

Abstract
Salvinorin A (SalA) is a potent and selective agonist of the kappa-opioid receptor (KOR), but its instability has frustrated medicinal chemistry efforts. Treatment of SalA with weak bases like DBU leads to C8 epimerization with loss of receptor affinity and signaling potency. Here we show that replacement of C20 with H and replacement of O6 with CH2 stabilizes the SalA scaffold relative to its C8 epimer, so much so that epimerization is completely supressed. This new compound, O6C-20-nor-SalA, retains high potency for agonism of KOR.
Keywords: Salvinorin, Kappa opioid receptor, Total Synthesis, Heck reaction, Conformational analysis
Graphical abstract
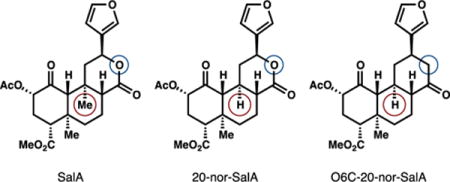
Salvinorin A (SalA) is a promising lead compound for the selective agonism of kappa-opioid receptors (KOR) in preference to mu- or delta-opioid receptors (MOR, DOR).1,2,3,4 The SalA scaffold is unstable, however, and epimerizes at C8 to a 240-fold less potent isomer, favored 2.5 to 1.5,6,7,8 We recently identified angular methyl C20 as a main contributor to scaffold instability due to a 1,3-diaxial interaction with the C12 hydrogen, which is relieved by epimerization.9 We found that deletion of C20 reversed the stability of the SalA scaffold and caused the potent isomer to predominate (1:2.5). This new lead compound, 20-nor-SalA, retained the high potency and selectivity of SalA itself, and its facile synthesis allowed the generation of new aryl analogs using a carboxylate-directed Heck reaction.9 We hypothesized that further scaffold stabilization might be possible by removing the restriction of the small dihedral angle (0° ideal)10 enforced by the C-ring lactone. Here we show that O→C replacement (lactone→cyclo-hexanone), combined with C20 deletion, stabilizes the SalA scaffold to the point where no epimerization is observed under conditions that equilibrate SalA. This new lead compound also retains the high affinity and potency of both SalA and 20-nor-SalA.
Although the structural complexity of 20-nor-SalA is equivalent to that of SalA, its synthetic complexity is markedly reduced – a relationship we identified through dynamic retrosynthetic analysis.9 Furthermore, 20-nor-SalA is significantly more stable, resulting in reduced epimerization of its C8 carbon. Epimerization is not, however, completely eliminated. We wondered if part of the driving force for C8 epimerization derives from the embedded ester, which strains the ring system due to semi-planarity, but cannot reach full planarity given the trans-ring junction (25° dihedral angle C12→C8, determined by x-ray crystallography, Ref. 9). Epimerization to a cis-fused junction relieves angle strain and allows better overlap of the O6 lonepair with the C17 carbonyl (7° dihedral angle C12→C8 in x-ray structure of 8-epi-1, Ref. 8). This destabilization of the more potent trans-ring fused isomer should not occur if a carbon atom replaced the O6 oxygen atom (similar atom replacement strategies have been used to impressive effect by Dale Boger,11 to whom this paper and issue of Biorg. Med. Chem. Lett. are dedicated). Instead, the trans-fused cyclohexanone should predominate over cis -based on a nearly ideal dihedral angle12 from C12→C8 and penalizing over-ring sterics from the aryl group in the C8 epimer.
Fortunately, this hypothesis was easy to test by synthesis. We could easily access intermediate 5 in only 6 steps from commercially available Hagemann’s ester (4), as reported previously.9 In our original route, introduction of the A-ring oxygen substituent was achieved by deprotonation with LDA, followed by oxidation with Davis’ oxaziridine. However, removal of sulfonamide byproducts proved difficult, so we instead explored the use of diatomic oxygen. Bubbling O2 gas through a solution of doubly-deprotonated 5, followed by addition of triethylphosphite delivered 6 in good yield. Acetylation and epimerization occurred uneventfully according to our reported procedure.
Introduction of the furyl substitutent by a Heck reaction would normally be challenging due to the low reactivity of hindered, unbiased alkenes and the competitive decomposition of 3-bromofuran. Its efficient attachment served a testament to the considerable rate acceleration effected by directivity from the nearby carboxylate.9 This carboxylic acid was transformed into a competent carbon nucleophile by derivatization to a β-keto-ester in two steps. First, reaction with carbonyl diimidazole (CDI) converted the carboxylic acid to its acyl imidazole. Second, magnesium chloride and triethylamine promoted addition of potassium methylmalonate into the acyl imidazole; decarboxylation delivered β-keto-ester 9. Even though the ester substituent is extraneous to the target structure (3), we thought it would increase the equilibrium concentration of the ketone-enol tautomer necessary to capture a furanyl cation.
In our previous work, strong Brønsted acids were ineffective to promote diastereoselective lactonization of 8 to the target compound, 20-nor-1. Instead, furan decomposition and stereoisomeric mixtures were observed. The cyclization of 9 to 10, in contrast, was promoted with super Brønsted acids like triflimide (Tf2NH), and 2:1 preference for the targeted diastereomer was observed. Finally, selective removal of the β-keto methyl ester was achieved using conditions reported by Krapcho.13
The propensity of 3 to epimerize was evaluated by subjecting it to conditions that we had found to cleanly epimerize SalA (1). Using only 5 mM DBU, 1 slowly reacted at 50 °C in acetonitrile-d3, as shown in Figure 3. This treatment only produced 8-epi-SalA and was easy to follow. More forcing conditions led to competing pathways like acetate solvolysis. In stark contrast to lactone 1, ketone 3–which should be more acidic–showed no change in its spectrum over 7 days. Even after 22 days, the only peaks to drift in the 1H NMR spectrum corresponded to DBU, which was affected by increased water content (see SI).
Figure 3.
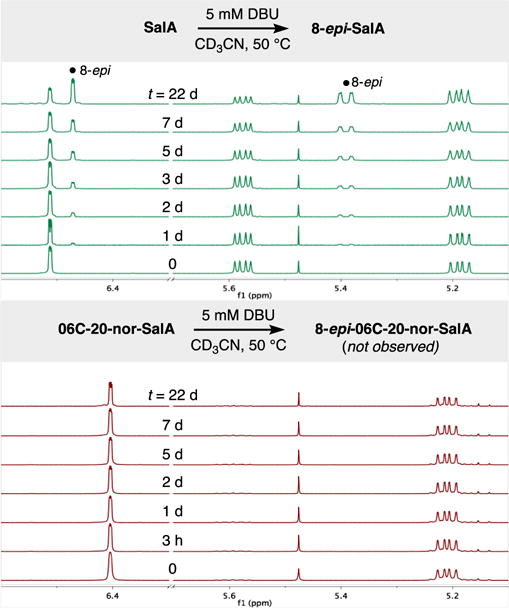
Treatment of 1 and 3 with 5 mM DBU in CD3CN at 50 °C, monitored over twenty-two days by 1H NMR.
Replacement of the SalA lactone with a cyclohexanone (O6→CH2) has never been investigated. Given the effects of this substitution on ring conformation, added to the conformational effects of C20 deletion, we thought potency might be affected substantially. Nevertheless, compound 3 demonstrated strong agonism of the KOR, with an EC50 for cAMP accumulation of 3.3 nM. This value is comparable to U69,593, a benchmark KOR agonist, and therefore parent analog 1. Like SalA itself, 3 did not strongly bias signaling 14 for the Gprotein pathway or βarrestin recruitment.
We conclude that cyclic ketone substitution of the SalA lactone has minimal effect on potency of KOR agonism even though it alters and stabilizes the scaffold geometry. Previous studies have explored the effect of carbonyl removal: a C-ring tetrahydropyran and dihydropyran largely retain affinity, but lose 5-14 fold potency. Reduction to the lactol results in 15-fold affinity loss, but retention of potency.7 Nevertheless, modifications to the native SalA scaffold must still contend with chemical instability and C8 epimerization. In contrast, we show here that cyclohexanone 3 relieves instability and prevents epimerization, yet maintains KOR agonism. Furthermore, replacement of the lactone by a cyclohexanone provides two other potential benefits. First, the ketone provides an additional and orthogonal functional group handle for scaffold diversification. Second, it retains the structural complexity of SalA and 1, while opening new retrosynthetic possibilities for streamlined synthesis.9 Both benefits provide a trajectory for property optimization of the salvinorin scaffold.
Supplementary Material
Figure 1.
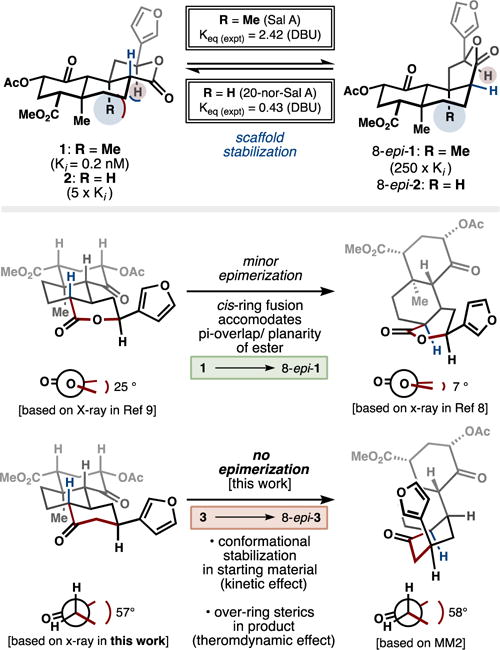
Effects of substitution patterns on conformations and configurations of the SalA scaffold.
Figure 2.
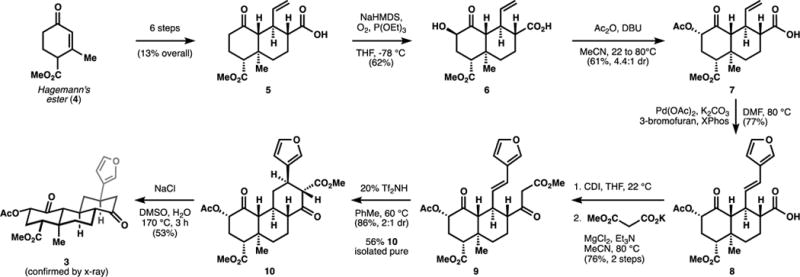
Short synthesis of O6C-20-nor-SalA.
Figure 4.

Activation of KOR in the βarrestin2-KOR enzyme fragment complementation assay (DiscoveRx) and cAMP Cisbio assay. Data represent the mean ± S.E.M. of N=3 assays performed in duplicate.
Acknowledgments
This paper is dedicated to Professor Dale L. Boger for his many contributions to chemistry and education. We thank the NIH (DA 038694, DA033073 and R35 GM122606) for financial support. We are grateful to Dr. Curtis Moore for x-ray crystallographic analysis of 3.15
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Orton E, Liu R. Trans Perioper Pain Med. 2014;1:9. [PMC free article] [PubMed] [Google Scholar]
- 2.Prisinzano TE, Rothman RB. Chem Rev. 2008;108:1732. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan F, Roth BL. Life Sci. 2004;75:2615. doi: 10.1016/j.lfs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Roth BL, Baner K, Westkaemper R, Seibert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdés LJJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J Org Chem. 1984;49:4716. [Google Scholar]
- 6.Brown L PhD. Dissertation. University of Michigan; 1984. [Google Scholar]
- 7.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munro TA, Duncan KK, Staples RJ, Xu W, Liu-Chen L-Y, Béguin C, Carlezon WA, Jr, Cohen BM. Beilstein J Org Chem. 2007;3:1. doi: 10.1186/1860-5397-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roach JJ, Sasano Y, Schmid CL, Zaidi S, Katrich V, Stevens RC, Bohn LM, Shenvi RA. ACS Cent Sci. 2017 doi: 10.26434/chemrxiv.5318188. ChemRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham RJ, Ghersi A, Petrillo G, Sancassan F. J Chem Soc, Perkins Trans. 1997;2:1279. [Google Scholar]
- 11.Boger DL. J Org Chem. doi: 10.1021/acs.joc.7b02088. [DOI] [Google Scholar]
- 12.Garbisch EW, Jr, Griffith MG. J Am Chem Soc. 1968;90:6543. [Google Scholar]
- 13.Krapcho PA, Jahngen EGE, Jr, Lovey AJ, Short FW. Tetrahedron Lett. 1974;15:1091. [Google Scholar]
- 14.Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, Cameron MD, Bannister TD, Bohn LM. Cell. 2017;171:1165. doi: 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cambridge Crystallographic Data Centre deposition number: CCDC 1818713
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.