jiang WZ, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucl Acids Res. 2011;39(1):359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brinegar K, Yetisen A, Choi S, et al. The commercialization of genome-editing technologies. Crit Rev Biotechnol. 2017;37(7):924–932. [DOI] [PubMed] [Google Scholar]
- [35].Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ledford H. How the US CRISPR patent probe will play out. Nat News [Internet]. 2016. March 10;531(7593):149 [cited 2017 Dec 29]. Available from:: http://www.nature.com.ezproxy.library.uwa.edu.au/news/how-the-us-crispr-patent-probe-will-play-out-1.19519 [DOI] [PubMed] [Google Scholar]
- [37].Cohen J. Ding, ding, ding! CRISPR patent fight enters next round [Internet]. 2017. July 26, Am 9: 00 [cited 2017 Dec 29]. Available from: http://www.sciencemag.org.ezproxy.library.uwa.edu.au/news/2017/07/ding-ding-ding-crispr-patent-fight-enters-next-round
- [38].CRISPR COMMERCIALIZATION RISK | REGENHEALTHSOLUTIONS [Internet]. [cited 2017. December 29]. Available from: http://www.regenhealthsolutions.info/4/crispr_commercialization_risk_780838.html
- [39].Wolter F, Puchta H. Knocking out consumer concerns and regulator’s rules: efficient use of CRISPR/Cas ribonucleoprotein complexes for genome editing in cereals. Genome Biol. 2017;18:43. [DOI] [PMC free article] [PubMed] [Google Scholar]