

Abstract
The ubiquitin ligase, Itch, is required to prevent autoinflammatory disease in mice and humans. Itch-deficient mice develop lethal pulmonary inflammation characterized by the production of Th2 cytokines (for example, interleukin-4 (IL-4)); however, the contribution of Itch to immune defense against respiratory pathogens has not been determined. We found that Itch-deficient mice were highly susceptible to intranasal infection with the respiratory pathogen Klebsiella pneumoniae. Infected Itch-deficient mice exhibited increased immune cell infiltration, cytokine levels and bacterial burden in the respiratory tract compared with control mice. However, numbers of resident alveolar macrophages were reduced in the lungs from Itch-deficient mice both before and after infection. High levels of Th2 cytokines in the respiratory tract correlated with deceased alveolar macrophages, and genetic ablation of IL-4 restored alveolar macrophages and host defense to K. pneumoniae in Itch-deficient mice, suggesting that loss of alveolar macrophages occurred as a consequence of Th2 inflammation. Adoptive transfer of Itch−/− CD4+ T cells into Rag−/− mice was sufficient to drive reduction in numbers of Itch-replete alveolar macrophages. Finally, we found that Stat6 signaling downstream of the IL-4 receptor directly reduced fitness of alveolar macrophages when these cells were exposed to the Itch−/− inflamed respiratory tract. These data suggest that Th2 inflammation directly impairs alveolar macrophage fitness in Itch−/− mice, and elucidate a previously unappreciated link between Th2 cells, alveolar macrophages and susceptibility to bacterial infection.
Keywords: alveolar macrophage, bacterial pneumonia, Itch, Th2 cell
Introduction
The ubiquitin ligase, Itch, regulates the immune system through facilitating the covalent attachment of ubiquitin molecules to specific substrates.1,2 Patients with a loss-of-function mutation in the Itch gene develop autoantibodies as well as nonspecific inflammatory infiltration of the gastrointestinal tract, liver and lungs.3 In addition, patients deficient in Itch, as well as patients with a mutation in an activator of Itch, ATM kinase, exhibit increased susceptibility to bacterial respiratory infections.3,4,5 Much like humans, mice with a genetic inversion resulting in loss of Itch function (Itchy mutant mice, here called Itch−/− mice) develop a lethal autoimmune disease that is characterized by aberrant CD4+ T-cell activation and differentiation into the Th2 lineage, lung and skin inflammation, anti-nuclear antibodies, and excess interleukin-4 (IL-4) production.2,6,7,8 Previous research has partly elucidated the mechanisms by which Itch deficiency causes inappropriate immune activation and autoinflammation;7,8,9,10,11,12 however, the mechanism by which Itch contributes to protective immunity in the context of respiratory bacterial infection is virtually unknown.
K lebsiella pneumoniae is a Gram-negative bacteria that can infect the respiratory tract, causing significant morbidity among immunocompromised individuals (including patients deficient in the Itch activator ATM kinase) and in patients who have been intubated.5,13 In mice, experimental intranasal infection with K. pneumoniae can lead to lethal pneumonia;14,15 however, a sublethal dose of K. pneumoniae is cleared by the immune system during the first week of infection, depending largely on innate immune cells, including neutrophils and alveolar macrophages.16,17,18
Alveolar macrophages are important regulators of lung inflammation, both dampening responses to environmental antigens and acting as first responders to invading pathogens, including K. pneumoniae. 19,20,21,22,23 Histologic analyses of lungs from Itch-deficient mice have identified large, multinucleated alveolar macrophages within alveolar spaces as a key pathological feature;6 thus, one mechanism through which Itch deficiency could influence immunity to respiratory infection is through an effect on alveolar macrophages. Importantly, macrophage survival and function are regulated by the Th2 cytokine IL-4,24,25 which is highly expressed in Itch−/− mice.7
In this report, we sought to define the consequences of Itch deficiency on host defense against respiratory challenge with K. pneumoniae. We found that Itch-deficient mice exhibited markedly increased susceptibility to bacterial pneumonia, displaying increased bacterial titers, immune cell infiltration, cytokine production and decreased survival compared with control mice. We found that Th2 inflammation correlated with reduced numbers, reduced phagocytic capacity and altered morphology of alveolar macrophages in Itch-deficient mice. Importantly, IL-4 was required for loss of alveolar macrophages and susceptibility to infection. Furthermore, we found that Itch−/− CD4+ T cells were sufficient to drive attrition of alveolar macrophages in Rag1 −/− mice, obviating the requirement for Itch deficiency in any other cell type. Finally, we showed that the numbers of alveolar macrophages in Itch−/− mice were regulated directly by cell-intrinsic IL-4 receptor alpha chain (IL-4Rα)–Stat6 signaling. Together, these data support that Itch deficiency leads to an accumulation of IL-4 producing T cells in the lung, resulting in reduced numbers and function of alveolar macrophages, development of lung inflammation, and increased susceptibility to respiratory bacterial infection.
Materials and methods
Mice
WT, Itch−/−, IL4−/−, Itch−/−IL4−/− and Rag1−/− mice were bred in house at the Children’s Hospital of Philadelphia. Lungs from Stat6−/− mice were a generous gift from Taku Kimbayashi’s laboratory at the University of Pennsylvania. Mice were used between 8 and 14 weeks of age, and within experiments they were matched for age and sex. For intranasal injections, mice were anesthetized with an intraperitoneal injection of ketamine and xylazine, and then 50 μl liquid containing 103 colony-forming unit (CFU) was instilled into the nose. Animal housing, care and experimental procedures were carried out in compliance with the Animal Care and Use Committee.
Tissue collection and processing
To collect the bronchoalveolar lavage (BAL), the trachea was cannulated and 0.8 ml phosphate-buffered saline (PBS) was injected into the airways and then recovered, repeated three times with the same 0.8 ml volume. Cells and fluid were separated by centrifugation at 1000g for 5 min. BAL fluids were stored at −80 °C, and cells were immediately transferred to cytospin slides.
Lungs were perfused by flushing 10 ml cold PBS through the right heart. Lungs were minced and digested in collagenase (1000 U/l type 1A and 1125 U/l type 1, Sigma. Atlanta, GA, USA) at 37 °C for 30 min. Single-cell suspensions were generated by disrupting the digested lung through a cell strainer. Red blood cells were lysed, and then live lung cells were enumerated by trypan blue exclusion using a hemocytometer.
Flow cytometry and antibodies
Myeloid cells in single-cell lung suspensions were identified by surface staining: lung cells were stained with fixable live-dead dye (Life Technologies, Chicago, IL, USA) and then surface stained for 30 min with the following antibodies in the presence of Fc Block (BD, clone 2.4G2). All antibodies were purchased from BioLegend (SanDiego, CA, USA) unless otherwise noted: anti-mouse CD45 (clone 30F11), SiglecF (BD, clone E502440), CD11c (clone N418), CD11b (clone M1/70), Ly6G (clone IA8) and Ly6C (clone HK1.4). For intracellular cytokine staining of T cells, lung cell suspensions were stimulated with 30 ng/ml phorbol 12-myristate 13-acetate (Millipore, Chicago, IL, USA) and 1 μg/ml ionomycin (Abcam, Boston, MA, USA) in the presence of Brefeldin A (GolgiPlug from BD, New York, NY, USA) for 4 h, then cells were stained with fixable live-dead dye and surface stained for 30 min with the following antibodies in the presence Fc Block: CD4 (clone GK1.5), CD3 (clone 17A2) or TCRb (clone H57597), and CD8 (clone 53-6.7). Cells were then fixed and permeablized with BD Cytofix/Cytoperm kit, (BD, New York, NY, USA) and stained for interferon gamma (IFNγ; clone XMG1.2), IL-4 (clone 11B11) and IL-5 (clone TRFK5) for 1 h. All flow cytometry data was collected at the CHOP flow cytometry core with an LSR Fortessa (BD), and data were analyzed with FlowJo software (Treestar, Ashland, OR, USA).
Cell sorting and adoptive transfer
CD4+ T cells were isolated by negative magnetic selection as previously described.26 In brief, spleen single-cell suspensions from WT, Itch−/− or Itch−/−IL4−/− mice were incubated with a mixture of hybridoma supernatants containing antibodies directed to MHC class II (M5/114) and CD8 (BioLegend, clone 536.7), followed by magnetic bead-conjugated goat anti-rat Ig (Qiagen, Carol Stream, IL, USA), then magnetic separation was carried out using a DynaMag-2 magnet (Invitrogen, Carlsbad, CA, USA). A total of 4 × 106 purified CD4+ T cells were injected intravenous into Rag1−/−-recipient mice.
For flow cytometric sorting of alveolar macrophages, lung cell suspensions from WT or Stat6−/− mice were stained with CD45, CD11c and SiglecF. CD45+SiglecF+CD11c+ cells were sorted at the CHOP flow cytometry core using a MoFlo Astrios (BD) cell sorter. Two days before adoptive transfer of alveolar macrophages, recipient mice received a 50 μl intranasal dose of clodronate liposomes (Encapsula Nano Sciences, Brentwood, TN, USA) to deplete endogenous macrophages. Two days later, 50 μl cell suspension containing 100000–300000 purified alveolar macrophages were transferred intranasally.
Cytokine analysis
Multiplex cytokine analysis from BAL of infected WT, Itch−/−, IL4−/− and Itch−/−IL4−/− mice was performed using the Q-Plex Mouse Cytokine Screen IR 16-plex system (Quansys, Logan, UT, USA). For cytokine gene expression in whole lung homogenates from uninfected mice, lung was homogenized in Trizol using an electric homogenizer. RNA was extracted and precipitated with chloroform and isopropanol followed by washing with 75% ethanol. Complementary DNA (cDNA) was made using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). Quantitative PCR of cDNA was analyzed with TaqMan and primer/probes for IL-4, IL-5, granulocyte–macrophage colony-stimulating factor (GM-CSF) and β-actin.
BAL cytospin
Cells from BAL were adhered to glass slides by centrifugation. Slides were stained with a modified Giemsa stain (Differential Quik stain kit, Polysciences Inc., Warrington, PA, USA). Slides were visualized on the Leica (Chicago, IL, USA) DM4000B upright scope and pictures were taken with a Spot RT/SE Slider camera (Sterling Heights, MI, USA) at the Children's Hospital of Philadelphia pathology core.
K. pneumoniae infections and determination of CFU
K. pneumoniae serotype 2 (American Type Culture Collection strain 43816; Manassas, VA, USA) was grown as previously described.27 In brief, K. pneumoniae was grown in tryptic soy broth (TSB) for 16 h at 37 °C with shaking. Next, fresh TSB cultures were inoculated and grown for 2 h at 37 °C with shaking. Cultures were washed two times with PBS and then OD 600 was determined. Bacterial titers were calculated using the formula 1 OD=4 × 108 CFU/ml. For infection, concentration was adjusted to give a dose of 103 CFU per 50 μl. Titers were verified by plating serial dilutions on MacConkey agar plates overnight at 37 °C.
Titers from lungs of infected animals were determined by homogenizing lung in 2–4 ml PBS using an electric homogenizer for 30 s and plating serial dilutions of the homogenate on MacConkey agar plates for overnight culture at 37 °C.
Histology
Lungs were perfused and BAL was collected as detailed above. The trachea was cannulated and lungs were inflated with formalin to a constant pressure, trachea was tied and inflated lungs were fixed for 48 h. Paraffin-embedded lung sections were stained with hematoxylin and eosin. Slides were visualized on the Leica DM4000B upright scope and pictures were taken with a Spot RT/SE Slider camera.
In vivo phagocytosis assay
Mice were anesthetized with an intraperitoneal injection of ketamine and xylazine, and then 50 μl of PBS containing 4 mg/ml pHRodo Green E. coli Bioparticles (Life Technologies) was instilled into the nostrils. After 1 h, mice were killed, and BAL and lung cells were collected. Green fluorescence was analyzed in alveolar macrophages from lung and BAL by flow cytometry.
Statistics
Statistics were calculated using Prism6 for Mac OS X software (GraphPad, SanDiego, CA, USA). *, ** or *** denotes P⩽0.05, P⩽0.01 or P⩽0.001, respectively.
Results
Itch deficiency leads to impaired host defense against bacterial pneumonia
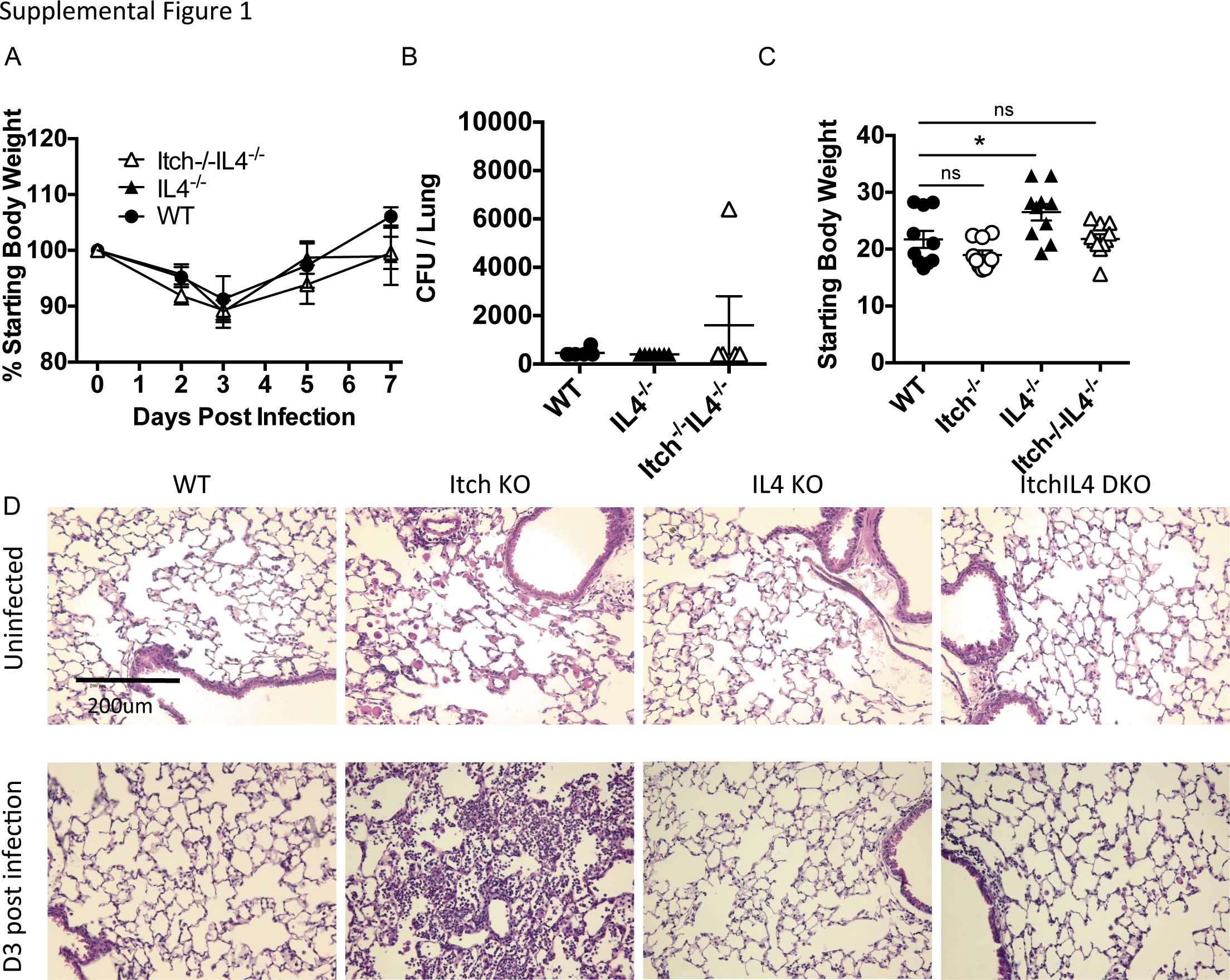
Itch has been shown to be an important negative regulator of immune cell function and pulmonary inflammation; however, the role of Itch in host defense has not been determined. Because of its prominent role in the respiratory immune response, we hypothesized that Itch would have an important role in host defense to respiratory infection. To probe the role of Itch in pulmonary immune defense, we used a model of experimental bacterial pneumonia; we intranasally infected Itch−/− mice or control mice with the Gram-negative bacterium K. pneumoniae, then measured weight loss, survival and parameters of inflammation. We found that Itch−/− mice were highly susceptible to K. pneumoniae infection, exhibiting decreased survival and increased weight loss compared with wild-type (WT) controls (Figures 1a and b). All Itch−/− mice died by 4 days post infection, at a time point consistent with a defect in innate immunity. Importantly, at 3 days post infection, lungs from Itch−/− mice contained markedly increased bacterial loads compared with controls, suggesting that these mice were unable to mount sufficient innate immune responses to control the invading bacteria (Figure 1c). In contrast, WT mice survived, and by 7 days post infection, they had regained full body weight and cleared the infection (Supplementary Table 1A and B). Of note, as Itch−/− age, they stop gaining weight due to the onset of inflammatory disease. Large differences in starting body weight could influence the outcome of infection; however, we infected mice at a young enough age that the differences in body weights were small and not statistically significant (Supplementary Table 1C). Despite the inability to control bacteria, we found that inflammatory cytokines associated with responses to infection were significantly increased in the airways of Itch−/− mice, including IL-1b, IL-1a, MIP-1a and MCP-1 (Figure 1d). In addition, numbers of infiltrating immune cells, including neutrophils, monocytes and eosinophils, were increased in Itch−/− mice compared with controls, suggesting that recruitment of immune cells in response to bacterial infection was intact (Figure 1e). Histology of infected lungs from Itch−/− mice showed dense neutrophilic and eosinophilic infiltrate as well as large macrophages within the airspaces, but not in close contact with alveolar epithelial cells (Supplementary Table 1D). Interestingly, total numbers of alveolar macrophages isolated from lung cell suspensions (identified as CD11c+SiglecF+) were significantly lower in Itch−/− mice after infection (Figure 1e), correlating with impaired bacterial control, despite increased numbers of infiltrating immune cell types. These data suggest that Itch is required for maintenance of numbers of resident alveolar macrophages during infection.
Figure 1.
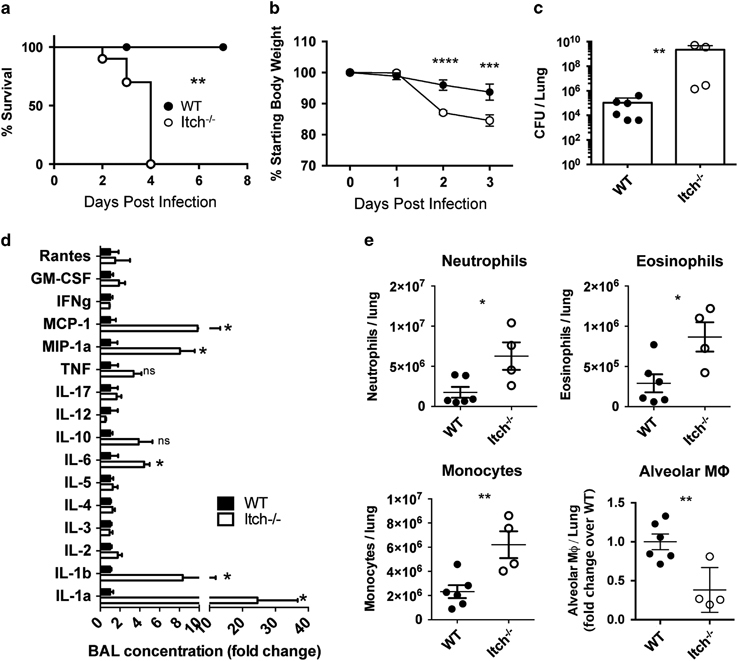
Itch−/− mice exhibit increased susceptibility to bacterial pneumonia. Itch−/− and WT mice were infected intranasally with 103 CFU K. pneumoniae. (a, b) Survival and weight loss (n=8–10, compiled from three independent experiments). (c) Bacterial colony-forming units (CFU) in lung homogenate on day 3 post infection. (d) BAL cytokines were quantified using the Quansys 16-cytokine enzyme-linked immunosorbent assay array. (e) Myeloid cells were identified from lung single-cell suspensions using flow cytometry. Neutrophils were CD45+SiglecF−CD11b+Ly6G+, eosinophils were CD45+SiglecF+CD11c−, monocytes were CD45+, SiglecF−,Ly6G−CD11b+Ly6C+ and alveolar macrophages were CD45+CD11bloCD11c+SiglecF+. (c–e) n=4–6, compiled from two independent experiments. Each dot represents an individual mouse. Significance was determined for survival, weight loss and cytokine/bacterial burden/myeloid cell populations as follows: Log-rank test, two-way analysis of variance and unpaired t-test were used, respectively. *, **, *** or **** denotes P≤0.05, P≤0.01, P≤ 0.001 or P<0.0001, respectively.
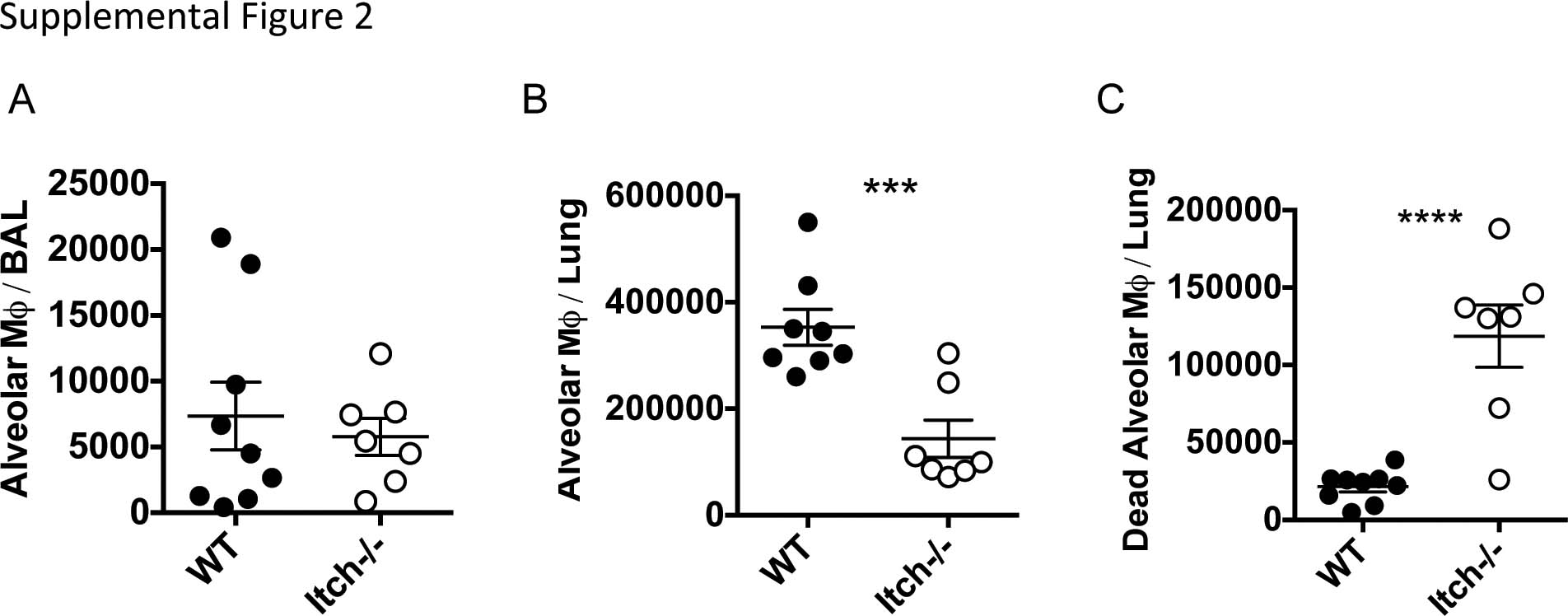
Alveolar macrophages are resident immune sentinels in the lower respiratory tract that reside in the alveolar airspaces, tightly adhered to the alveolar epithelial cells.28 Because these cells are required for effective host defense against K. pneumoniae infection,20 we wanted to determine whether Itch deficiency caused a change in the number or function of these cells before infection. Analyzing both BAL cells and lung cell suspensions by flow cytometry, we found that although the BAL from WT or Itch−/− mice contained comparable numbers of alveolar macrophages (Supplementary Table 2A), lungs from WT mice contained approximately twice as many alveolar macrophages as lungs from Itch−/− mice (Figures 2a and b). Importantly, we are able to detect approximately 50-fold more alveolar macrophages in the lung cell suspensions than in the BAL (Supplementary Table 2A and B), likely because alveolar macrophages tightly adhere to alveolar epithelial cells, thus requiring enzymatic digestion and mechanical disruption to be released from the lung tissue. Interestingly, we observed that a significant number of alveolar macrophages from Itch−/− lungs stained brightly positive for the viability dye, suggesting that these cells were dead and failed to be cleared by other phagocytic cells or by expulsion through the muco-ciliary elevator (Supplementary Table 2C). These cells were excluded in the dead cell gate from the enumeration of total alveolar macrophages by flow cytometry. We also observed an increase in the numbers of CD11c−SiglecF+ eosinophils in lungs from Itch−/− mice (Figure 2b), which is consistent with previous literature29 and with the known role for Itch in limiting Th2-mediated inflammation in the lung.7
Figure 2.
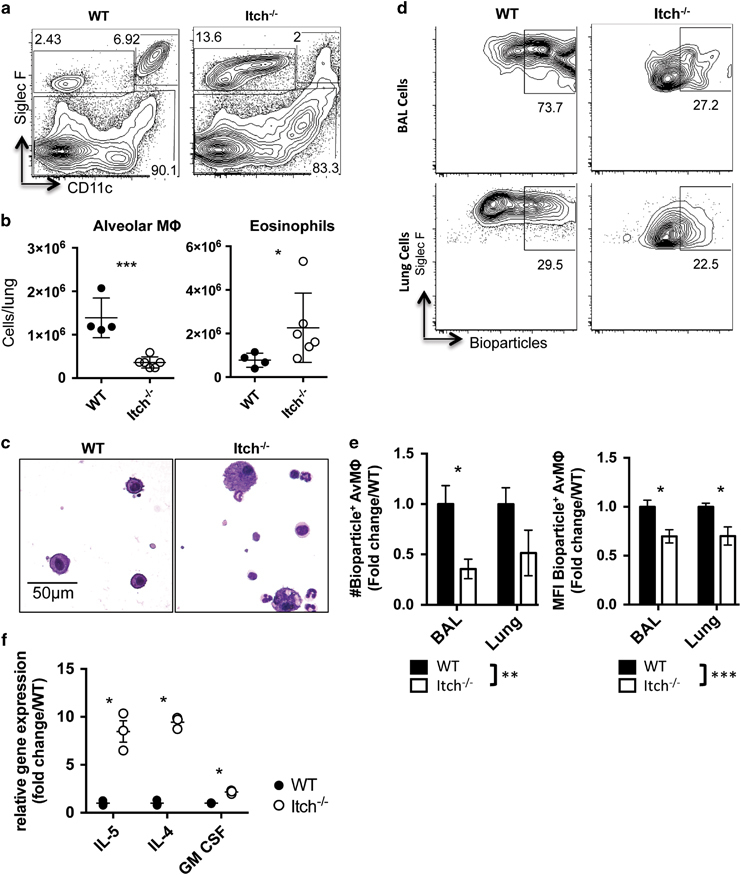
Reduced numbers of alveolar macrophages in Itch−/− mice correlate with Th2 cytokines. BAL and lung were collected from uninfected WT and Itch−/− mice. (a) Representative flow cytometry plots and (b) quantification of alveolar macrophages and eosinophils in lung cell suspensions. Flow cytometry plots are gated on live, singlet, CD45+. Alveolar macrophages are CD11c+CD11bloSiglecF+, and eosinophils are CD11c−SiglecF+ (n=4–6, compiled from two independent experiments). (c) Representative BAL cell cytospin slides stained with modified Giemsa stain and visualized at × 40. (d) Representative flow cytometry plots showing uptake of E. coli bioparticles in BAL and lung alveolar macrophages after intranasal administration. (e) Quantification of numbers and mean fluorescence intensity of bioparticle positive BAL and lung alveolar macrophages. n=7–9, data are compiled from three independent experiments. Significance was calculated by two-way analysis of variance. (f) Cytokine gene expression in whole lung homogenate was quantified by QPCR (n=3). Dots represent individual mice. Significance was calculated using an unpaired t-test. *, **, or *** denote P≤0.05, P≤0.01, or P≤0.001, respectively.
In addition to a reduction in the numbers of alveolar macrophages, we observed that the remaining alveolar macrophages in Itch−/− mice exhibited an altered morphology; namely, alveolar macrophages collected from the BAL or observed in histological lung sections of Itch−/− mice were enlarged and vacuolated (Figure 2c; Supplementary Table 1D). We next wanted to determine whether these remaining alveolar macrophages were capable of phagocytosing bacteria, therefore we administered bioparticles (inactive E. coli that was conjugated to a pH sensitive dye) intranasally to WT or Itch−/− mice and assessed uptake after 1 h by flow cytometry. Because the particles would emit green fluorescence only if exposed to low pH, fluorescence would only be observed if the particles were inside the phagolysosome. We observed that lungs from Itch−/− mice contained fewer total bioparticle+ alveolar macrophages, and in addition, we found that those that did ingest the particles exhibited reduced mean fluorescence intensity compared with WT (Figures 2d and e), suggesting that the alveolar macrophages from Itch−/− mice are functionally defective as well as reduced in number. As expected, gene expression of the Th2 cytokines IL-4 and IL-5 were elevated in whole lung homogenate from Itch−/− mice, inversely correlating with reduced numbers of alveolar macrophages (Figure 2f). The cytokine GM-CSF is required for homeostasis and function of alveolar macrophages,30,31 and GM-CSF-deficient mice exhibit enlarged and vacuolated alveolar macrophages. Importantly, rather than being reduced, the level of GM-CSF was elevated in the Itch-deficient respiratory tract (Figure 2f), suggesting that a lack of GM-CSF could not explain the reduced numbers and altered morphology of alveolar macrophages in Itch−/− mice.
IL-4 is required for reduced alveolar macrophages and impaired host defense in Itch−/− mice
Because we found that increased Th2 cytokines correlated with reduced numbers and function of alveolar macrophages in Itch−/− mice, and because Th2 cytokines like IL-4 can have profound effects on macrophages,24,25 we wanted to determine whether IL-4 was required for the loss of alveolar macrophages in Itch deficiency. Therefore, we examined lungs from mice that were doubly deficient in Itch and IL-4 (Itch−/−IL4−/− mice) as compared with IL-4−/− mice. We observed that the percentages, numbers, morphology and phagocytic capacity of alveolar macrophages in the lungs of Itch−/−IL4−/− mice were similar to IL-4−/− controls (Figures 3a–e). In addition, the levels of IL-5 and GM-CSF in the respiratory tract of Itch−/−IL4−/− mice were not significantly higher than controls (Figure 3f). These data support that aberrant IL-4 activity drives the reduced numbers and altered appearance of alveolar macrophages in Itch−/− mice.
Figure 3.
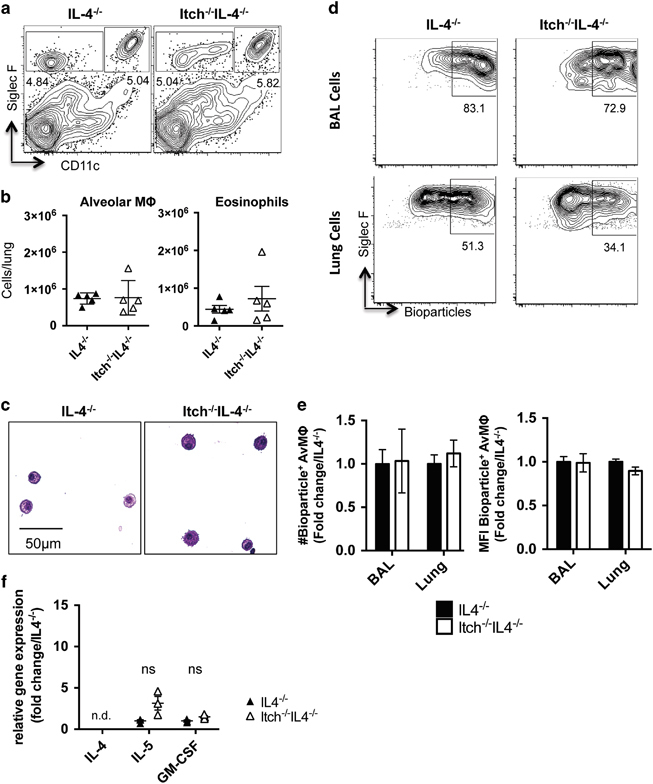
Numbers of alveolar macrophages are restored in Itch−/− mice lacking IL-4. BAL and lung were collected from uninfected IL-4−/− and Itch−/−IL4−/− mice. (a) Representative flow cytometry plots and (b) quantification of alveolar macrophages and eosinophils in lung cell suspensions. Flow cytometry plots are gated on live, singlet, CD45+. Alveolar macrophages are CD11c+CD11bloSiglecF+, and eosinophils are CD11c−SiglecF+ (n=5, compiled from two independent experiments). (c) Representative BAL cell cytospin slides stained with modified Giemsa stain and visualized at × 40. (d) Representative flow cytometry plots showing uptake of E. coli bioparticles in BAL and lung alveolar macrophages after intranasal administration. (e) Quantification of numbers and mean fluorescence intensity of bioparticle positive BAL and lung alveolar macrophages. n=5–6, data are compiled from two independent experiments. Significance was calculated by two-way analysis of variance. (f) Cytokine gene expression in whole lung homogenate was quantified by QPCR (n=3). Dots represent individual mice. Significance was calculated using an unpaired t-test.
To determine whether IL-4 deficiency, resulting in an intact alveolar macrophage compartment, would improve immune defenses against respiratory infection in Itch−/− mice, we infected Itch−/−IL4−/− and IL-4−/− control mice with K. pneumoniae, and we observed that in the absence of IL-4, Itch−/− mice largely survived to day 7 and exhibited survival, weight loss and bacterial burden that was comparable to controls (Figures 4a–c). There was considerably more death in mice that were deficient in IL-4 compared with WT, suggesting that regulated levels of IL-4 may have a protective role during this infection. In addition, there was a trend that Itch−/−IL4−/− mice displayed slightly higher bacterial loads at day 3. Thus, although IL-4 is the dominant driver of increased susceptibility in Itch−/− mice, there are likely other factors that also contribute to impaired immunity in these mice. Despite this, surviving mice had fully regained body weight and cleared the infection by 7 days post infection (Supplementary Table 1A and B). We next evaluated the inflammatory cytokine and infiltrating leukocyte profile in the respiratory tract. We found that some of the Itch−/−IL4−/− mice (two out of six examined for cytokine) displayed highly increased BAL cytokine compared with IL4−/− controls; however, these increases were not consistent and did not reach statistical significance (Figure 4d). Although many of the same cytokines were elevated in both Itch−/− BAL and Itch−/−IL4−/− BAL fluids after infection (comparing Figure 1d with Figure 4d), two cytokines were conspicuously different, namely, IL-1a and RANTES. It is possible that these cytokines reflect a difference in productive inflammation (that is, leading to bacterial clearance) vs damaging inflammation that may cause weight loss and death. Importantly, we saw no changes in the numbers of infiltrating inflammatory cells between IL4−/− and Itch−/−IL4−/− mice either by flow cytometry or by histology (Figure 4e; Supplementary Table 1D). Interestingly, the IL-4−/− mice exhibit increased starting body size compared with WT mice, but this did not appear to impact disease susceptibility (Supplementary Table 1C). Taken together, these data support that Itch is required for protective innate immune responses to K. pneumoniae through its critical role in controlling inappropriate IL-4 activity.
Figure 4.
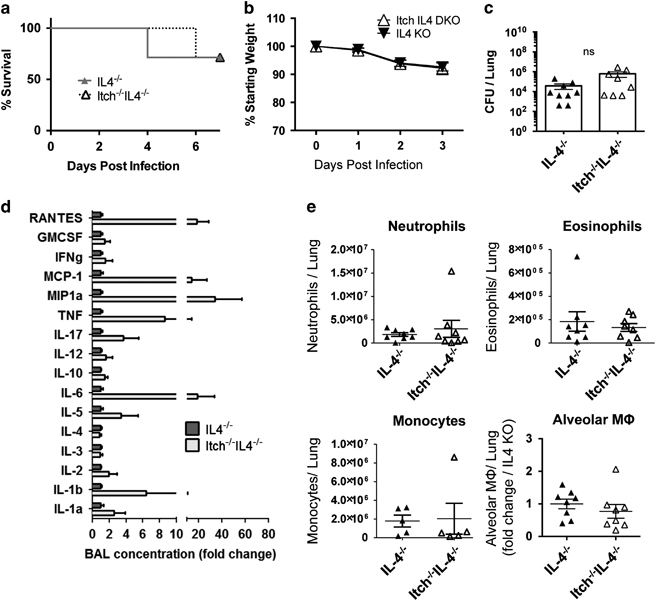
Itch−/−IL-4−/− mice exhibit comparable bacterial control and alveolar macrophages as IL-4−/− control mice. IL4−/− and Itch−/−IL4−/− mice were infected intranasally with 103 CFU K. pneumoniae. (a) Survival (n=7–8, combined from two independent experiments), (b) Weight loss (n=10–11) and (c) bacterial colony-forming units (CFU) in lung homogenate on day 3 post infection (n=8, compiled from three independent experiments). (d) BAL cytokines were quantified using the Quansys 16-cytokine enzyme-linked immunosorbent assay array (n=6, compiled from three independent experiments). (e) Myeloid cells were identified from lung single-cell suspensions using flow cytometry. Neutrophils were CD45+SiglecF−CD11b+Ly6G+, eosinophils were CD45+SiglecF+CD11c−, monocytes were CD45+, SiglecF−, Ly6G−CD11b+Ly6C+ and alveolar macrophages were CD45+CD11c+CD11bloSiglecF+ (n=5–8, compiled from three independent experiments). Dots represent individual mice. Significance was determined for survival, weight loss and cytokine/bacterial burden/myeloid cell populations as follows: Log-rank test, two-way analysis of variance and unpaired t-test were used, respectively.
Itch−/− CD4+ T cells are sufficient to drive loss of alveolar macrophages
Based on our findings above, it was clear that IL-4 was required to drive loss of alveolar macrophages and impaired host defense in Itch−/− mice, but it was unclear which cell types(s) were controlled by Itch to limit IL4/IL4-receptor signaling and preserve numbers of alveolar macrophages. Loss of Itch in CD4+ T cells leads to aberrant Th2 differentiation and Th2 cytokine production (for example, IL-4, IL-5),7 which could impact the alveolar macrophages. Alternatively, Itch could have a role in regulating how cells respond to Th2 cytokines, as Itch has been shown to critically regulate several IL-4-sensitive immune cell types, including B cells32 macrophages11,33,34 and additional types of T cells (for example, Tfh and Tregs).7,8,9,10,12,35 To determine whether IL-4 production by Itch-deficient CD4+ T cells would be sufficient to drive the loss of Itch-replete alveolar macrophages, we transferred total splenic CD4+ T cells (containing normal mixtures of conventional and regulatory T cells) from WT, Itch−/− and Itch−/−IL4−/− mice into Rag1−/− mice, and then evaluated the frequency and numbers of alveolar macrophages 8 weeks after transfer (Figure 5a). We first checked to see whether the transferred T cells could be detected in the respiratory tract and if they were capable of producing cytokines. To test this, we stimulated lung cell suspensions in vitro and evaluated cytokine production by intracellular cytokine staining. As expected, all transferred CD4+ T cells were able to produce IFNγ after stimulation, but only IL-4 sufficient Itch−/− CD4+ T cells resembled Th2 cells, capable of producing IL-4 and IL-5 (Figure 5b). Flow cytometric evaluation of alveolar macrophages in lung cell suspensions revealed that Itch-replete macrophages exposed to Itch−/− CD4+ T cells but not WT or Itch−/−IL4−/− CD4+ T cells were reduced in frequency and number (Figure 5c). Furthermore, alveolar macrophages from mice that had received Itch−/− CD4+ T cells were significantly increased in their size and granularity, as measured by forward- and side-scatter parameters using flow cytometry (Figure 5d). Finally, BAL alveolar macrophages from mice that had received Itch−/− CD4+ T cells exhibited morphologic changes and resembled those seen in Itch−/− mice, with altered characteristics that included increased size and vacuolation (Figure 5e). These data indicated that the reduced frequency and numbers of alveolar macrophages in Itch−/− mice occurred downstream of aberrant IL-4 production by Itch−/− CD4+ T cells, and that these responses to aberrant Th2 cells did not require the presence of B cells, CD8+ T cells or Itch deficiency in any other cell type, including alveolar macrophages.
Figure 5.
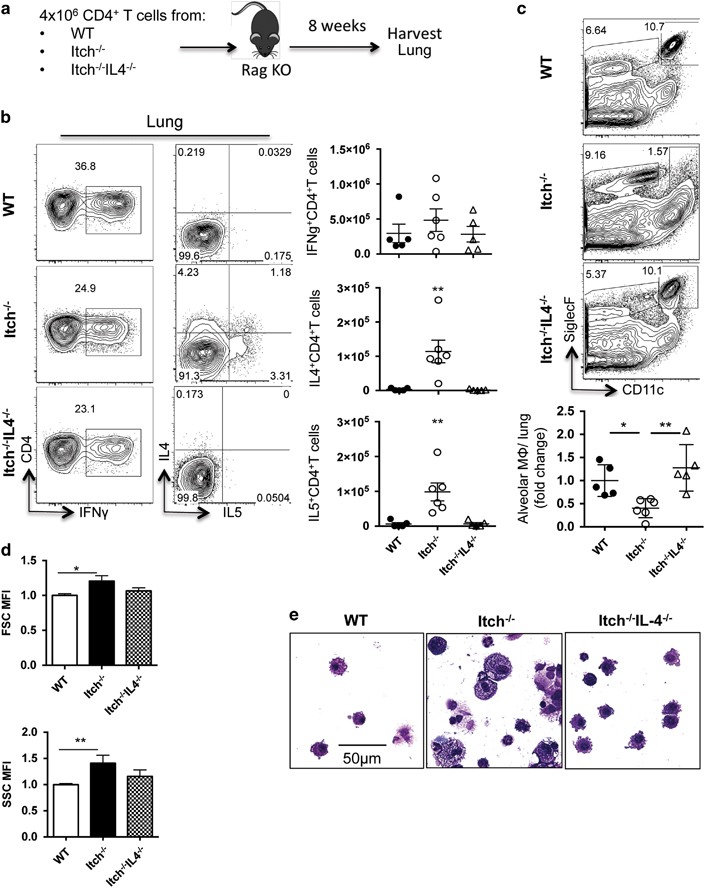
Itch−/− CD4+ T cells are sufficient to drive loss of alveolar macrophages in an IL-4-dependent manner. (a) Diagram describing experimental design. A total of 4 × 106 purified spleen CD4+ T cells from WT, Itch−/− or Itch−/−IL4−/− mice were transferred intravenously to Rag−/− mice, then BAL and lungs were collected 8 weeks later. (b) Lung cell suspensions were stimulated with PMA and ionomycin for 4 h in the presence of Brefeldin A, and then cells were stained for surface markers and intracellular cytokines. Representative flow cytometry plots and quantifications of absolute numbers are shown. Cells are gated on live, singlet, CD3+CD4+. Dots represent individual mice (n=5–6). (c) Alveolar macrophages were identified from lung cell suspensions by flow cytometry. Representative flow cytometry plots and quantification of alveolar macrophages are shown. Flow plots were gated on live, singlet, CD45+, and alveolar macrophages were CD11c+CD11bloSiglecF+. (d) The mean fluorescence intensity of the forward- and side-scatter parameters (FSC and SSC, respectively) was calculated for alveolar macrophages using the geometric mean formula on FlowJo software. Alveolar macrophage numbers, FSC and SSC were divided by the average WT numbers within each experiment to calculate fold change per experiment, normalizing for experimental variability (n=5–6, compiled from two independent experiments). (e) Representative BAL cytospins stained with modified Giemsa stain and visualized at × 40. Significance was calculated using a one-way analysis of variance. * or ** denote P≤0.05 or P≤0.01, respectively.
Stat6 signaling within alveolar macrophages directly influences persistence in the respiratory tract of Itch−/− mice
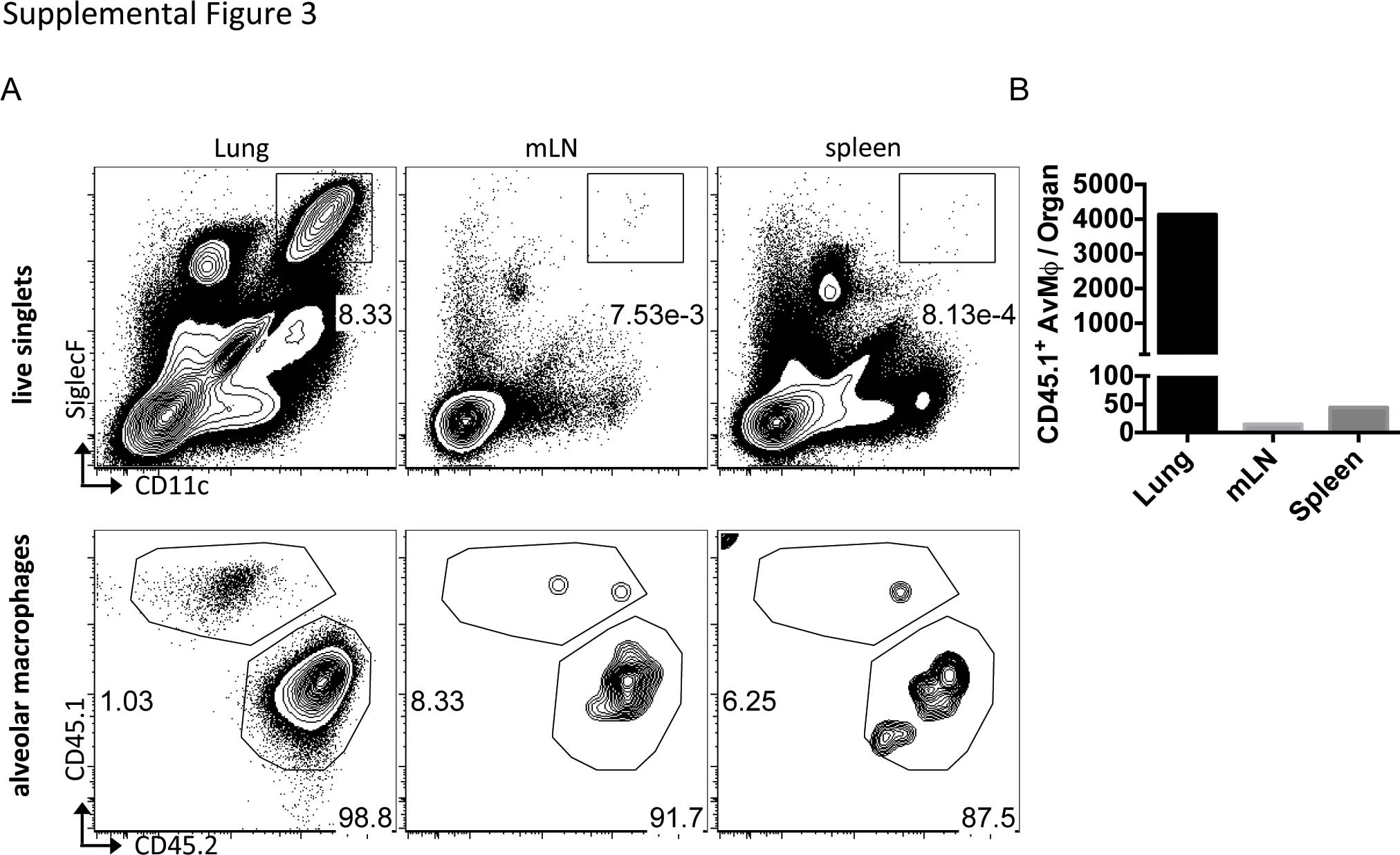
Having determined that IL-4 from Itch−/− CD4+ T cells was sufficient to drive reduced numbers of alveolar macrophages, we next sought to test whether IL-4 mediated this phenotype via signaling directly on alveolar macrophages or through indirect effects of inflammation caused by IL-4 signaling in other cell types. IL-4/IL-4 receptor signaling is mediated by Stat6 and is a potent modulator of macrophage survival, proliferation and function both in vivo and in vitro.24,25 Thus, we hypothesized that in Itch−/− mice, IL-4 receptor signaling on alveolar macrophages might directly cause the reduction in numbers of alveolar macrophages. To test this hypothesis, we developed an alveolar macrophage transfer system so we could transfer Stat6−/− alveolar macrophages into an Itch−/− respiratory tract to see whether the cells would survive better than Stat6 replete cells. We first tested the experimental system for engraftment efficiency and organ specificity using WT mice. We administered clodrosomes intranasaly, then 3 days later, we transferred WT (CD45.1) alveolar macrophages into WT (CD45.2) mice. At 7 days post transfer, we looked for transferred cells in the lung, spleen and mediastinal lymph node (mLN) by flow cytometry. We found that transferred cells were detectable in the lung at a rate of ~1% of the total alveolar macrophage population. Less than 50 transferred cells were detected in the spleen or mLN, supporting that the majority of cells remain in the respiratory tract (Supplementary Table 3).
To test the role of Stat6 in survival of alveolar macrophages, we co-transferred cell trace violet labeled, congenically marked, alveolar macrophages from WT (CD45.1) or Stat6−/− (CD45.2) mice into Itch-deficient or WT mice (CD45.2), intranasally. Recipient mice were pretreated with intranasal clodronate liposomes 3 days before transfer of alveolar macrophages to improve the engraftment. One week later, we analyzed both the host and the transferred alveolar macrophages for frequency, size and complexity by flow cytometry (Figure 6a). We were able to detect cell trace positive transferred alveolar macrophages at a frequency of ~1–5% of the endogenous alveolar macrophages. Importantly, we retrieved fewer WT alveolar macrophages from Itch−/− mice than from WT mice, but equal numbers of Stat6−/− macrophages from WT and Itch−/− lungs (Figure 6b), suggesting that the Itch−/− lung environment was toxic to WT macrophages. When we directly compared WT vs Stat6−/− macrophages in the same host, we found that Stat6−/− alveolar macrophages out-competed their WT competitors in the Itch−/− host, but not the WT host (Figure 6c). These data show that IL-4R signaling directly controls alveolar macrophage fitness in Itch−/− mice. Taken together, these data support that IL-4 from Th2 cells impairs maintenance of the alveolar macrophage population in Itch−/− mice, correlating with defects in host defense against pulmonary bacterial infection, and linking Th2 inflammation to increased susceptibility to bacterial pneumonia.
Figure 6.
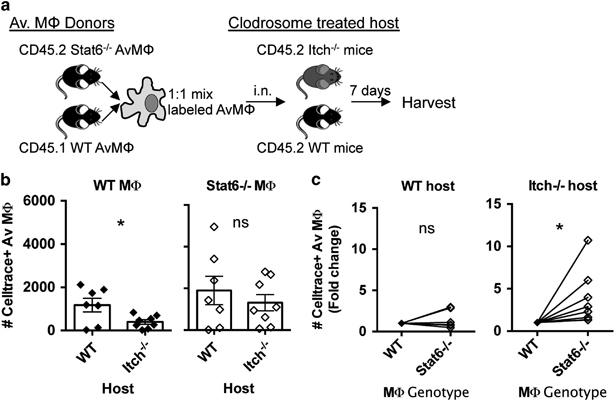
Stat6−/− alveolar macrophages exhibit improved fitness in the Itch−/− lung environment. (a) Diagram describing experimental design. Alveolar macrophages (AM) were sorted from lungs of donor mice and labeled with cell trace violet, then intranasally transferred to recipient mice that had been pretreated with clodrosomes. Recipient lungs were collected 7 days post transfer. (b) Transferred AM were identified as CD11c+SiglecF+CellTrace violet+. Total numbers of recovered Stat6−/− or WT alveolar macrophages in either WT or Itch−/− host are displayed. (c) The same data from b are displayed in a different way: numbers of Stat6−/− and WT alveolar macrophages detected from the same host are compared in a paired manner. n=7–8, compiled from three independent experiments. Significance was determined by (b) unpaired t-test and (c) paired t-test. * denote P≤0.05, respectively.
Discussion
In this report, we demonstrated that Itch deficiency leads to a profound host susceptibility to bacterial pneumonia after experimental infection with K. pneumoniae. Mortality was correlated with increased bacterial loads and decreased numbers and function of alveolar macrophages in the lung, pointing to a failure of innate immune responses to control bacterial infection. The defects in bacterial control and alveolar macrophage numbers and function were rescued by genetic ablation of IL-4 in Itch−/− mice. Adoptive transfer of IL-4-producing Itch−/− CD4+ T cells was sufficient to drive loss of alveolar macrophages in Rag1−/− mice, and intact IL-4R/Stat6 signaling on alveolar macrophages was necessary to lead to decreased alveolar macrophage survival in the Itch−/− respiratory tract.
Firstly, in our study, we found that Itch−/− mice were markedly susceptible to respiratory bacterial infection. Mice exhibited increased death, correlated with increased bacterial loads in the lung 3 days post infection. The increased bacteria at this early time point (that is, before the onset of adaptive immune responses) strongly suggest that Itch−/− mice mount a defective innate immune response. Of the immune cells and cytokines that we measured on day 3 post infection, numbers of alveolar macrophages were the only immune parameter that was reduced in the respiratory tract. Importantly, reduced numbers of alveolar macrophages were evident in Itch−/− lungs prior to infection, and the remaining alveolar macrophages displayed impaired phagocytic function in vivo. These observations suggested that Itch played an important role in maintenance of these resident immune sentinels.
Alveolar macrophages can become dysfunctional in the absence of GM-CSF or subsequent to inflammation. GM-CSF levels were intact in Itch−/− lung cells, suggesting that a defect in GM-CSF was not the reason alveolar macrophages were reduced in number. Thus, we removed Th2 inflammation by crossing Itch−/− mice to IL-4−/− mice. The removal of IL-4 restored alveolar macrophages and immune protection from K. pneumoniae infection. Because alveolar macrophages are required for clearance of K. pneumoniae infection,17,20 the reduction in the size and functional capacity of this important innate immune population downstream of aberrant Th2 inflammation in Itch−/− mice offers a possible explanation for the defect in bacterial control. Importantly, our results clearly demonstrate that activated Th2 cells are required to drive increased susceptibility to respiratory bacterial infection in Itch−/− mice, which is consistent with evidence that Th2 inflammation and IL-4 are linked with increased susceptibility to secondary bacterial infection during recovery after a previous lung insult.36,37 Although the correlation between alveolar macrophages, bacterial control and host protection is striking, it is important to note that we did not definitively show that the defect in macrophages is responsible for the failure of Itch−/− mice to clear the infection, and further studies would be needed to establish this link.
In addition, this report demonstrates that Itch deficiency in CD4+ T cells is sufficient to drive lung inflammation in Itch−/− mice, accompanied by reduced numbers and altered morphology of alveolar macrophages. These findings show that, although Itch functions in many cell types, including macrophages and B cells, Itch deficiency in T cells, yielding activation of IL-4-producing Th2 cells, has a dominant effect on alveolar macrophages that does not require Itch deficiency in any other cell type. This is an important point because lung inflammation and altered morphology of alveolar macrophages can occur due to neutralization of GM-CSF by anti-GM-CSF autoantibodies;38 however, as Itch−/− T cells could drive this phenotype in Rag1−/− mice, we can rule out a requirement for autoantibody in the development of lung inflammation and alveolar macrophage deficiency in Itch−/− mice. Importantly, we have not ruled out additional cell-intrinsic roles for Itch in macrophages in other contexts. Indeed, Itch has been shown to regulate inflammatory signaling pathways in bone marrow-derived macrophages downstream of cytokines and toll-like receptors in vitro;11,33,34 therefore, one would predict that Itch deficiency in macrophages would have a major role to have in host defense in vivo. However, our paper clearly demonstrates that the presence of overwhelming Th2 inflammation in the Itch−/− mice has strong effects on alveolar macrophages that effectively mask any additional cell-intrinsic role(s) for Itch. Future studies aimed at understanding the role of Itch in macrophages in vivo should employ a mouse model with a conditional deletion of Itch in myeloid cells only (for example, CD11ccre or LysMcre), so as to avoid the confounding effects of Th2 inflammation.
It is important to note that, although we have demonstrated a reduction in alveolar macrophage cell numbers in Itch−/− mice, we have not identified the cause of the loss of alveolar macrophage numbers. It is possible that these cells are dying more or are failing to repopulate after normal turnover. Our observation that many alveolar macrophages from Itch−/− mice have compromised membranes, allowing them to take up the viability dye, suggests that these cells dying at an increased rate. Interestingly, recent reports have implicated both Toll-like receptor (TLR) and IL-1R signaling in inflammatory cell death of alveolar macrophages,39,40 which supports the hypothesis that alveolar macrophages exhibit increased death in Itch−/− lungs, which have high levels of IL-1 in the BAL after infection with the TLR4 ligand-containing bacteria, K. pneumonia. In addition, if these cells are undergoing inflammatory cell death, this could explain why dying alveolar macrophages are detected in the lungs from Itch−/− mice, rather than quickly being cleared. Further experiments would be required to definitively demonstrate that the cells are dying at an increased rate, and use of TUNEL and caspase staining could be used to determine the type of cell death that is occurring (that is, apoptosis, necrosis and pyroptosis).
Interestingly, Itch−/− T cells that were not competent to produce IL-4 did not produce increased IFNγ or IL-5 (compared with WT CD4+ T cells) after adoptive transfer, which is consistent with the idea that aberrant IL-4 production due to loss of inhibition by Itch in CD4+ T cells begins a feed-forward inflammatory loop. The effects of IL-4 on macrophage function have been extensively studied in vitro, and IL-4 is one of the signals capable of driving polarization of alternatively activated macrophages (AAM)41 as well as promoting foamy morphology.42 Importantly, AAMs are associated with impaired bacterial clearance, and have been demonstrated to display impaired bacterial phagocytosis and killing after exposure to IL-4,41,43,44 supporting the idea that chronic Th2 inflammation, caused by Itch−/− CD4+ T cells, could directly lead to altered function and morphology of alveolar macrophages. However, because Itch−/−IL4−/− T cells fail to produce both IL-5 and IL-4 (and possibly other inflammatory mediators not measured in this report, for example, IL-13), it is possible that that other cytokines produced by Th2 cells or other cells responding to Th2 cells could contribute to reduced numbers of alveolar macrophages.
We partially addressed this issue by adoptive transfer of Stat6−/− alveolar macrophages, which are unable to signal through the IL-4Rα, into Itch−/− mice. Results from these experiments supported that Stat6−/− alveolar macrophages, which lacked the ability to signal through the IL-4Rα, were better able to survive and persist in the Itch−/− inflamed respiratory tract; thus, cytokine signaling directly on the alveolar macrophages was important in contributing to the observed reduction in cell numbers. It is important to note that IL-4Rα/Stat6 signaling is also used by the Th2 cytokine IL-13; thus, we have not determined whether IL-4 or IL-13 is responsible for this effect. Although it is not explicitly examined in this study, IL-13 is also elevated in Itch−/− mice,45 and it is possible that both IL-4 and IL-13 contribute to the reduction in alveolar macrophages. In addition, these experiments are limited by the efficiency of alveolar macrophage transfer, as transferred cells make up only 1–5% of the alveolar macrophage pool. To study the functional effects (that is, host defense against respiratory infection) of removing IL-4 signaling from alveolar macrophages in Itch−/− mice, future studies would use Itch−/− mice crossed to mice in which IL-4 signaling could be conditionally deleted in macrophages (for example, IL-4Rflox or Stat6flox × LysMcre or CD11cre mice).
Taken together, our data point to a direct role of T-cell-derived Th2 cytokines in impairment of the resident population of alveolar macrophages in Itch−/− mice. This report provides a previously unappreciated link between aberrantly activated Th2 cells, lethal lung inflammation and host defense against bacterial pneumonia. This network may broadly apply to situations in which Th2 cytokines are elicited in the lung (for example, asthma and wound healing), resulting in a negative impact on alveolar macrophages and host defense.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We give special thanks to Junjie Mei in the Worthen lab for assistance with the K. pneumoniae infection model, as well as to Theresa Leichner in the Kimbayashi lab for providing Stat6−/− mice. In addition, we thank Stephanie Sprout for outstanding technical help. This research was funded by the following sources: The National Institutes of Health, (R01AI093566 and R01AI114515) and the American Asthma Foundation (AAF 13-0020).
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2016.69
References
- 1.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 2.Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet. 1998;18:143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 3.Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santini S, Stagni V, Giambruno R, Fianco G, Di Benedetto A, Mottolese M, et al. ATM kinase activity modulates ITCH E3-ubiquitin ligase activity. Oncogene. 2014;33:1113–1123. doi: 10.1038/onc.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schroeder SA, Zielen S. Infections of the respiratory system in patients with ataxia-telangiectasia. Pediatr Pulmonol. 2014;49:389–399. doi: 10.1002/ppul.22817. [DOI] [PubMed] [Google Scholar]
- 6.Hustad CM, Perry WL, Siracusa LD, Rasberry C, Cobb L, Cattanach BM, et al. Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus. Genetics. 1995;140:255–265. doi: 10.1093/genetics/140.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- 8.Parravicini V, Field AC, Tomlinson PD, Basson MA, Zamoyska R. Itch-/- alphabeta and gammadelta T cells independently contribute to autoimmunity in Itchy mice. Blood. 2008;111:4273–7282. doi: 10.1182/blood-2007-10-115667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9:254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- 10.Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–253. doi: 10.1038/ni1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed N, Zeng M, Sinha I, Polin L, Wei WZ, Rathinam C, et al. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat Immunol. 2011;12:1176–1183. doi: 10.1038/ni.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enzler T, Chang X, Facchinetti V, Melino G, Karin M, Su B, et al. MEKK1 binds HECT E3 ligase Itch by its amino-terminal RING motif to regulate Th2 cytokine gene expression. J Immunol. 2009;183:3831–3838. doi: 10.4049/jimmunol.0803412. [DOI] [PubMed] [Google Scholar]
- 13.Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berendt RF. Relationship of method of administration to respiratory virulence of Klebsiella pneumoniae for mice and squirrel monkeys. Infect Immun. 1978;20:581–583. doi: 10.1128/iai.20.2.581-583.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawlor MS, Hsu J, Rick PD, Miller VL. Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol Microbiol. 2005;58:1054–1073. doi: 10.1111/j.1365-2958.2005.04918.x. [DOI] [PubMed] [Google Scholar]
- 16.Xiong H, Carter RA, Leiner IM, Tang YW, Chen L, Kreiswirth BN, et al. Distinct Contributions of Neutrophils and CCR2+ Monocytes to Pulmonary Clearance of Different Klebsiella pneumoniae Strains. Infect Immun. 2015;83:3418–3427. doi: 10.1128/IAI.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kostina E, Ofek I, Crouch E, Friedman R, Sirota L, Klinger G, et al. Noncapsulated Klebsiella pneumoniae bearing mannose-containing O antigens is rapidly eradicated from mouse lung and triggers cytokine production by macrophages following opsonization with surfactant protein D. Infect Immun. 2005;73:8282–8290. doi: 10.1128/IAI.73.12.8282-8290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steichen AL, Binstock BJ, Mishra BB, Sharma J. C-type lectin receptor Clec4d plays a protective role in resolution of Gram-negative pneumonia. J Leukoc Biol. 2013;94:393–398. doi: 10.1189/jlb.1212622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000;173:39–51. doi: 10.1034/j.1600-065X.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- 20.Broug-Holub E, Toews GB, van Iwaarden JF, Strieter RM, Kunkel SL, Paine R, 3rd, et al. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun. 1997;65:1139–1146. doi: 10.1128/iai.65.4.1139-1146.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015;36:307–314. doi: 10.1016/j.it.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Holt PG. Down-regulation of immune responses in the lower respiratory tract: the role of alveolar macrophages. Clin Exp Immunol. 1986;63:261–270. [PMC free article] [PubMed] [Google Scholar]
- 23.Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- 24.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor beta, interleukin-4, and interleukin-10. Ann NY Acad Sci. 1993;685:713–739. doi: 10.1111/j.1749-6632.1993.tb35934.x. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008;112:111–119. doi: 10.1182/blood-2007-10-118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schurr JR, Young E, Byrne P, Steele C, Shellito JE, Kolls JK. Central role of toll-like receptor 4 signaling and host defense in experimental pneumonia caused by Gram-negative bacteria. Infect Immun. 2005;73:532–545. doi: 10.1128/IAI.73.1.532-545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 2014;506:503–506. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramon HE, Riling CR, Bradfield J, Yang B, Hakonarson H, Oliver PM. The ubiquitin ligase adaptor Ndfip1 regulates T cell-mediated gastrointestinal inflammation and inflammatory bowel disease susceptibility. Mucosal Immunol. 2011;4:314–324. doi: 10.1038/mi.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian F, Han Y, Song J, Lei J, Yan X, Xie N, et al. Pulmonary resident neutrophils regulate the production of GM-CSF and alveolar macrophages. FEBS J. 2016;283:1465–1474. doi: 10.1111/febs.13684. [DOI] [PubMed] [Google Scholar]
- 31.Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Ann Rev Physiol. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Veselits M, O'Neill S, Hou P, Reddi AL, Berlin I, et al. Ubiquitinylation of Ig beta dictates the endocytic fate of the B cell antigen receptor. J Immunol. 2007;179:4435–4443. doi: 10.4049/jimmunol.179.7.4435. [DOI] [PubMed] [Google Scholar]
- 33.Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol. 2009;19:1255–1263. doi: 10.1016/j.cub.2009.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Wu C, Matesic LE, Li X, Wang Z, Boyce BF, et al. Ubiquitin E3 ligase Itch negatively regulates osteoclast formation by promoting deubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem. 2013;288:22359–22368. doi: 10.1074/jbc.M112.442459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao N, Eto D, Elly C, Peng G, Crotty S, Liu YC. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol. 2014;15:657–666. doi: 10.1038/ni.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen WH, Toapanta FR, Shirey KA, Zhang L, Giannelou A, Page C, et al. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett. 2012;141:227–234. doi: 10.1016/j.imlet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Z, Zhang J, Zhang X, Li D, Wang H, Xu X, et al. Interleukin 4 deficiency reverses development of secondary pseudomonas aeruginosa pneumonia during sepsis-associated immunosuppression. J Infect Dis. 2015;211:1616–1627. doi: 10.1093/infdis/jiu668. [DOI] [PubMed] [Google Scholar]
- 38.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135:223–235. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, et al. Lipopolysaccharide induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to interleukin-1alpha release. Immunity. 2015;42:640–653. doi: 10.1016/j.immuni.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, et al. TLR4-upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. 2016;6:31663. doi: 10.1038/srep31663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Ann Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siracusa MC, Reece JJ, Urban JF, Jr, Scott AL. Dynamics of lung macrophage activation in response to helminth infection. J Leukoc Biol. 2008;84:1422–1433. doi: 10.1189/jlb.0308199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benoit M, Desnues B, Mege J-L. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 44.Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, et al. M2 Macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than M1 cells in vitro. J Immunol. 2009;182:4415–4422. doi: 10.4049/jimmunol.0713732. [DOI] [PubMed] [Google Scholar]
- 45.Marino A, Menghini R, Fabrizi M, Casagrande V, Mavilio M, Stoehr R, et al. ITCH deficiency protects from diet-induced obesity. Diabetes. 2014;63:550–561. doi: 10.2337/db13-0802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.