

Abstract
Inherited mutations of transthyretin (TTR) destabilize its structure, leading to aggregation and familial amyloid disease. Although numerous crystal structures of WT and mutant TTRs have been determined, they have failed to yield a comprehensive structural explanation for destabilization by pathogenic mutations. To identify structural and dynamic variations that are not readily observed in the crystal structures, we used NMR to study WT TTR and three kinetically and/or thermodynamically destabilized pathogenic variants (V30M, L55P, and V122I). Sequence-corrected chemical shifts reveal important structural differences between WT and mutant TTR. The L55P mutation linked to aggressive early onset cardiomyopathy and polyneuropathy induces substantial structural perturbations in both the DAGH and CBEF β-sheets whereas the V30M polyneuropathy-linked substitution perturbs primarily the CBEF sheet. In both variants, the structural perturbations propagate across the entire width of the β-sheets from the site of mutation. Structural changes caused by the V122I cardiomyopathy-associated mutation are restricted to the immediate vicinity of the mutation site, directly perturbing the subunit interfaces. NMR relaxation dispersion measurements show that WT TTR and the three pathogenic variants undergo millisecond time scale conformational fluctuations to populate a common excited state with altered structure in the subunit interfaces. The excited state is most highly populated in L55P. The combined application of chemical shift analysis and relaxation dispersion to these pathogenic variants reveals differences in ground state structure and in the population of a transient excited state that potentially facilitates tetramer dissociation, providing new insights into the molecular mechanism by which mutations promote TTR amyloidosis.
Keywords: protein aggregation, amyloidogenesis, chemical shift, NMR relaxation dispersion, protein dynamics
For Table of Contents Use Only
Introduction
Transthyretin (TTR) is a 55kDa tetrameric protein that functions as a carrier of holoretinol binding protein and thyroxine (T4) in the blood and cerebrospinal fluid. Of the 127 amino acids that make up the TTR sequence, familial amyloidogenic mutations have currently been documented for at least 77 sites (http://www.amyloidosismutations.com). These mutations destabilize the native tetrameric quaternary structure of TTR, facilitating dissociation and monomer misfolding that results in aggregation into numerous structures including amyloid.1–3 Dissociation of the TTR tetramer is the rate-limiting step for the process of aggregation that appears to drive degeneration. The specific site and nature of each mutation results in a spectrum of amyloid phenotypes with distinct tissue tropisms. The most common include TTR sequences that deposit in the heart, resulting in Familial Amyloid Cardiomyopathy (FAC), in the peripheral and autonomic nervous system, resulting in Familial Amyloid Polyneuropathy (FAP), and the central nervous system, which results in Cerebral Amyloid Angiopathy (CAA). To understand the basis for these tissue specificities or tropisms, Sekijima et al.4 conducted an analysis of the kinetic and thermodynamic stabilities of a panel of recombinant transthyretin mutants by measuring urea denaturation midpoints and rates of tetramer dissociation. In this way, transthyretin mutants could be separated into those that are kinetically destabilized, i.e. the tetramer dissociates faster and hence the aggregation rate increases, or are thermodynamically destabilized, where once dissociated, the monomer misfolds more completely into the misfolded amyloidogenic intermediate.
The V122I mutation is the most common amyloidogenic variant, presenting as a cardiac phenotype in patients of African descent.5 The V122I tetramer is kinetically destabilized, dissociating approximately two-fold more rapidly than wild-type (WT) TTR resulting in an increase in the rate of fibril formation.6 The most common pathogenic mutation associated with polyneuropathy, V30M, is kinetically stable but is thermodynamically destabilized relative to WT TTR and is strongly amyloidogenic.4, 6 The L55P TTR tetramer is both kinetically and thermodynamically destabilized and is associated with aggressive, early-onset polyneuropathy and cardiomyopathy phenotypes.4, 6, 7
To date, more than 200 structures of normal and amyloidogenic variants of TTR and their ligand complexes have been determined using x-ray crystallography.8, 9 With a few exceptions, these structures are highly similar and provide few insights into the mechanism by which mutations destabilize TTR. Detailed comparisons have been drawn between crystal structures of transthyretin and its pathogenic mutants since the first structure of a pathogenic TTR mutant was determined over twenty years ago,10 with the aim of identifying structural defects that could explain pathogenicity. Each of the structures shows TTR assembled as a native homotetramer, around a central channel that accommodates the two T4 binding sites (Fig. 1A). Each subunit folds as a β-sandwich, with two β-sheets comprising the DAGH strands and CBEF strands, and with a short helix at the C-terminal end of the E-strand. The ‘strong’ dimer interface is formed by antiparallel arrangement of the H and H’ strands and F and F’ strands to form an extended β-sandwich, which is stabilized by backbone hydrogen bonding between the H and H’ strands; the F and F’ strands do not undergo backbone-backbone H-bonding (Fig. 1B). The F and F’ interface is further stabilized by insertion of F87 into a hydrophobic pocket in the opposing subunit. These dimers associate through the ‘weak’ dimer-dimer interface to form a tetramer through association of the AB loops and GH loops of diagonally opposite subunits, with interfaces stabilized through hydrophobic interactions and intermolecular hydrogen bonds.
Figure 1.
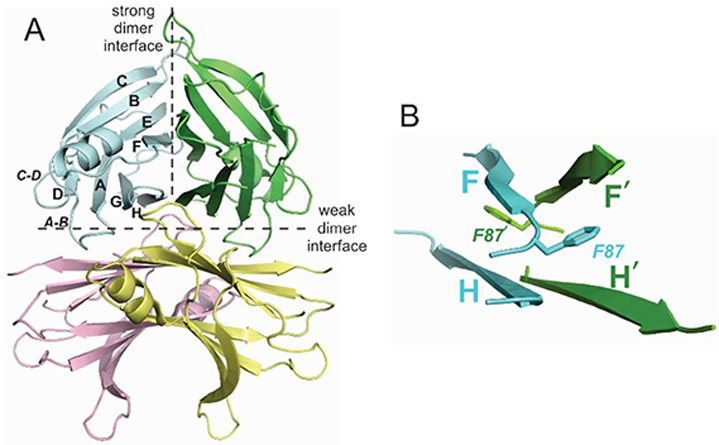
A) Structure of wild type transthyretin tetramer (PDB ID 5CNH). Protomers are shown in cyan, green, yellow and pink and the strong and weak dimer interfaces are indicated by hatched lines. B) Structure of strong dimer interface showing antiparallel arrangement of F and F’, H and H’ β-strands and location of the F87 side chain, which projects into a hydrophobic pocket in the neighboring subunit.
The absence of major differences between the x-ray structures of WT and mutant TTR suggests that the perturbations caused by the pathogenic mutations alter the dynamic and energetic landscapes of the protein, allowing it to access higher energy conformations prone to dissociation and aggregation. Regarding this hypothesis, solution NMR spectroscopy offers important advantages over other structural methods for studying the destabilizing effects of TTR mutations. Chemical shifts are a rich source of structural information, with the hydrogen, carbon and nitrogen nuclei potentially acting as probes that are sensitive to different structural perturbations.11 Furthermore, the solution NMR experiments required for chemical shift assignment may be carried out under near physiological conditions (temperature, pH, salt), free from the constraints of a crystal lattice that may affect the observed conformation of the protein. Additionally, if destabilization is a result of population of unstable higher energy conformations, these dynamic effects may be investigated using NMR relaxation methods.
Here, we have compared backbone chemical shifts of WT TTR homotetramers with those of the pathogenic V30M, L55P, and V122I mutant homotetramers and conducted 15N CPMG relaxation dispersion experiments to identify structural and dynamic differences between kinetically and thermodynamically destabilized mutant tetramers. The data reveal subtle but important changes that are not readily observed in the crystal structures. Our results show that the mutants are destabilized by highly specific structural and dynamic changes that explain the observed kinetic and thermodynamic stability differences.
MATERIALS AND METHODS
Sample preparation
TTR and its pathogenic mutants (mature sequence after signal sequence cleavage, without an affinity tag) were expressed in E. coli and purified using a 4-step protocol that includes a salt-cut step and three chromatographic steps. A pET29b(+) vector (EMD Millipore) encoding the desired TTR sequence was transformed into B121 Star (DE3) cells (ThermoFisher Scientific) and plated onto LB-agar plates with kanamycin. A single colony was grown in 1 mL LB media supplemented with kanamycin at 37 °C. The culture was adapted to growth in a deuterated background by stepwise addition of M9 media prepared in 2H2O, supplemented with 15N ammonium sulfate, and with 13C glucose for samples used for triple resonance experiments. 50ml cultures were inoculated with the adapted cells and grown overnight at 37°C. This culture was then used to inoculate a 950 mL culture at 37 °C, and cell growth was monitored by the optical density at 600 nm (OD600). When the OD600 reached 0.8, isopropyl β-D-1- thiogalactopyranoside (IPTG, final concentration of 1 mM) was added to induce over-expression of TTR and, after induction of 24–36 h, the cells were harvested by centrifugation (10 min at 13,700×g). This procedure produces TTR samples of approximately 80% deuteration, which was sufficient for both assignment and relaxation experiments.
Purification of TTR was performed at room temperature. All buffers were flushed with argon to avoid oxidation during protein purification. Fresh cell pellets were resuspended in 50 mM Tris, 150 mM NaCl (pH 7.5; 15 ml buffer/L of culture) and frozen at −80 °C. Resuspended cells were thawed and lysed by sonication at 4 °C in the presence of a protease inhibitor (1 mM PMSF). The sonication was carried out using 80% of the output power, with 2 sec on and 2 sec off, for 1 min, then cells were rested for 3 min to maintain a low temperature. This procedure was repeated a total of 6 times. Lysed cells were centrifuged for 30 min at 16,000×g. Ammonium sulfate (final concentration of 242 g/L) was slowly added to the supernatant with rigorous stirring at 4 °C for 15 min to precipitate the majority of the cellular proteins. The solution was centrifuged at 12,000 g for 15 min at 4 C. The pellet was discarded and the supernatant was supplemented with additional ammonium sulfate to a final concentration of 365 g/L, with rigorous stirring at 4 °C for 15 min. The solution was centrifuged at 12,000 g for 15 min at 4 C. The pellet was resuspended in 20–30 mL of anion exchange buffer A (25 mM Tris and 1 mM EDTA, pH 8.0) and dialyzed against 4 L of buffer A for 2 h at room temperature. After dialysis, the sample was filtered and applied to a 50-mL Source 15Q anion exchange column (Amersham Biosciences, Piscataway, NJ) equilibrated with buffer A. TTR was eluted using a linear gradient of NaCl (160 mL; 50–350 mM) followed by an NaCl wash (50 mL; 350 mM). Fractions containing TTR were pooled and further purified by repeating the anion exchange step with a linear NaCl gradient (100 mL; 50–350 mM) and a 350 mM NaCl wash (100 mL). Eluted TTR was purified using a 300 mL Superdex 75 gel filtration column (Amersham Biosciences, Piscataway, NJ) in SEC buffer (10 mM sodium phosphate, 100 mM KCl, and 1 mM EDTA, pH 7.6) to remove contaminants and any soluble aggregates. These 3 steps together yielded pure TTR (as measured by SDS-PAGE with Coomassie blue staining) in SEC buffer. Concentrations of purified TTR solutions were determined spectrophotometrically, using a molar extinction coefficient ε280 of 18450 M−1 cm−1. TTR solutions were stored at 25 °C and were used within a week of purification. The identity and purity of recombinant TTR produced using this protocol was confirmed by SDS-PAGE and high-resolution LC-ESI MS analyses. No significant impurities were identified in the purified TTR, whose purity was estimated to be > 98% based on SDS-PAGE followed by Coomassie blue staining, and no covalent modification or non-covalent small molecule ligands were present.
NMR Spectroscopy
NMR spectra were acquired using Bruker Avance spectrometers operating at 500, 750, 800, and 900 MHz and processed using NMRPipe12 and NMRFAM-SPARKY.13 V30M TTR was assigned using standard TROSY-based HNCA and HNCACB spectra14–16 acquired on samples of 0.8 mM 2H, 13C, 15N-labeled protein in 10 mM sodium phosphate, 100 mM KCl, 5% 2H20 at pH 7 and 25 °C. Assignments for the other TTR constructs at pH7 and 298K were made from TROSY-HNCA spectra using the V30M spectrum as a reference. Assignments were then transferred to spectra collected at 37 °C and pH 6 using standard TROSY-HNCO experiments14 acquired at intermediate temperatures and pH values. All assignment data were acquired at 750 MHz. Weighted average 15n/1hn chemical shift differences were calculated as .
Relaxation compensated, TROSY-based constant-time Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments for amide 15N17, 18 were performed for wild-type TTR and each of the three mutants at 500, 800 MHz, and for wild-type and L55P at 900 MHz. Data were acquired in a fully interleaved manner using 60% non-uniform sampling (NUS) in the indirect dimension and 32 scans per sampling point19. Relaxation dispersion experiments were carried out at 37 °C using 0.3 mM TTR samples in 10 mM sodium phosphate buffer, pH 6, containing 100 mM KCl and 5% 2H2O. Total relaxation time in the CPMG experiment was 40 ms. Heat compensation was achieved by placing a compensatory 40 ms CPMG block after data acquisition such that each experiment included an identical number of total pulses. Relaxation dispersion data were processed and reconstructed using NMRPipe,12 MDDNMR20 and FuDA (http://pound.med.utoronto.ca), and were fitted to the Carver-Richards equation21 using the program GLOVE.22 Global exchange rates and minor state populations were determined by simultaneously fitting the dispersion curves for each TTR variant. Errors in intensities were set to 4% for 500 MHz data and estimated using repeat experiments for the high-field data, which exhibited more severe exchange broadening. Uncertainties in the fitted exchange parameters were estimated using Monte Carlo simulations.
RESULTS
Comparison of wild type and pathogenic TTR structures
We selected three pathogenic mutants for the NMR studies based on their kinetic and thermodynamic stabilities.4 These are V30M, which has similar kinetic stability to WT TTR but lower thermodynamic stability, V122I, which has similar thermodynamic stability to WT TTR but is kinetically destabilized, and L55P, which is both kinetically and thermodynamically destabilized. Comparison of structures 1BMZ (WT),23 4TL4 (V30M),24 3DJZ (L55P)25 and 1TTR (V122I),26 all of which were crystallized without bound ligand at pHs close to pH 7, show that structural perturbations caused by the mutations are minimal, with backbone RMSD (for structured residues) relative to WT of 0.28, 0.23 and 0.38 Å for V30M, V122I and L55P, respectively. Small structural variations observed in the FG loops and terminal residues have been attributed to flexibility and differences in crystal packing and seem to be unrelated to amyloidogenicity.8, 9
Analysis of chemical shift differences
1H-15N TROSY-HSQC spectra of the wild-type and mutant proteins were acquired under identical conditions and are shown in Fig 2.
Figure 2.
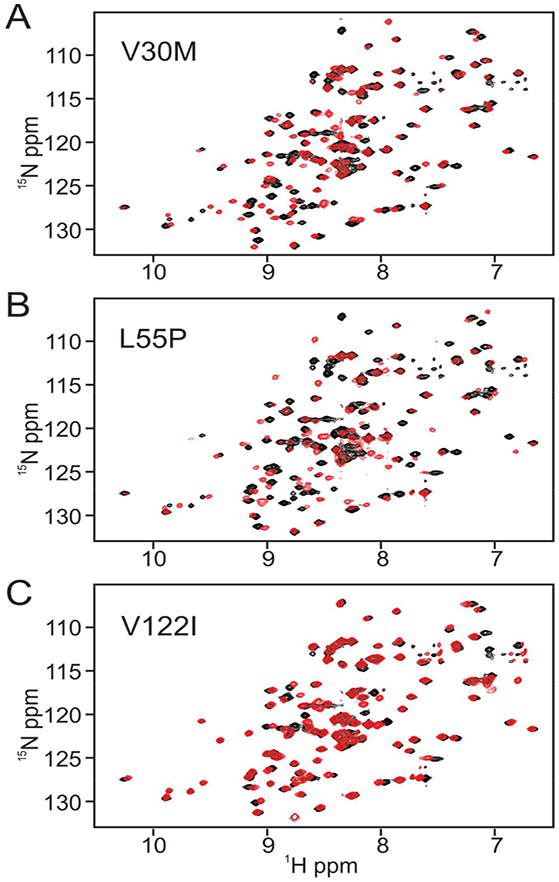
Superposition of 1H-15N TROSY-HSQC spectra of wild type (black cross peaks) and variant TTR (red cross peaks). A. wild type and V30M, B. wild type and L55P, C. wild type and V122I. All spectra were acquired at 750 MHz under identical conditions (10 mM sodium phosphate, 100 mM KCl, 5% 2H20 at pH7 and 298K).
The V30M and L55P spectra show substantial chemical shift changes for a number of cross peaks, whereas the spectrum of V122I appears largely unchanged from wild type. Backbone resonance assignments for wild type TTR and the V30M and L55P variants have been reported previously.27, 28 Since the published assignments were made under solution conditions different from those used in the current study, independent assignments for wild type TTR and the pathogenic mutants were made from triple resonance spectra. Sequence-corrected secondary shifts (observed 13Cα, amide 15N and amide proton 1Hn chemical shift minus sequence-corrected random coil shift29) were calculated to identify subtle changes in the backbone structure of the mutants, including changes at the mutation site. Differences between WT and sequence corrected chemical shifts of the mutants are shown in Table S1 and Fig. S1 and are mapped to the TTR structure in Fig. 3.
Figure 3.
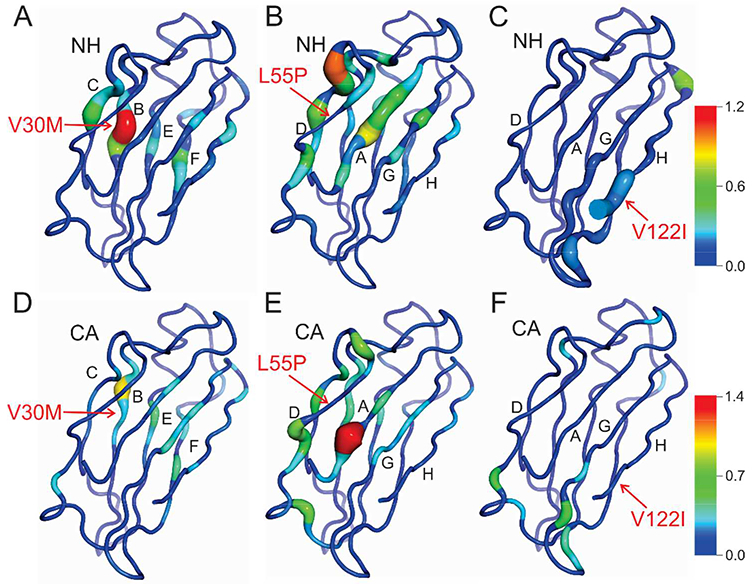
Differences in sequence corrected chemical shifts between wild-type TTR and pathogenic mutants mapped to the X-ray structure. A-C) Weighted average 15N/1Hn chemical shift differences between wild-type and A) V30M, B) L55P, and C) V122I TTR. The sites of the mutations are indicated by arrows. The protein backbone is depicted as a tube whose thickness varies in proportion to the magnitude of the chemical shift difference; the backbone color gradient ranges from dark blue (Δδav = 0 ppm) to red (Δδav = 1.2 ppm). D-F) Differences in 13Cα chemical shift between wild-type and D) V30M, E) L55P, and F) V122I TTR. The backbone color gradient varies from dark blue (Δδ 13Cα = 0 ppm) to red (Δδ 13Cα = 1.4 ppm).
Together these chemical shifts provide a complementary description of structural changes, since each nucleus is sensitive to different structural perturbations.30, 31 13Cα and 15N chemical shifts are sensitive to local sequence variation and backbone dihedral angle changes: 13Cα shifts are sensitive to φ and ψ angles for the current residue, and 15N shifts reflect the φ angle of the current residue and ψ angle of the preceding one. The 15N chemical shift is highly sensitive to hydrogen bonding to the carbonyl group of the peptide bond and somewhat less sensitive to direct hydrogen bonding to the NH group.31 1Hn shifts are sensitive to changes in backbone dihedral angles, but are also strongly affected by hydrogen bonding, ring current effects and local charge. The mutant showing the greatest overall change in chemical shifts relative to WT is L55P, while V122I shows very few shift differences, which are localized to the site of mutation. Importantly, detailed analysis of the data for each of the mutants reveals specific local and global structural changes that are not apparent from the X-ray structures.
Thermodynamic destabilization by V30M
The V30M mutation is located on strand B, where its side chain is packed into the hydrophobic core of the protomer. The crystal structure of V30M TTR is very similar to that of the WT protein except for a slight expansion of the hydrophobic core due to movement of the CBEF β-sheet away from the DAGH sheet 10, 32 Based on the NMR data, it is clear that replacement of the short β-branched valine side chain with the longer unbranched methionine side chain also alters the local backbone conformation, as indicated by ~0.3–0.9 ppm changes in 13Cα chemical shift, relative to WT, of residues in the B strand (V28-H31) that span the site of mutation (Fig. 3D). Changes in 13Cα chemical shift are also observed for V16, T49, I73, and A109, the side chains of which form a hydrophobic cluster that surrounds the M30 side chain, indicating changes in backbone conformation that propagate from the site of mutation.
The largest 15N and 1HN chemical shift perturbations are associated with residues located in the immediate vicinity of the V30M substitution (Fig. 3A, Fig. 4A). The 15N resonance of M30 shifts upfield by 6.1 ppm relative to wild-type TTR; after correcting for the differences in the Val and Met random coil shifts, this represents a 5.9 ppm change in the secondary chemical shift of residue 30. The large magnitude of this shift suggests both backbone dihedral angle changes at residues 29 and 30 (supported also by the changes in 13Cα chemical shift) and perturbation of the direct M30 NH - G47 CO and indirect A29 CO - D74 NH hydrogen bonds.30, 31 The backbone 15N resonance of the neighboring H31 is shifted 2.5 ppm upfield in the V30M mutant, suggesting that the M30 CO – G47 NH hydrogen bond may also be weakened in the mutant TTR. Weakening of the hydrogen bonds to G47 is further supported by the upfield shifts observed for the 15N resonances of G47 and K48 in V30M relative to wild type TTR. In addition to changes in hydrogen bonding between the N-terminal region of strand B and residues G47-T49 on strand C, the conformational perturbations introduced by the V30M substitution are transmitted across the CBEF β-sheet to E72 and I73 on strand E and to E92 and V93 in β-strand F (Fig. 4A). Substantial changes in 15N, 1HN, and 13Cα shifts are observed for E92 and V93 (Fig. 4A), suggesting changes in both backbone conformation and hydrogen bonding interactions. These residues are located in the strong dimer interface, approximately 10 Å from the site of mutation. The large change in 1H chemical shift (0.4 ppm) observed for the E92 amide is particularly interesting, since this residue forms both intra- and inter-subunit hydrogen bonds with the hydroxyl group of Y116.
Figure 4.
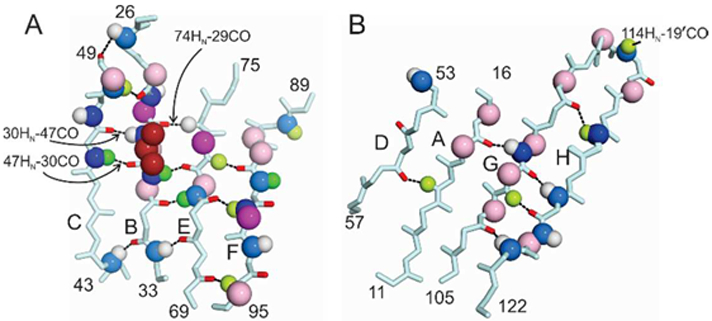
Chemical shift differences between wild-type and V30M TTR for residues in A) the CBEF β-sheet and B) the DAGH β-sheet. Chemical shift differences are indicated by colored spheres mapped to the atom involved. Dark blue, Δδ 15N > 1.0 ppm; light blue, 0.5 < Δδ 15N < 1.0 ppm; dark green, Δδ 1Hn > 0.2 ppm; pale green, 0.1 < Δδ 1Hn < 0.2 ppm; white, 0 < Δδ 1Hn < 0.1 ppm; magenta, Δδ 13Cα > 0.4 ppm; pale pink, 0.2 < Δδ 13Cα < 0.4 ppm. Hydrogen bonds formed by residues that exhibit relaxation dispersion are indicated by dotted lines, with the carbonyl oxygens colored red. The mutated side chain (V30) is shown as dark red spheres in panel A. Hydrogen bonds that are perturbed at the site of mutation are labeled.
The 15N, 1HN, and 13Cα chemical shifts of several residues in the DAGH β-sheet are also perturbed by the V30M substitution (Fig. 3, Fig. 4B), but the number of residues involved and the magnitude of the shift differences are substantially smaller than those observed for the CBEF β-sheet. These perturbations propagate from the center of strand A, opposite the site of mutation, across strands G and H to the strong dimer interface, affecting both backbone conformation (13Cα shifts) and hydrogen bonding interactions (1Hn and 15N shifts). In particular, the hydrogen bond network between T106 - A109 (strand G) and T119 – V121 (strand H) is perturbed by the V30M mutation. Additional chemical shift perturbations are observed for residues P113- S115 in the GH loop, which forms part of the weak dimer interface.
Kinetic and thermodynamic destabilization by L55P
The L55P mutation is located on the D-strand at an edge of the DAGH β-sheet. X-ray structures show that this mutation disrupts β-strand D and breaks two cross sheet hydrogen bonds to β-strand A (V14 Hn – L55CO and V14 CO – L55 HN) that are present in the wild type TTR structure.25, 33 These hydrogen bonds are also disrupted in solution, as is evident from large 1HN and 15N chemical shift changes for V14 and K15 in the L55P mutant (Fig.5A, Fig. S1). However, given the magnitude and extent of the chemical shift perturbations relative to wild type TTR, the structural changes caused by the L55P substitution appear to be more extensive in solution than observed in the X-ray structure. As for V30M, it is likely that the conformational changes in the crystal are limited by direct and water-mediated contacts between β-strands B, C, and D and residues in the BC, DE, and FG loops of a neighboring molecule in the crystal lattice.
Figure 5.
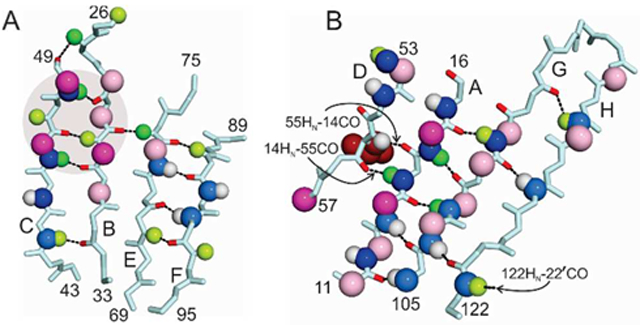
Chemical shift differences between wild-type and L55P TTR for residues in A) the CBEF β-sheet and B) the DAGH β-sheet. Chemical shift differences are indicated by colored spheres mapped to the atom involved using the same color code and chemical shift ranges as in Fig. 4. Dark blue, Δδ 15N > 1.0 ppm; light blue, 0.5 < Δδ 15N < 1.0 ppm; dark green, Δδ 1Hn > 0.2 ppm; pale green, 0.1 < Δδ 1Hn < 0.2 ppm; white, 0 < Δδ 1Hn < 0.1 ppm; magenta, Δδ 13Cα > 0.4 ppm; pale pink, 0.2 < Δδ 13Cα < 0.4 ppm. Hydrogen bonds formed by residues that exhibit relaxation dispersion are indicated by dotted lines, with the carbonyl oxygens colored red. The mutated side chain (L55) is shown as dark red spheres in panel B and critical hydrogen bonds are labeled. Residues in stands B and C that contact the L55 side chain are indicated by the shaded circle in panel A.
Loss of hydrogen bonding between strands D and A is accompanied by a rearrangement of the protein backbone in strand A, as indicated by the large change (1.4 ppm) in the 13Cα chemical shift of M13 and substantial 13Cα shift changes for residues P11, L12, and K15 (Fig. 3E, Fig. 5A). The structural perturbations in strand A propagate across the entire DAGH β-sheet to T119 in the strong dimer interface and to V122, which shows changes in 15N and 1HN chemical shift that suggest altered hydrogen bonding to G22 CO in a neighboring subunit across the weak dimer interface.
The L55P mutation also perturbs the backbone structure (13Ca chemical shift changes) and hydrogen bonding (15N and 1HN shifts) in strand C, which packs directly against the mutated side chain. These perturbations propagate through the hydrogen bonding network of the CBEF β-sheet to the strong dimer interface. The L55P mutation perturbs resonances of the entire CD loop and part of the DE loop, with large differences in 15N and/or 13Cα shifts observed for G47, T49, S50, S52, G57, L58 and T59 relative to the wild type protein. The structural changes in the CD loop are propagated, likely through changes in packing and in hydrogen bonding interactions between V16 HN - A25 CO and T49 HN - N27 CO, to the AB loop, which makes intermolecular contacts across the weak dimer interface. The changes in 15N, 1HN, and 13Cα chemical shifts of G57, L58, and T59 between L55P and WT TTR are consistent with the conformational differences observed in the X-ray structures, which reveal small differences in the backbone structure and disruption of a hydrogen bond between the C10 sulfhydryl and the G57 NH found in the WT protein. Structural perturbations across the CBEF sheet are strikingly similar to those seen in V30M, but the magnitude of the chemical shift changes are smaller.
Kinetic destabilization by V122I
The V122I mutation is located at the C-terminus of the H-strand. The side chain projects into a hydrophobic pocket formed by the side chains of Y105 and A120 on the same protomer and F87 and Y114 on the neighboring subunit (Fig.6). Comparison of the crystal structures of V122I (1TTR) and WT TTR (5CNH) indicate very modest (< 0.5 Å) structural changes in the immediate vicinity of the mutation site, including a slight shift in the side chain of Y114.26
Figure 6.
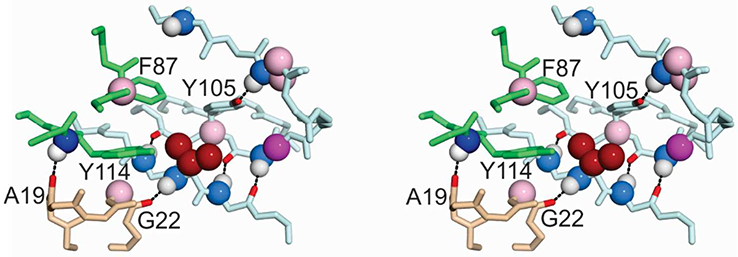
Stereo view showing chemical shift differences between wild-type and V122I TTR for residues located near the site of mutation. The mutated side chain (V122) is located on the cyan subunit and shown as dark red spheres. Parts of the neighboring subunits across the strong dimer interface (green) and the weak dimer interface (tan) are shown. Chemical shift differences are indicated by colored spheres mapped to the atom involved using the same color code and chemical shift ranges as in Fig. 4. Dark blue, Δδ 15N > 1.0 ppm; light blue, 0.5 < Δδ 15N < 1.0 ppm; dark green, Δδ 1Hn > 0.2 ppm; pale green, 0.1 < Δδ 1Hn < 0.2 ppm; white, 0 < Δδ 1Hn < 0.1 ppm; magenta, Δδ 13Cα > 0.4 ppm; pale pink, 0.2 < Δδ 13Cα < 0.4 ppm. Hydrogen bonds formed by residues that exhibit relaxation dispersion are indicated by dotted lines, with the carbonyl oxygens and Y105 hydroxyl atom colored red.
In contrast to V30M and L55P, the chemical shifts of V122I and WT TTR are very similar, with only a small number of residues showing significant differences (Fig. 3C, Fig. 3F, Fig. S1). These residues are clustered near the site of mutation, both within the protomer bearing the mutation and also in neighboring subunits where residues proximal to V/I122 exhibit small 15N, 1HN, or 13Cα chemical shift changes (Fig. 6). Most of the chemical shift changes between V122I and wild-type TTR are very small, suggesting that structural perturbations caused by the mutation are minor, consistent with the similarity in their crystal structures. The structural perturbations appear to propagate from the site of mutation through subtle changes in the hydrophobic core that are required to accommodate the bulky I122 side chain, as well as through altered hydrogen bonding interactions between the H and G β-strands and between the Y105 side chain hydroxyl and the N98 backbone amide, an interaction that is thought to stabilize the FG loop.34 The effects of the mutation are also transmitted to neighboring subunits, as manifest in changes in the 13Cα chemical shifts of G22 and F87, and the 15N chemical shift of Y114 (Fig. 6, Fig. S1). The side chains of F87 and Y114 pack against V122 in the neighboring subunit across the strong dimer interface, while the A19 and G22 carbonyl oxygen atoms are hydrogen bonded to the Y114 and V122 amides, respectively, the only two inter-protomer hydrogen bonds in the weak dimer-dimer interface. Thus, the V122I mutation causes small and highly localized structural perturbations that are transmitted to neighboring subunits across both the strong and weak subunit interfaces.
15N relaxation dispersion experiments
The chemical shift analysis described above provides insights into differences in structure and hydrogen bonding in the ground states of WT TTR and the amyloidogenic variants. To probe μs-ms time scale fluctuations of the ground state conformation, 15N R2 relaxation dispersion measurements were performed for WT and the variant TTRs. In preliminary experiments using a number of independently prepared samples of WT and V30M TTR, a consistent subset of residues exhibited dispersion but the magnitude of the exchange contributions (Rex) was found to be variable depending on expression and purification procedures and solution conditions. To circumvent this problem and allow reliable comparisons of the relaxation data for WT, V30M, L55P, and V122I, a matched set of samples was expressed and purified under identical, carefully controlled conditions (see Materials and Methods). For each TTR variant, 15N R2 dispersion data were acquired simultaneously at spectrometer frequencies of 500 and 800 MHz at pH 6.0 and 37 °C. Representative relaxation dispersion curves are shown in Fig. 7 and dispersion profiles for all residues that undergo dispersion are shown in Figs. S2 – S5. Apart from the residues shown in these figures, the relaxation rates for all other residues, including those located near the sites of mutation, are independent of the CPMG pulse spacing, i.e. their dispersion profiles are flat. Due to sample aggregation during acquisition, the 800 MHz data for L55P were too noisy for analysis. However, dispersion curves of adequate quality were obtained for matched WT/L55P samples at 500 and 900 MHz.
Figure 7.
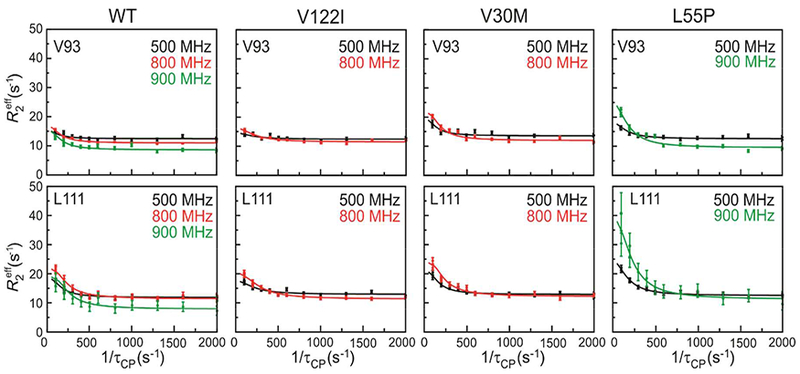
15N relaxation dispersion profiles for V93 and L111 in WT, V122I, V30M, and L55P TTR measured at 500 MHz (black), 800 MHz (red), and 900 MHz (green). Solid lines show the best fit to a global two-site exchange model.
15N relaxation dispersion was observed for a conserved subset of residues in WT TTR and in the V30M, L55P, and V122I variants: E89 and V93 in β-strand F; A108, A109, L110, and L111 in β-strand G; and S115, S117, T118, and V121 in β-strand H (Fig. 8; a stereo version of this figure is shown as Fig. S6). The amide nitrogens in strands F and H that exhibit dispersion belong to peptide groups that form direct hydrogen bonds to strands F’ and H’ across the strong dimer interface or, in the case of V93, are hydrogen bonded to the hydroxyl of Y116 in strand H’ of the neighboring subunit. The conformational fluctuations in strand H are transmitted, apparently via the inter-strand hydrogen bond network, to peptide groups in strand G. Relaxation dispersion was also observed for the 15N resonance of A19 for all TTR variants, showing the presence of μs-ms fluctuations in the weak dimer interface.
Figure 8.
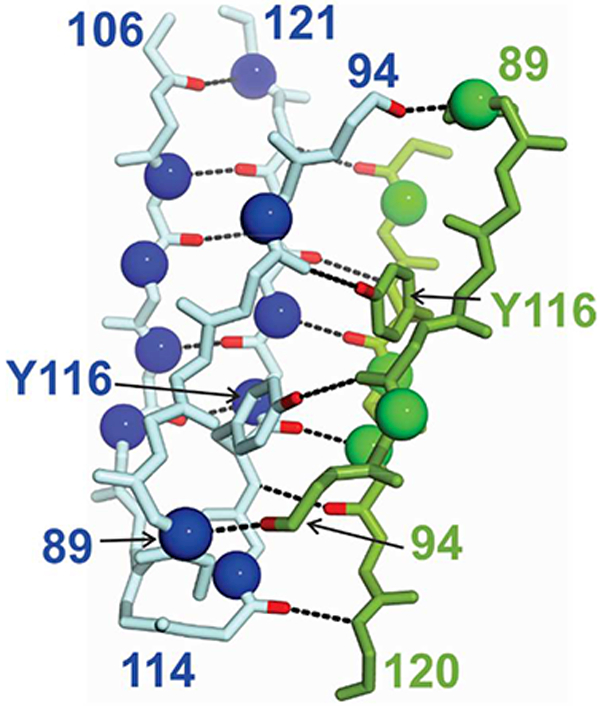
Location of residues in β-strands G and H that exhibit 15N relaxation dispersion in all TTR variants studied, mapped to the TTR structure. For clarity, only the H strand of the green subunit is shown. The nitrogen atoms of the residues that display dispersion are shown as blue or green spheres. Hydrogen bonds are indicated by dotted lines and the carbonyl atoms involved are colored red. The side chain of Y116, which hydrogen bonds to the E92 carbonyl, is shown.
The relaxation dispersion data were fitted to a two-site exchange model using the Carver and Richards equations21 to determine the exchange rate constant (kex), the populations of the two states (pa and pb), and the chemical shift difference between them (Δϖ, in ppm). For each TTR variant, the dispersion data were fitted to global values of kex and pb (Table 1).
Table 1.
Kinetic parameters derived from relaxation dispersion measurements
| TTR variant | kex (s−1) | Pb (%) |
|---|---|---|
| WT | 410 ± 23 | 3.9 ± 0.2 |
| V30M | 380 ± 33 | 4.4 ± 0.3 |
| L55P | 550 ± 34 | 10.6 ± 3 |
| V122I | 660 ± 39 | 3.3 ± 0.4 |
The same subset of residues exhibits 15N relaxation dispersion in WT TTR and all of the variants, suggesting that each of the proteins undergoes conformational exchange between its ground state and a common excited state. This is supported by the fitted Δϖ values for wild type, V122I, and L55P, which are very similar for any given residue in the subunit interfaces (Table S2). The Δϖ values for residues in strand G of L55P differ somewhat from those of wild type and V122I, which likely reflects differences in the structure of the DAGH sheet. The cross peaks for residues 89, 117, and 118 of V30M are broader and weaker than for other variants; the dispersion profiles are consequently very noisy and were excluded from the analysis. However, Δϖ for other residues in the subunit interfaces (A19, V93, and S115) are similar to those of the other proteins (Table S2). While all of the proteins appear to adopt a similar excited state, the kinetics with which it is formed and its equilibrium population differs for each variant (Table 1).
DISCUSSION
The transthyretin aggregation pathway has been elucidated through extensive biochemical studies. Aggregation is initiated by dissociation of the TTR tetramer to form a misfolded monomeric intermediate,1–3, 35 which rapidly aggregates by a downhill polymerization mechanism.36, 37 While the overall mechanistic pathway is well established, the mechanism by which pathogenic mutations promote tetramer dissociation and monomer unfolding are poorly understood at the molecular level. The numerous X-ray structures of wild type, non-amyloidogenic, and amyloidogenic variant human TTRs are virtually identical and have failed to reveal structural changes that might explain amyloidogenicity8, 9
Our motivation for analyzing the chemical shift data for the amyloidogenic V30M, L55P, and V122I mutants, each of which is destabilized by a different combination of kinetic and thermodynamic effects, was to provide a different perspective from that available from X-ray crystallography and to clarify the significance of the minor structural differences observed in the crystal structures. The chemical shift data suggest that the pathogenic mutations introduce a number of subtle but significant structural changes to the TTR ground state that are either not observed in the crystal structures due to constraints of crystal packing, or are so small as to not be readily observed in the X-ray structures.
The V30M and L55P substitutions induce substantial 15N, 1HN, and 13Cα chemical shift perturbations relative to wild-type (Fig. S1), which propagate across the β-sheets from the site of mutation to the strong dimer interface (Figs. 3–5). Although the V30 and L55 side chains are in contact in the wild type structure and are part of the same hydrophobic cluster, the two mutations perturb the TTR protomer structure in different ways. The V30M substitution perturbs the CBEF β-sheet but has a much smaller effect on residues in the DAGH sheet, whereas the L55P mutation induces substantial perturbations into both β-sheets. Both the X-ray structures and NMR chemical shifts show that wild-type TTR and the V122I variant adopt very similar conformations, with structural changes limited to the immediate vicinity of the mutation (Fig.3, Fig 6).
The observation that the structure and hydrogen bonding in the CBEF β-sheet of V30M is perturbed to a greater extent than that of the DAGH sheet is consistent with the differences in sheet-to-sheet packing observed in the X-ray structures of wild type and V30M TTR.10, 32 The substantial changes in 13Cα chemical shifts for a subset of residues in strands B, E, and F indicate changes, mostly subtle and not discernible in the X-ray structures, in backbone dihedral angles across the central region of the CBEF sheet (Fig. 4). The largest changes in local backbone conformation occur at the site of mutation, as indicated by the large changes in the A29 13Cα and M30 15N chemical shifts; a small backbone conformational difference at this site is confirmed by comparison of wild-type (1BMZ, 4L1T, 5CN3, 5CNH) and V30M (1TTC, 3KGS, 4PWE, 4TL4) structures in the Protein Data Bank. Changes in 15N and 1HN shifts show that the structural changes are accompanied by perturbation of the cross sheet hydrogen bonding network, propagating across the entire central region of the CBEF β-sheet. The upfield secondary 15N chemical shift of M30 relative to V30 in wild-type TTR is much larger (5.9 ppm, corrected for differences in the intrinsic Val/Met chemical shift29) than the 2–3 ppm expected30 based on the differences in backbone dihedral angles of residues 29 and 30 in the X-ray structures. Density functional theory indicates that the peptide 15N resonance is exquisitely sensitive to hydrogen bond formation, shifting strongly upfield by as much as ~5.5 ppm upon weakening or rupture of the direct (to the NH) and/or indirect (to the CO) hydrogen bonds.’31 Thus the difference between the observed and structure-based estimate of the M30 15N shift suggests additional contributions from weakening of the indirect A29 CO - D74 NH and direct M30 NH - G47 CO hydrogen bonds in the V30M mutant. The perturbations of the CBEF sheet indicated by the chemical shifts are in accord with hydrogen - deuterium exchange measurements which show that the V30M mutation destabilizes the B and E strands.27
The L55P substitution results in larger and more extensive chemical shift perturbations than V30M, affecting residues in both the DAGH and CBEF β-sheets (Fig. 3, Fig. 5, Fig. S1). In X-ray structures of the L55P variant, residues 54–56 move away from strand A, disrupting strand D and its hydrogen bonding interactions with strand A.25, 33 Strand D is also disrupted in solution, as evidenced by the large changes in 13Cα, 15N, and 1HN chemical shifts for residues 52–59 in the L55P variant. Whereas strand A adopts very similar conformations in the crystal structures, the large chemical shift differences observed for residues L12-L17, encompassing the entire length of the β-strand, show that in solution the strand A conformation varies substantially in L55P and WT TTR. This conformational perturbation is transmitted through the hydrogen bond network to strands G and H. The NMR data reveal much more substantial perturbations of the structure of and hydrogen bonding within the DAGH sheet than is evident from the X-ray structures, probably because the secondary and tertiary structure is stabilized in the crystal by direct and water-mediated contacts between β-strands B, C, and D and neighboring molecules in the crystal lattice.
The extent of perturbation of the NMR spectra of the TTR variants provides qualitative insights into their relative thermodynamic stabilities. The similarity of the chemical shifts of wild-type and V122I TTR indicates that the mutation can be accommodated with only minor changes in structure, consistent with their similar thermodynamic stabilities.4, 38 In contrast, the highly pathogenic V30M and L55P mutations result in extensive perturbation of the TTR structure and accordingly these variants are strongly destabilized relative to WT TTR.4, 38, 39
Although V30M is thermodynamically less stable than L55P, the L55P variant is more strongly amyloidogenic and is associated with more aggressive, early onset disease.4 Our present NMR studies provide new molecular level insights into potential factors that make L55P so aggregation prone. Among the three amyloidogenic TTR variants investigated in this work, only the L55P substitution results in substantial perturbation of the DAGH β-sheet (Fig. 3, Fig 5). Several recent studies implicate destabilization of the DAGH sheet as a driving force for TTR aggregation and amyloid formation. NMR experiments reveal millisecond time scale structural fluctuations in the DAGH β-sheet of a monomeric TTR variant (M-TTR) that become exaggerated as the pH is lowered to form an aggregation competent, amyloidogenic state.40 In contrast, the CBEF sheet retains a stable, native-like fold, both at neutral pH and under conditions that favor amyloid formation.40, 41 Solid state NMR confirms that the CBEF sheet remains intact in TTR amyloid and suggests that disruption of the strand D – strand A interactions play an important role in amyloid formation by V30M and L55P TTR.42, 43 Our chemical shift data show that the L55P mutation perturbs the structure of the DAGH β-sheet to a much greater extent than V30M, suggesting a plausible explanation for the enhanced amyloidogenicity of L55P despite its greater thermodynamic stability.
While the chemical shifts provide insights into the molecular basis by which the V30M, L55P, and V122I mutations destabilize the TTR protomer and facilitate local unfolding, aggregation and amyloid formation, they provide little insight into the primary step in amyloidosis, dissociation of the TTR tetramer. There is considerable evidence that aggregation of folded globular proteins is initiated by transient formation of aggregation-prone states that are populated by thermal fluctuations of the native (ground) state.40, 44, 45 To obtain insights into processes that may promote dissociation of the TTR tetramer, we performed NMR relaxation dispersion experiments to probe ms time scale conformational fluctuations that populate higher energy excited states. Nuclei that undergo relaxation dispersion report on conformational exchange between the ground state and excited state(s) in which they experience a different chemical shift (Δϖ). Remarkably, a completely conserved subset of residues exhibits 15N R2 dispersion in wild-type TTR and in all three pathogenic variants, suggesting that all four proteins transiently populate a similar excited state in which the 15N chemical shifts differ from those in the ground state. Of the residues that exhibit dispersion, the E89, V93, S115, S117, and T118 nitrogen atoms belong to peptide groups in β-strands F and H that are directly involved in backbone or side chain (Y116 hydroxyl) hydrogen bonding interactions across the strong dimer interface (Fig. 8). The magnitude of Δϖ for each residue is similar for each of the TTR variants (Table S2), confirming that each variant adopts a similar excited state structure with a perturbed subunit interface. 15N relaxation dispersion is also observed for A19, which hydrogen bonds to Y114 NH across the weak dimer interface; again, the chemical shift differences between ground and excited state are the same for each variant (other than L55P where cross peak overlap precluded quantitative analysis of the data). Conformational fluctuations are also observed for residues in the G strand (Fig. 8) but Δϖ values are more variable here, which may reflect differences in the structure and stability of the DAGH sheet in the various mutants.
The observation that, through thermal fluctuations, each of the TTR variants transiently samples a common excited state with altered structure in the strong and weak dimer interfaces hints at a possible role in tetramer dissociation. Indeed it is notable that, of the TTR variants studied, the L55P tetramer forms the highest population of the excited stated (10.6%, Table 1), forms the least stable tetramer, and has the highest pH optimum (pH 5.0) for fibril formation.4 Wild type, V122I, and V30M TTR have lower excited state populations (3.9, 3.3, and 4.4%, respectively, Table 1), form stable tetramers, and have lower optimal pH for fibril formation (pH 4.2, 4.2, and 4.4, respectively).4 Although V30M has a strong propensity to aggregate, the V30M tetramer is at least as stable as wild type TTR, consistent with its similar excited-state population; however, the V30M monomer, once formed, is thermodynamically destabilized and readily forms aggregates.4, 38 V122I is an apparent exception to the trend, in that its excited state population is comparable to wild type TTR yet its tetramer is destabilized and dissociates 2-fold more rapidly.6, 38 This apparent discrepancy may reflect ground-state destabilization; the mutated side chain directly contacts F87 and Y114 in a neighboring protomer and, based on chemical shift changes, causes localized structural perturbations in neighboring subunits across both the strong and weak dimer interfaces (Fig. 6). We note also that the rate of exchange between ground and excited states appears to be faster for V122I (Table 1). In future work it will be interesting to examine additional TTR variants to seek more quantitative insights into the nature of the excited state and the factors that mediate tetramer dissociation.
A previous report of 15N relaxation dispersion experiments on WT TTR and the V30M and L55P mutants is in qualitative agreement with our current results, revealing structural fluctuations in the F, G, and H strands and the AB loop that mediate the propensity to form fibrils.46 However, the published data exhibit considerable variation in the location of the dispersing residues and in the magnitude of the exchange contributions (Rex) for the different TTR variants, similar to the results we obtained in preliminary experiments when expression and purification protocols were not sufficiently well controlled. Despite differences in pH, the reported exchange rates (kex) are the same as ours within experimental uncertainties but the excited state populations are higher.46 In addition, data for the V30M and L55P mutants were acquired at a different temperature from wild type, making quantitative comparison difficult. As detailed in the experimental section, great care was taken in the present work to produce carefully matched TTR samples and acquire relaxation dispersion data under identical solution conditions, which likely accounts for the differences between the previous results and those reported here.
Our current data demonstrate the value of using multiple complementary structural methods to interpret and validate observations on subtle changes in protein structure. The plethora of X-ray crystallographic studies on TTR have provided a huge dataset for detailed analysis of structural differences between wild-type TTR and its pathogenic mutants; however for most mutants, the structural differences are very subtle and are smaller than the differences that arise from crystal packing.8, 9 Indeed, for the structures of both V30M and V122I, our data suggest that the destabilizing effect of the mutation is partly obscured by crystal contacts close to the mutation site. Solution NMR methods are not subject to the constraints imposed by a crystal lattice, and provide important and complementary structural data to that available from crystallographic methods. The backbone 15N, 1HN, and 13Cα chemical shifts are exquisitely sensitive to subtle differences between the ground state structures of the wild type and mutant TTR tetramers, revealing pathways by which structural perturbations propagate from the site of mutation to distant regions of the protein structure. Complementary R2 relaxation dispersion experiments provide new insights into thermally accessible excited states, in which there are structural perturbations in the subunit interfaces that potentially facilitate tetramer dissociation. The combined application of these NMR methods to the pathogenic V30M, L55P, and V122I mutants reveal differences in ground state structure and in the population of a transient excited state, advancing our understanding of the molecular mechanisms by which mutations promote transthyretin amyloidosis.
Supplementary Material
Acknowledgements
We thank Gerard Kroon and Phillip Aoto for expert assistance with NMR experiments and Euvel Manlapaz for technical support.
Funding
This work was supported by National Institutes of Health grants DK34909 (P.E.W.) and DK46335 (J.W.K.) and the Skaggs Institute for Chemical Biology (P.E.W. and J.W.K.)
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS publications website at DOI: xxx. Six figures showing all data for the chemical shift differences between mutant and wild-type TTR; relaxation dispersion data for wild type, V30M, L55P and V122I mutant proteins; and a stereo version of Figure 8, and a table showing values of 15N Δϖ (ppm) derived from fits of relaxation dispersion data for wild type, V122I, L55P, and V30M TTR using kex and pb values listed in Table 1.
ACCESSION CODES
Backbone resonance assignments have been deposited in the Biological Magnetic Resonance Data Bank under accession numbers 27513 (L55P), 27514 (wild type TTR), 27515 (V30M), and 27516 (V122I).
References
- [1].Colon W, and Kelly JW (1992) Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro, Biochemistry 31, 8654–8660. [DOI] [PubMed] [Google Scholar]
- [2].Lai Z, Colon W, and Kelly JW (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid, Biochemistry 35, 6470–6482. [DOI] [PubMed] [Google Scholar]
- [3].Quintas A, Vaz DC, Cardoso I, Saraiva MJ, and Brito RM (2001) Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants, J. Biol. Chem 276, 27207–27213. [DOI] [PubMed] [Google Scholar]
- [4].Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, and Kelly JW (2005) The biological and chemical basis for tissue-selective amyloid disease, Cell 121, 73–85. [DOI] [PubMed] [Google Scholar]
- [5].Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, and Buxbaum JN (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans, N. Engl. J. Med 336, 466–473. [DOI] [PubMed] [Google Scholar]
- [6].Hammarstrom P, Jiang X, Hurshman AR, Powers ET, and Kelly JW (2002) Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity, Proc. Natl. Acad. Sci. U.S.A 99 Suppl 4, 16427–16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jacobson DR, McFarlin DE, Kane I, and Buxbaum JN (1992) Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement, Hum Genet 89, 353–356. [DOI] [PubMed] [Google Scholar]
- [8].Hörnberg A, Eneqvist T, Olofsson A, Lundgren E, and Sauer-Eriksson AE (2000) A comparative analysis of 23 structures of the amyloidogenic protein transthyretin, J. Mol. Biol 302, 649–669. [DOI] [PubMed] [Google Scholar]
- [9].Palaninathan SK (2012) Nearly 200 X-ray crystal structures of transthyretin: what do they tell us about this protein and the design of drugs for TTR amyloidoses?, Curr. Med. Chem 19, 2324–2342. [DOI] [PubMed] [Google Scholar]
- [10].Hamilton JA, Steinrauf LK, Liepnieks J, Benson MD, Holmgren G, Sandgren O, and Steen L (1992) Alteration in molecular structure which results in disease: the Met-30 variant of human plasma transthyretin, Biochim. Biophys. Acta. Mol. Basis Disease 1139, 9–16. [DOI] [PubMed] [Google Scholar]
- [11].Wishart DS, and Case DA (2001) Use of chemical shifts in macromolecular structure determination, Methods Enzymol 338, 3–34. [DOI] [PubMed] [Google Scholar]
- [12].Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, and Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- [13].Lee W, Tonelli M, and Markley JL (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy, Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kay LE, Ikura M, Tschudin R, and Bax A (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins, J. Magn. Reson 89, 496–514. [DOI] [PubMed] [Google Scholar]
- [15].Grzesiek S, and Bax A (1992) Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein, J. Magn. Reson 96, 432–440. [Google Scholar]
- [16].Wittekind M, and Mueller L (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins, J. Magn. Reson 101, 201–205. [Google Scholar]
- [17].Loria JP, Rance M, and Palmer AG III. (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy, J. Am. Chem. Soc 121, 2331–2332. [Google Scholar]
- [18].Loria JP, Rance M, and Palmer AG (1999) A TROSY CPMG sequence for characterizing chemical exchange in large proteins, J. Biomol. NMR 15, 151–155. [DOI] [PubMed] [Google Scholar]
- [19].Aoto PC, Fenwick RB, Kroon GJA, and Wright PE (2014) Accurate scoring of non-uniform sampling schemes for quantitative NMR, J. Magn. Reson 246, 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Orekhov VY, and Jaravine VA (2011) Analysis of non-uniformly sampled spectra with multi-dimensional decomposition, Prog. NMR Spect 59, 271–292. [DOI] [PubMed] [Google Scholar]
- [21].Carver JP, and Richards RE (1972) General 2-site solution for chemical exchange produced dependence of T2 upon Carr-Purcell pulse separation, J. Magn. Reson 6, 89–105. [Google Scholar]
- [22].Sugase K, Konuma T, Lansing JC, and Wright PE (2013) Fast and accurate fitting of relaxation dispersion data using the flexible software package GLOVE, J. Biomol. NMR 56, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, and Kelly JW (1998) Inhibiting transthyretin conformational changes that lead to amyloid fibril formation, Proc. Natl. Acad. Sci. USA 95, 12956–12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Saelices L, Johnson LM, Liang WY, Sawaya MR, Cascio D, Ruchala P, Whitelegge J, Jiang L, Riek R, and Eisenberg DS (2015) Uncovering the Mechanism of Aggregation of Human Transthyretin, J. Biol. Chem 290, 28932–28943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cendron L, Trovato A, Seno F, Folli C, Alfieri B, Zanotti G, and Berni R (2009) Amyloidogenic potential of transthyretin variants: insights from structural and computational analyses, J. Biol. Chem 284, 25832–25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Damas AM, Ribeiro S, Lamzin VS, Palha JA, and Saraiva MJ (1996) Structure of the Val122Ile Variant Transthyretin - a Cardiomyopathic Mutant, Acta Cryst. D 52, 966–972. [DOI] [PubMed] [Google Scholar]
- [27].Liu K, Kelly JW, and Wemmer DE (2002) Native State Hydrogen Exchange Study of Suppressor and Pathogenic Variants of Transthyretin, J. Mol. Biol 320, 821–832. [DOI] [PubMed] [Google Scholar]
- [28].Liu K, Cho HS, Hoyt DW, Nguyen TN, Olds P, Kelly JW, and Wemmer DE (2000) Deuterium-proton exchange on the native wild-type transthyretin tetramer identifies the stable core of the individual subunits and indicates mobility at the subunit interface, J. Mol. Biol 303, 555–565. [DOI] [PubMed] [Google Scholar]
- [29].Nielsen JT, and Mulder FAA (2018) POTENCI: prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins, J. Biomol. NMR [DOI] [PubMed] [Google Scholar]
- [30].Wishart DS, and Nip AM (1998) Protein chemical shift analysis: a practical guide, Biochem. Cell Biol 76, 153–163. [DOI] [PubMed] [Google Scholar]
- [31].Xu XP, and Case DA (2002) Probing multiple effects on 15N, 13Cα, 13Cβ, and 13C’ chemical shifts in peptides using density functional theory, Biopolymers 65, 408–423. [DOI] [PubMed] [Google Scholar]
- [32].Hamilton JA, Steinrauf LK, Braden BC, Liepnieks J, Benson MD, Holmgren G, Sandgren O, and Steen L (1993) The X-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30→Met variant to 1.7 Å resolution, J. Biol. Chem 268, 2416–2424. [PubMed] [Google Scholar]
- [33].Sebastiᾶo MP, Saraiva MJ, and Damas AM (1998) The Crystal Structure of Amyloidogenic Leu55-Pro Transthyretin Variant Reveals a Possible Pathway for Transthyretin Polymerization into Amyloid Fibrils, J. Biol. Chem 273, 24715–24722. [DOI] [PubMed] [Google Scholar]
- [34].Blake CC, Geisow MJ, Oatley SJ, Rerat B, and Rerat C (1978) Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A, J. Mol. Biol 121, 339–356. [DOI] [PubMed] [Google Scholar]
- [35].McCutchen SL, Colon W, and Kelly JW (1993) Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity, Biochemistry 32, 12119–12127. [DOI] [PubMed] [Google Scholar]
- [36].Jiang X, Smith CS, Petrassi HM, Hammarström P, White JT, Sacchettini JC, and Kelly JW (2001) An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured, Biochemistry 40, 11442–11452. [DOI] [PubMed] [Google Scholar]
- [37].Hurshman AR, White JT, Powers ET, and Kelly JW (2004) Transthyretin Aggregation under Partially Denaturing Conditions Is a Downhill Polymerization, Biochemistry 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- [38].Hurshman Babbes AR, Powers ET, and Kelly JW (2008) Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis, Biochemistry 47, 6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shnyrov VL, Villar E, Zhadan GG, Sanchez-Ruiz JM, Quintas A, Saraiva M. J. ú., and Brito RMM (2000) Comparative calorimetric study of non-amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin, Biophys. Chem 88, 61–67. [DOI] [PubMed] [Google Scholar]
- [40].Lim KH, Dyson HJ, Kelly JW, and Wright PE (2013) Localized structural fluctuations promote amyloidogenic conformations in transthyretin, J. Mol. Biol 425, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lim KH, Dasari AKR, Hung I, Gan Z, Kelly JW, and Wemmer DE (2016) Structural Changes Associated with Transthyretin Misfolding and Amyloid Formation Revealed by Solution and Solid-State NMR, Biochemistry 55, 1941–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lim KH, Dasari AKR, Ma R, Hung I, Gan Z, Kelly JW, and Fitzgerald MC (2017) Pathogenic Mutations Induce Partial Structural Changes in the Native β-Sheet Structure of Transthyretin and Accelerate Aggregation, Biochemistry 56, 4808–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lim KH, Dasari AKR, Hung I, Gan Z, Kelly JW, Wright PE, and Wemmer DE (2016) Solid-State NMR Studies Reveal Native-like β-Sheet Structures in Transthyretin Amyloid, Biochemistry 55, 5272–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chiti F, and Dobson CM (2009) Amyloid formation by globular proteins under native conditions, Nat. Chem. Biol 5, 15–22. [DOI] [PubMed] [Google Scholar]
- [45].Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundström P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, and Kay LE (2012) Structure of an intermediate state in protein folding and aggregation, Science 336, 362–366. [DOI] [PubMed] [Google Scholar]
- [46].Das JK, Mall SS, Bej A, and Mukherjee S (2014) Conformational Flexibility Tunes the Propensity of Transthyretin to Form Fibrils Through Non-Native Intermediate States, Angew. Chem. Int. Ed. Engl 53, 12781–12784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.