

Abstract
Lessons Learned.
OPB‐111077 is a novel inhibitor of STAT3 and mitochondrial oxidative phosphorylation that exhibited promising anticancer activity in preclinical models.
In this first‐in‐human phase I study of OPB‐111077 in unselected advanced cancers, treatment‐emergent adverse events, most frequently nausea, fatigue, and vomiting, were generally mild to moderate in intensity and could be medically managed.
Overall, only modest clinical activity was observed after OPB‐111077 given as monotherapy. Notable antitumor activity was seen in a subject with diffuse large B‐cell lymphoma.
Background.
OPB‐111077 is a novel inhibitor of STAT3 and mitochondrial oxidative phosphorylation with promising anticancer activity in preclinical models.
Methods.
Open‐label, phase I trial of OPB‐111077 in advanced cancers with no available therapy of documented benefit. Initial dose escalation in unselected subjects was followed by dose expansion. Patients received oral OPB‐111077 daily in 28‐day cycles until loss of clinical benefit.
Results.
Eighteen subjects enrolled in dose escalation, and 127 in dose expansion. Dose‐limiting toxicities were observed at 300 mg and 400 mg QD; maximum tolerated dose was defined as 250 mg QD. Frequently reported treatment‐emergent adverse events (TEAEs) included nausea, fatigue, and vomiting. TEAEs were generally mild to moderate and could be medically managed. OPB‐111077 reached micromolar drug concentrations, had an elimination half‐life of approximately 1 day, and reached steady‐state by day 8. A durable partial response was observed in one subject with diffuse large B‐cell lymphoma. Seven subjects with diverse tumor types had stable disease or minor responses for at least eight treatment cycles (224 days).
Conclusion.
OPB‐111077 is generally well tolerated, and its pharmacokinetic profile is sufficient for further clinical development. Notable clinical activity was observed in a subject with diffuse large B‐cell lymphoma. Overall, modest efficacy was observed against unselected tumors.
Abstract
经验总结
OPB‐111077是STAT3和线粒体氧化磷酸化的新型抑制剂, 在临床前模型中显示出有极具前景的抗癌活性。
在针对非选择性晚期癌症患者进行的这一首项OPB‐111077人体I期研究中, 治疗期出现的不良事件(最常见的是恶心、疲劳和呕吐)的强度通常为轻度至中度, 并且可以进行医学管理。
总体而言, 进行OPB‐111077单药治疗后仅观察到中度临床活性。在弥漫性大B细胞淋巴瘤受试者中观察到明显的抗肿瘤活性。
摘要
背景.OPB‐111077是STAT3和线粒体氧化磷酸化的新型抑制剂, 在临床前模型中显示出有极具前景的抗癌活性。
方法. 本项是OPB‐111077治疗无可用的获益记载疗法的晚期癌症的开放标签I期试验。在非选择性受试者中经过初始剂量递增研究后, 进行剂量扩展研究。患者每日接受OPB‐111077口服给药, 以28天为一周期, 直至丧失临床获益。
结果.18例受试者进入剂量递增研究, 127例受试者进行剂量扩展研究。在300 mg和400 mg QD剂量下观察到剂量限制性毒性;最大耐受性剂量定义为250 mg QD。经常报告的治疗期出现的不良事件(TEAE)包括恶心、疲劳和呕吐。TEAE通常为轻度至中度, 并且可以进行医学管理。OPB‐111077达到微摩尔药物浓度, 消除半衰期约为1天, 至第8天达到稳态。在一例弥漫性大B细胞淋巴瘤受试者中观察到部分缓解持续时间较长。7例肿瘤类型不同的受试者病情稳定或出现轻微缓解达至少8个治疗周期(224天)。
结论.OPB‐111077通常具有良好的耐受性, 其药代动力学特征足以支持进一步的临床开发。在一例弥漫性大B细胞淋巴瘤受试者中观察到明显的临床活性。总体而言, 观察到对非选择性肿瘤有中度疗效。
Discussion
Previous studies showed that STAT3 can promote tumorigenesis by at least two mechanisms: first, by acting as a nuclear transcription factor that alters expression of pro‐tumorigenic gene‐regulatory networks [1], [2], [3], [4], [5], [6], [7], [8]; and second, by acting as a regulator of oxidative phosphorylation (OXPHOS) via interaction with complexes I and II of the mitochondrial electron transport chain [9], [10], [11], [12], [13]. OPB‐111077 is an oral, small‐molecule, new chemical agent that has been shown to be a potent, high‐specificity inhibitor of STAT3 in preclinical models (Otsuka Pharmaceuticals, unpublished). The current study assessed the safety, pharmacokinetics, and preliminary antitumor activity of OPB‐111077 in patients with unselected advanced cancers that, based on prior genetic and molecular studies, were thought to have potential for a therapeutic response to an anti‐STAT3 agent.
Results demonstrated that OPB‐111077 was generally well tolerated in human subjects with advanced cancers. During dose escalation, none of 10 subjects receiving OPB‐111077 at or below the 250‐mg maximum tolerated dose developed a dose‐limiting toxicity or other serious TEAE. Most reported TEAEs were reversible and could be medically managed. Common TEAEs included nausea, vomiting, and fatigue. Following single‐ and multiple‐dose daily oral OPB‐111077 in the range of 100–400 mg, OPB‐111077 reached micromolar levels, which was in the range that correlated with antitumor activity in preclinical models.
In the study's efficacy evaluations, one ongoing subject with diffuse large B‐cell lymphoma achieved a partial response per RECIST, Version 1.1. Seven subjects maintained stable disease for at least eight treatment cycles (gastric cancer, cholangiocarcinoma, prostate cancer, renal cell carcinoma, KRAS‐mutant colon cancer, esthesioneuroblastoma, and B‐cell lymphoma; Fig. 1). Nine (60%) and thirty (32.3%) subjects had best overall responses of stable disease during the dose escalation and expansion stages, respectively.
Figure 1.

Maximum change in measured tumor size versus duration of OPB‐111077 therapy per patient. (A): Maximum percent change in tumor size in 101 subjects who completed postbaseline tumor measurements in stage 2. (B): Days of OPB‐1110777 treatment in the same patients. Note that the bars in the top graph and the bars in the bottom graph represent the subjects in the same order.
Abbreviation: NSCLC, non‐small cell lung cancer.
Anticancer activity of OPB‐111077 was therefore, in general, modest in this study, and further development of the drug will require identification of specific sensitive cancer subtypes. Given that OPB‐111077 blocks OXPHOS, it seems feasible, for instance, that cancer cells sensitive to metabolic inhibition might be sensitive to OPB‐111077 therapy. Notably, chemotherapy‐resistant and cancer stem cells have been reported to be dependent on OXPHOS [14], [15], and drug combinations with OPB‐111077 can be envisioned to exploit this observation [16], [17]. It is important to note, however, that it remains unclear whether effects other than those on OXPHOS were required for the observed anticancer effect of OPB‐111077 in the current study population of advanced cancer patients.
In conclusion, OPB‐111077 can be administered safely. Its pharmacokinetic profile is acceptable for further clinical development. Although notable clinical activity was observed in a subject with diffuse large B‐cell lymphoma, monotherapy demonstrated minimal clinical activity against unselected tumors overall. Research continues to identify drivers of OPB‐111077 clinical activity and synergistic combinations.
Trial Information
- Disease
Advanced cancer
- Stage of Disease/Treatment
Metastatic/advanced
- Prior Therapy
No designated number of regimens
- Stage 1
Phase I ‐ Dose escalation
- Stage 2
Phase I ‐ Dose expansion
- Primary Endpoint
Safety
- Primary Endpoint
Tolerability
- Secondary Endpoint
Pharmacokinetics
- Secondary Endpoint
Pharmacodynamic
- Secondary Endpoint
Efficacy
- Secondary Endpoint
Maximum tolerated dose
- Secondary Endpoint
Recommended phase II dose
- Secondary Endpoint
Food‐effect substudy
- Investigator's Analysis
Drug tolerable, hints of efficacy
Drug Information for Phase I Dose Escalation (Stage 1)
- Generic/Working Name
OPB‐111077
- Trade Name
NA
- Company Name
Otsuka Pharmaceutical
- Drug Type
Small molecule
- Drug Class
STAT3 and OXPHOS inhibitor
- Dose
100–400 mg per flat dose
- Route
p.o.
- Schedule of Administration
Every morning under fasting conditions in 28‐day cycles.
Drug Information for Phase I Expansion (Stage 2)
- Generic/Working Name
OPB‐111077
- Trade Name
NA
- Company Name
Otsuka Pharmaceuticals
- Drug Type
Small molecule
- Drug Class
STAT3 and OXPHOS inhibitor
- Dose
250 mg per flat dose
- Route
p.o.
Patient Characteristics for Phase I Dose Escalation (Stage 1)
- Number of Patients, Male
12
- Number of Patients, Female
6
- Stage
All advanced
- Age
Mean years (standard deviation [SD]) = 64.4 (11.4)
- Performance Status: ECOG
-
0 — 7
1 — 11
2 — 0
3 — 0
Unknown — 0
- Other
- Prior surgery, n (%)
15 (83.3)
- Prior radiotherapy, n (%)
9 (50.0)
- Prior chemotherapy/hormone, n (%)
16 (88.9)
Patient Characteristics for Phase I Expansion (Stage 2)
- Number of Patients, Male
59
- Number of Patients, Female
68
- Age
Mean age (SD) = 60.9 (11.6)
- Performance Status: ECOG
-
0 — 35
1 — 86
2 — 6
3 — 0
Unknown — 0
- Other
- Prior surgery, n (%)
107 (84.3)
- Prior radiotherapy, n (%)
69 (54.3)
- Prior chemotherapy/hormone, n (%)
127 (100)
Primary Assessment Method for Phase I Dose Escalation (Stage 1)
- Title
Dose escalation (stage 1)
- Number of Patients Screened
19
- Number of Patients Enrolled
18
- Number of Patients Evaluable for Toxicity
18
- Number of Patients Evaluated for Efficacy
15
- Evaluation Method
RECIST 1.1
- Response Assessment CR
n = 0 (0%)
- Response Assessment PR
n = 0 (0%)
- Response Assessment SD
n = 10 (67%)
- Response Assessment PD
n = 5 (33%)
- Response Assessment OTHER
n = 0 (0%)
Primary Assessment Method for Phase I Expansion (Stage 2)
- Title
Expansion (stage 2)
- Number of Patients Screened
145
- Number of Patients Enrolled
127
- Number of Patients Evaluable for Toxicity
127
- Number of Patients Evaluated for Efficacy
93
- Evaluation Method
RECIST 1.1
- Response Assessment CR
n = 0 (0%)
- Response Assessment PR
n = 0 (0%)
- Response Assessment SD
n = 30 (32%)
- Response Assessment PD
n = 54 (58%)
- Response Assessment OTHER
n = 9 (10%)
Adverse Events

Treatment‐emergent adverse events reported in ≥10% of subjects in stage 2 (expansion); n = 127.
Abbreviations: GGT, gamma‐glutamyltransferase; NC/NA, no change from baseline/no adverse event.
Serious Adverse Events

One cardiac event (grade 3 right ventricular dysfunction) was judged possibly related. The rest were unrelated to study medication.
Pharmacokinetics/Pharmacodynamics

Abbreviations: AUC∞, area under the concentration‐time curve from time 0 to infinity; Cmax, peak (maximal) concentration of drug in plasma; hr, hours; NA, nonapplicable; SD, standard deviation; tmax, time to maximum (peak) plasma concentration; T½, terminal phase elimination half‐life.
Assessment, Analysis, and Discussion
- Completion
Study completed
- Investigator's Assessment
Drug tolerable, hints of efficacy
Previous studies have shown that STAT3 promotes tumorigenesis by at least two mechanisms: first, by acting as a nuclear transcription factor that alters expression of pro‐tumorigenic gene‐regulatory networks [1], [2], [3], [4], [5], [6], [7], [8]; and second, by acting as a regulator of oxidative phosphorylation (OXPHOS) via interaction with complexes I and II of the mitochondrial electron transport chain [9], [10], [11], [12], [13]. Given this broad involvement in tumorigenesis, significant effort has been devoted to the discovery of small‐molecule STAT3 inhibitors [18], [19], [20]. One agent that showed early promise was OPB‐31121, which binds with high affinity to STAT3 (Kd = 10 nM), blocks STAT3 activity without upstream kinase inhibition, and inhibits growth of diverse malignant cells at nanomolar concentrations in various preclinical models [21], [22]. A phase 1 study, however, found that the bioavailability of OPB‐31121 was low, and further development of the agent was discontinued [23].
The same phase I study found that the primary metabolite of OPB‐31121, designated OPB‐111077, accumulated at higher tissue levels [23]. Subsequent in vitro studies found that OPB‐111077 had significant growth inhibitory effect in a variety of cancer models (Otsuka Pharmaceuticals, unpublished). Following up on these observations, the current study was designed to assess safety, pharmacokinetics, and preliminary antitumor activity of OPB‐111077 in patients with advanced cancers that, based on prior genetic and molecular studies, were thought to have potential for a therapeutic response to an anti‐STAT3 agent. The study consisted of a dose escalation phase (n = 18), followed by a dose expansion phase at the maximum tolerated dose (MTD; n = 127; Tables 1, 2, 3). All patients received oral OPB‐111077 once daily until loss of clinical benefit.
Table 1. Patient demographics and baseline characteristics.

Abbreviations: ECOG, Eastern Cooperative Oncology Group; NSCLC, non‐small cell lung cancer; SD, standard deviation.
Table 2. Subject disposition during dose escalation (stage 1).

As of the report cutoff date (May 27, 2015), one subject (subject 0010110) in the 300‐mg group was ongoing on treatment in the dose escalation stage.
Subjects who completed cycle 2 day 1 assessments were defined as completers.
Subjects who received any OPB‐111077 were included in safety analysis.
Subjects who received at least one cycle of OPB‐111077 were analyzed for efficacy.
Abbreviation: C2 D1, cycle 2 day 1.
Table 3. Subject disposition during dose expansion (stage 2).

Subjects who completed cycle 2 day 1 assessments were defined as completers.
Subjects who received any IMP were included in safety analysis.
Subjects who received at least one cycle of IMP were analyzed for efficacy.
Abbreviations: C2 D1, cycle 2 day 1; IMP, investigational medicinal product; NSCLC, non‐small cell lung cancer.
During dose escalation, no dose‐limiting toxicities (DLTs) were observed in the 100‐mg, 200‐mg, and 250‐mg dose cohorts (Table 4). Two DLTs were observed in the 300‐mg dose cohort (grade 3 dizziness and grade 3 nausea/vomiting) and in the 400‐mg dose cohort (two cases of grade 3 vomiting). The MTD and recommended phase II dose were thus determined to be 250 mg QD. All 18 subjects reported at least one treatment‐emergent adverse event (TEAE) during dose escalation, and 125 of 127 subjects (98.4%) reported at least one such event during dose expansion. Frequently reported TEAEs included nausea, vomiting, and fatigue (Table 5). At doses ≥300 mg, vomiting was dose limiting despite optimal antiemetic therapy, whereas nausea and vomiting at the MTD could be controlled by antiemetics. Grade 1 or 2 hypothyroidism was reported in 20% of study participants, nearly all of which (27 of 29 [93.1%] cases) were thought to be drug‐related. Four subjects (22%) in dose escalation and 45 (35.4%) in dose expansion had serious TEAEs. Frequently reported serious adverse events (SAEs) were cardiac, gastrointestinal, and respiratory disorders (Table 6). With the exception of a grade 3 cardiac event, none of these SAEs were considered related to study medication.
Table 4. Dose‐limiting toxicities as a function of OPB‐111077 dose.

Abbreviations: —, no data; DLT, dose‐limiting toxicity.
Table 5. Treatment‐emergent adverse events reported in ≥10% of subjects in stage 2 (expansion).
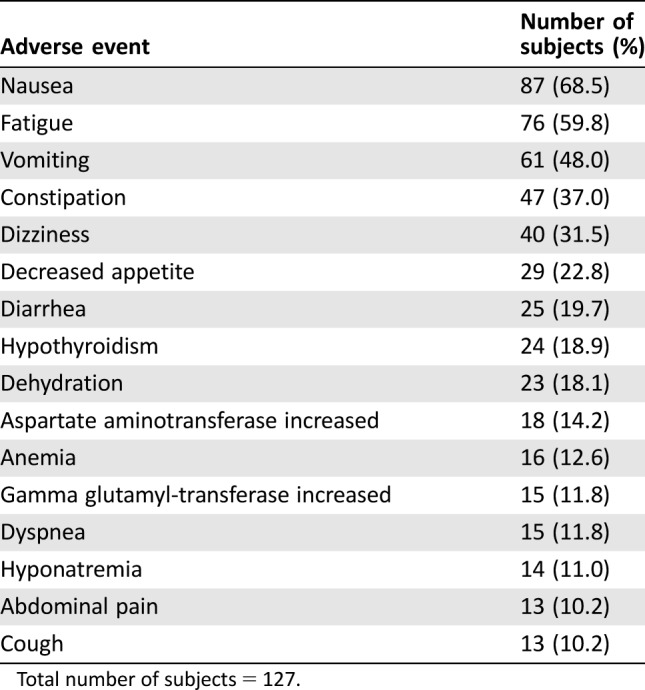
Total number of subjects = 127.
Table 6. Most frequently reported serious adverse events.
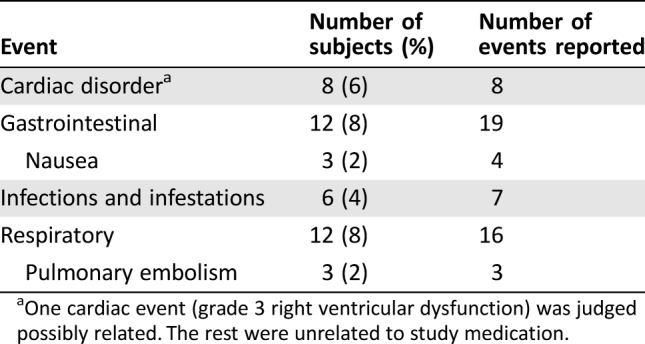
One cardiac event (grade 3 right ventricular dysfunction) was judged possibly related. The rest were unrelated to study medication.
OPB‐111077 exposure increased linearly after single and multiple ascending daily doses (Table 7). Tmax at the MTD was about 4 hours. Steady‐state concentrations were observed by day 8, consistent with the observed elimination half‐life of about 1 day. AUC0–24h and Cmax accumulation ratios were about 2–3.
Table 7. OPB‐111077 pharmacokinetics.

Shown are mean (SD) values, unless otherwise designated. Subjects received single doses of OPB‐111077 on cycle 1 day 1 and cycle 2 day 1; blood samples were collected over subsequent treatment‐free intervals until day 4 in each cycle.
Values are median (minimum–maximum).
Abbreviations: —, no data; AUC∞, area under the concentration‐time curve from time 0 to infinity; Cmax, peak (maximal) concentration of drug in plasma; hr, hours; NA, nonapplicable; SD, standard deviation; tmax, time to maximum (peak) plasma concentration; t1/2,z, terminal phase elimination half‐life.
Among 15 subjects in dose escalation evaluated for best overall response, 9 (60%) had stable disease, and 5 (33.3%) had progressive disease (at least eight cycles on study therapy) [24]. An additional subject continued treatment in the dose escalation stage as of the database cutoff date (May 27, 2015) and had stable disease on initiating his 26th cycle at 300 mg. In dose expansion, 93 subjects were assessed for efficacy and 89 were evaluated for best overall response. In best overall response analysis, 30 subjects (32.3%) had stable disease, 54 (58.1%) had progressive disease, 3 (3.2%) were not evaluable, and 2 (2.2%) were missing an assessment.
Maximum change in tumor size versus duration of OPB‐111077 therapy is shown in Figure 1. Eight subjects met protocol‐defined antitumor activity thresholds. These responses included an individual with diffuse large B‐cell lymphoma (with BCL2 amplification only on fluorescence in situ hybridization studies and no amplification of BCL6 or MYC) on OPB‐111077 for 17 months who achieved a partial response (Fig. 2). In addition, a renal cell carcinoma patient had 35% shrinkage in lung metastases but progressed with brain metastasis. Another seven subjects met stable disease criteria for at least eight treatment cycles. Tumor types and stable disease durations were as follows: gastric cancer (two subjects for 8 months), cholangiocarcinoma (8 months), prostate cancer (10 months), renal cell carcinoma (10 months; subject had 35% shrinkage of lung metastasis, but developed brain metastases), KRAS‐mutant colon cancer (14 months), and esthesioneuroblastoma (48 months).
Figure 2.

Partial response in diffuse large B‐cell lymphoma. A 78‐year‐old man with diffuse large B‐cell lymphoma diagnosed 3 years and six chemotherapy regimens before study entry. (A): Baseline computed tomography (CT) scan showing red arrow pointing to bulky left axillary mass. (B): End of cycle 2 positron emission tomography (PET) scan showing persistent left axillary glucose uptake in areas of lymphoma. (C): End of cycle 4 PET scan showing complete metabolic response. (D, E): End of cycle 14 PET and CT scans with red arrows pointing to ongoing complete metabolic response and nonpathologic residual nodes, respectively. After a gradual 90% decline in measured disease, at month 17 the subject's disease progressed distantly in the thigh, and study therapy was stopped.
Given its modest efficacy in this trial, further development of OPB‐111077 will require identification of specific cancer subtypes sensitive to STAT3 inhibition. Because OPB‐111077 blocks OXPHOS, it seems feasible that cancer cells sensitive to metabolic inhibition might be particularly sensitive to the drug. Chemotherapy‐resistant and cancer stem cells have both been reported to be dependent on OXPHOS [14], [15], and drug combinations with OPB‐111077 might build on this observation [16], [17]. Conversely, it remains unconfirmed what effects led to the observed anticancer effect of OPB‐111077 in the current trial.
This study was limited by the absence of pharmacodynamic confirmation of OPB‐111077 effect. An assay to assess inhibition of IL6‐stimulated Y705 phosphorylation of STAT3 in peripheral blood mononuclear cells has been developed using a phospho‐specific monoclonal antibody, but it was not used here due to the difficulty of maintaining analytic validity when the IL6‐stimulation protocol was deployed at trial sites. Bearing in mind this caveat, this first in‐human study of OPB‐111077 has shown that this new anti‐STAT3 agent can be administered safely and its pharmacokinetic profile is acceptable for further clinical development. Notable clinical activity was observed in a subject with diffuse large B‐cell lymphoma, although monotherapy had minimal clinical activity against unselected tumors overall. Research continues to identify drivers of OPB‐111077 clinical activity and synergistic combinations.
Figures and Tables
Acknowledgments
David Norris, Ph.D. (Ecosse Medical Communications, Falmouth, Massachusetts, USA), assisted in the medical writing of this manuscript.
Footnotes
ClinicalTrials.gov Identifier: NCT01711034
Sponsor(s): Otsuka Pharmaceutical Development and Commercialization, Princeton, New Jersey
Principal Investigator: Anthony Tolcher
IRB Approved: Yes
Disclosures
Anthony Tolcher: Otsuka Pharmaceutical Development & Commercialization, Inc. (RF); Geoffrey I. Shapiro: Eli Lilly & Co., Pfizer, G1 Therapeutics, Merck/EMD Serono, Otsuka, Roche (C/A), Eli Lilly & Co., Pfizer, Merck/EMD Serono (RF); Jordan Berlin: Otsuka (RF); Thomas Witzig: Otsuka (RF); Andrea Bullock: Taiho, Halozyme, Bayer (C/A); Edwin Rock: Otsuka (E); Agnes Elekes: Otsuka Pharmaceutical Development & Commercialization, Inc. (E); Chester Lin: Otsuka (E); Dusan Kostic: Otsuka (E); Naoto Ohi: Fujii Memorial Research Institute, Otsuka Pharmaceutical Co., Ltd., (E); Drew Rasco: Otsuka Pharmaceutical Development & Commercialization, Inc. (RF); Kyriakos P. Papadopoulos: Otsuka Pharmaceutical Development & Commercialization, Inc. (RF); Amita Patnaik: Otsuka Pharmaceutical Development & Commercialization, Inc. (RF); Lon Smith: Otsuka Pharmaceutical Development & Commercialization, Inc. (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol 2008;19:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu F, Mukhopadhyay S, Sehgal PB. Live cell imaging of interleukin‐6‐induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am J Physiol Cell Physiol 2007;293:C1374–C1382. [DOI] [PubMed] [Google Scholar]
- 3. Schroeder A, Herrmann A, Cherryholmes G et al. Loss of androgen receptor expression promotes a stem‐like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res 2014;74:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 2002;8:945–954. [PubMed] [Google Scholar]
- 5. Weaver AM, Silva CM. Signal transducer and activator of transcription 5b: A new target of breast tumor kinase/protein tyrosine kinase 6. Breast Cancer Res 2007;9:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Furqan M, Akinleye A, Mukhi N et al. STAT inhibitors for cancer therapy. J Hematol Oncol 2013;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett 2007;251:199–210. [DOI] [PubMed] [Google Scholar]
- 8. Bar‐Natan M, Nelson EA, Xiang M et al. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 2012;1:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meier JA, Larner AC. Toward a new STATe: The role of stats in mitochondrial function. Semin Immunol 2014;26:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wegrzyn J, Potla R, Chwae YJ et al. Function of mitochondrial STAT3 in cellular respiration. Science 2009;323:793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gough DJ, Corlett A, Schlessinger K et al. Mitochondrial STAT3 supports Ras‐dependent oncogenic transformation. Science 2009;324:1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mackenzie GG, Huang L, Alston N et al. Targeting mitochondrial STAT3 with the novel phospho‐valproic acid (MDC‐1112) inhibits pancreatic cancer growth in mice. PLoS One 2013;8:e61532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Genini D, Brambilla L, Laurini E et al. Mitochondrial dysfunction induced by a SH2 domain‐targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc Natl Acad Sci U S A 2017;114:E4924–E4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dando I, Dalla Pozza E, Biondani G et al. The metabolic landscape of cancer stem cells. IUBMB Life 2015;67:687–693. [DOI] [PubMed] [Google Scholar]
- 15. Zhang X, de Milito A, Olofsson MH et al. Targeting mitochondrial function to treat quiescent tumor cells in solid tumors. Int J Mol Sci 2015;16:27313–27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denise C, Paoli P, Calvani M et al. 5‐fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem‐like traits. Oncotarget 2015;6:41706–41721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vellinga TT, Borovski T, de Boer VC et al. SIRT1/PGC1alpha‐dependent increase in oxidative phosphorylation supports chemotherapy resistance of colon cancer. Clin Cancer Res 2015;21:2870–2879. [DOI] [PubMed] [Google Scholar]
- 18. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer 2009;9:798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (STAT3) protein. J Med Chem 2012;55:6645–6668. [DOI] [PubMed] [Google Scholar]
- 20. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov 2013;12:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brambilla L, Genini D, Laurini E et al. Hitting the right spot: Mechanism of action of OPB‐31121, a novel and potent inhibitor of the signal transducer and activator of transcription 3 (STAT3). Mol Oncol 2015;9:1194–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayakawa F, Sugimoto K, Harada Y et al. A novel STAT inhibitor, OPB‐31121, has a significant antitumor effect on leukemia with STAT‐addictive oncokinases. Blood Cancer J 2013;3:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oh DY, Lee SH, Han SW et al. Phase I study of OPB‐31121, an oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res Treat 2015;47:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]