

Abstract
Oxidative stress has been described as a prime driver of granulosa cell (GCs) death during follicular atresia. Increasing evidence suggests potential roles of melatonin in protecting GCs from oxidative injury, though the underlying mechanisms remain largely undetermined. Here we first proposed that the inhibition of autophagy through some novel regulators contributes to melatonin-mediated GCs survival under conditions of oxidative stress. Oxidant-induced loss of GCs viability was significantly reduced after melatonin administration, which was correlated with attenuated autophagic signals upon oxidative stimulation both in vivo and in vitro. Compared with melatonin treatment, suppression of autophagy displayed similar preventive effect on GCs death during oxidative stress, but melatonin provided no additional protection in GCs pretreated with autophagy inhibitors. Notably, we found that melatonin-directed regulation of autophagic death was independent of its antioxidation/radical scavenging ability. Further investigations identified FOXO1 as a critical downstream effector of melatonin in promoting GCs survival from oxidative stress-induced autophagy. Specifically, suppression of FOXO1 via the melatonin-phosphatidylinositol 3-kinase (PI3K)-AKT axis not only improved GCs resistance to oxidative stress, but also abolished the autophagic response, from genes expression to the formation of autophagic vacuoles. Moreover, the activation of SIRT1 signaling was required for melatonin-mediated deacetylation of FOXO1 and its interaction with ATG proteins, as well as the inhibition of autophagic death in GCs suffering oxidative stress. These findings reveal a brand new mechanism of melatonin in defense against oxidative damage to GCs by repressing FOXO1, which may be a potential therapeutic target for anovulatory disorders.
Abbreviations: Ac, acetylated; ADA, the 3 AKT phosphorylation sites of FOXO1 are mutated; AKT, thymoma viral proto-oncogene; ATG, autophagy-related; AVOs, acidic vesicular organelles; AOI, antioxidant inhibitors; BECN1, beclin 1; BP, blank plasmid; CAT, catalase; CCK-8, Cell Counting Kit-8; DBD, a FOXO1 mutant without DNA-binding activity, FOXO1N208A,H212R; EP300, E1A binding protein p300; FOXO1, forkhead box O1; GCs, granulosa cell; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione; MAP1LC3B, microtubule associated protein 1 light chain 3 beta; Mel, melatonin; MTOR, mechanistic target of rapamycin (serine/threonine kinase); NPC, non-plasmid control; 3-NP, 3-nitropropionic acid; PCOS, polycystic ovary syndrome; PCD, programmed cell death; P.E, pepstatin A and E64; PIK3C3, phosphoinositide-3-kinase, class 3; PMSG, pregnant mare serum gonadotropin; PI3K, class I phosphoinositide 3-kinase; POF, premature ovarian failure; ROS, reactive oxygen species; SOD, superoxide dismutase; SQSTM1, sequestosome 1; T-AOC, total antioxidation capability; TEM, transmission electron microscopy; WCL, whole-cell lysates; 3-MA, 3-methyladenine
Keywords: Melatonin, Autophagic death, FOXO1, Granulosa cells, Oxidative damage, Antioxidation-independent
Graphical abstract
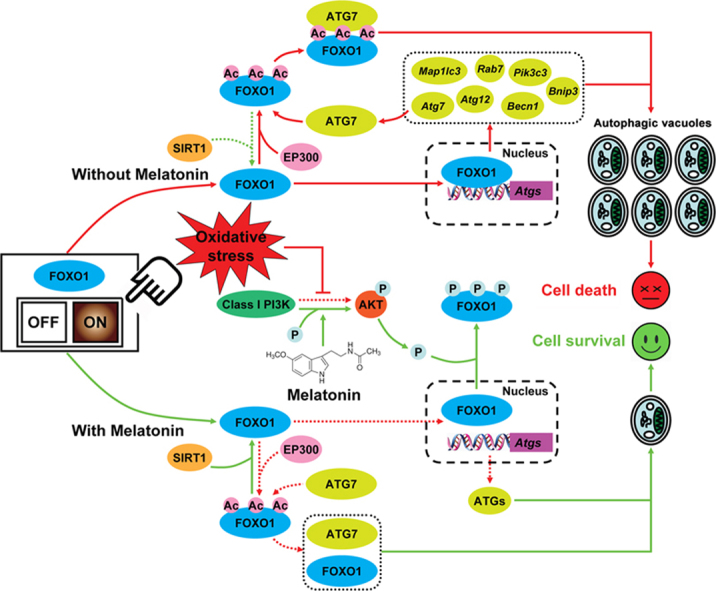
A schematic representation of melatonin-mediated autophagy regulation in GCs during oxidative stress.
Highlights
-
•
Melatonin inhibits oxidative damage in GC without scavenging oxidative stress itself.
-
•
Melatonin protects GC from oxidative damage via inhibiting autophagic cell death.
-
•
Inhibition of FOXO1-dependent autophagy by melatonin reduces oxidative damage in GC.
-
•
Suppression of autophagy through melatonin-PI3K-AKT-FOXO1 axis improves GC survival.
-
•
Melatonin reduces oxidative injury by inhibiting SIRT1-FOXO1-ATG7-dependent autophagy.
1. Introduction
In mammalian ovaries, more than 99% of the follicles are destroyed through a process known as atresia [1]. Previous studies suggested that granulosa cell (GCs) death dominates the progression of atretic degeneration [2]. Reactive oxygen species (ROS) are natural and unavoidable by-products of aerobic metabolism, but unlimited ROS generation gives rise to oxidative stress [3]. During follicular development, increased ROS levels are associated with accelerated metabolic rates in rapidly proliferating GCs [4]. ROS accumulation induces oxidative damage of ovarian GCs, hence setting the atretic program in motion, leading to pathogenesis of anovulatory disorders, such as polycystic ovary syndrome (PCOS) and premature ovarian failure (POF) [5]. Thus, elucidating the preventive mechanisms against oxidative stress-triggered GC death may provide plausible treatment strategies for reproductive failure caused by aberrant follicular atresia.
Autophagy, a highly conserved self-renewal process in eukaryotic cells, is characterized by the engulfment of cytoplasmic materials into double-membrane vesicles (autophagosomes) for subsequent degradation in lysosomes [6]. Generally, the autophagic machinery is necessary for the clearance of dysfunctional proteins and organelles [7]. However, under certain stressful conditions, autophagy is overstimulated to the extent that essential components for cell survival are digested [8]. For example, mammalian cells with normal ROS production usually display basal autophagic activity, whereas excessive autophagy has been suggested to induce self-destruction of cells suffering oxidative injury [9]. Recent observations from rodent ovaries showed elevated autophagic signals in GCs during follicular atresia [10], [11]. These findings were further confirmed by several reports which revealed a close correlation between oxidized low-density lipoprotein (oxLDL)-induced autophagy and GC death [12], [13], [14]. Notably, obese women with high levels of oxLDL exhibited ROS accumulation in the ovary, along with increased incidence of anovulatory infertility [12]. In fact, our earlier study has demonstrated a critical role of autophagy in promoting GC death upon oxidative stress [15]. Therefore, the discovery and identification of an antioxidant by targeting autophagy may provide benefits to GC survival against oxidative injury.
N-acetyl-5-methoxytryptamine (melatonin), an indoleamine originally discovered to be secreted by the pineal gland, performs versatile functions in regulating circadian rhythms, immune response, inflammation, carcinogenesis, and ROS scavenging [16], [17]. Recent evidence indicated potential effects of melatonin on autophagy suppression through its antioxidant properties [18], [19]. Further researches suggested that melatonin-mediated inhibition of autophagic death improves cellular resistance to noxious stimuli [20], [21]. Melatonin is ubiquitously distributed in every bodily compartment including in follicular fluid where its concentration is significantly higher than that in blood (36.5 ± 4.8 pg/ml vs. 10.0 ± 1.4 pg/ml) [22]. Correspondingly, the expression of melatonin receptors is detectable throughout the ovary [23], [24], but melatonin-binding sites have been observed more frequently in the granulosa layers of antral follicles [25]. Increased levels of follicular melatonin might protect GCs from free radical cytotoxicity, and thus maintaining the growth and development of healthy follicles [26]. In contrast, blocking melatonin production via pinealectomy accelerated the atretic process in mammalian ovaries [27]. Moreover, the reduction of follicular melatonin concentrations has been reported to induce anovulation in patients with PCOS [26]. However, few further clues exist regarding the role of autophagy in melatonin-mediated GC protection during oxidative stress.
FOXO1/FKHR is a pleiotropic transcription factor that modulates diverse cellular and physiological processes including proliferation, metabolism, differentiation, cell cycle, cell death, stress response and longevity [28], [29], [30], [31], [32]. The specific functions of FOXO1 are controlled by post-translational modifications (phosphorylation, acetylation, ubiquitination, and methylation), which in turn regulates its subcellular localization, protein-protein interactions, DNA-binding properties, protein stability and transcriptional activity in response to a wide range of external stimuli, such as growth factors, hormones, nutrients, cytokines and oxidative stress [33], [34]. Recently, evidence has emerged regarding the novel roles of FOXO1 in autophagy regulation upon stressful conditions, not only because FOXO1 promotes the expression of several autophagy-related genes, but also FOXO1 post-translational modifications are required for triggering the autophagic process [35], [36], [37]. Indeed, FOXO1 has been identified as a key inductor of autophagic death in GCs with oxidative damage [15]. Considering the capability of melatonin to inhibit autophagy upon oxidative stress [18], [19], we wonder whether melatonin-induced GC survival is correlated with the downregulation of FOXO1-dependent autophagy.
The present study suggested a primary role for autophagy suppression rather than antioxidation in melatonin-mediated GC protection through coordinating the PI3K-AKT-FOXO1 signaling cascades and SIRT1-FOXO1-ATG7 pathway. Our findings may provide new insights into the defense mechanisms of melatonin against oxidative injury.
2. Materials and methods
2.1. Reagents and antibodies
PBS (20012) was purchased from Gibco (Grand Island, NY, USA). Pregnant mare serum gonadotropin (PMSG) was purchased from Ningbo Second Hormone Factory (Ningbo, Zhejiang, China). Melatonin (S1204), pepstatin A (S7381), E64 (S7379), LY294002 (S1105), perifosine (S1037), Sirtinol (S2804), SRT1720 (S1129), 3-methyladenine (3-MA; S2767), and Z-VAD-FMK (S7023) were from Selleck Chemicals (Houston, TX, USA). 3-nitropropionic acid (3-NP; N5636), H2O2 (216763-100 ml), Tiron (89460), anti-MAP1LC3B (L7543) and anti-TUBA1A (T5168) were bought from Sigma-Aldrich (St. Louis, MO, USA). L-Buthionine sulfoximine (sc-200824), 3-Amino-1,2,4-triazole (sc-202016), diethyldithiocarbamic acid sodium salt trihydrate (sc-202576), mercaptosuccinic acid (sc-250305), carmustine (sc-204671), Luzindole (sc-202700), anti-SIRT1 (sc-74465), anti-EP300 (sc-585) and acetylated FOXO1 antibody (sc-49437) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Plasmids for FLAG-tagged FOXO1, including FOXO1-WT (Addgene, 12148), FOXO1-ADA/FOXO1T24A, S253D,S316A (Addgene, 12149), and FOXO1-DBD/FOXO1N208A,H212R (Addgene, 17555) were kindly contributed from Prof. Domenico Accili (Columbia University Medical Center). Antibodies against AKT (9272), phospho-AKT (4060), FOXO1 (2880), phospho-FOXO1 (9461), FLAG (2908), BECN1 (3495), MTOR (2983), ATG3 (3415), ATG5 (8540), ATG7 (2631), and ATG12 (4180) were obtained from Cell Signaling Technology (Beverly, MA, USA). SQSTM1 antibody (ab56416) was purchased from Abcam (Cambridge, MA, USA).
2.2. Animals and ethics
All mice procedures were performed in accordance with the guidelines of the Animal Research Institute Committee at Nanjing Agricultural University. Three-week-old female ICR mice (Qing Long Shan Co., Animal Breeding Center, Nanjing, China) were group-housed in a temperature-controlled (22 ± 2 ℃) room with a 12/12 h light/dark cycle (lights on from 7:00 a.m. to 7:00 p.m.), and had ad libitum access to water and food. Mice were divided into Control group, melatonin group, 3-NP group, and melatonin+ 3-NP group. Melatonin and 3-NP were dissolved in 0.9% saline containing 0.5% ethanol; 0.5% ethanol saline (v/v) is regarded as vehicle. Female ICR mice were injected intraperitoneally with vehicle or melatonin (15 mg/kg) at 8:00 a.m. once a day for 2 consecutive days, followed by 5 days of oxidative stimulation in ovarian GCs using a well-established in vivo model [38]. Briefly, from day 3 to day 7, mice received an additional intraperitoneal injection of 3-NP (50 mg/kg) or 0.5% ethanol saline at 8:00 p.m. each day, along with the daily administration of melatonin (15 mg/kg) or vehicle at 8:00 a.m. 24 h after the final injection, ovaries were collected for subsequent immunohistochemical staining, western blotting analysis or CCK-8 assay. The protocols of all animal experiments were approved by the Committee of Animal Research Institute, Nanjing Agricultural University, China.
2.3. Cell culture and treatments
Primary GCs were isolated from ovarian follicles and cultured as described previously [15], [38], [39], [40]. For drug administration, GCs pretreated with melatonin (10 μM) for 24 h were washed in PBS, and incubated with medium containing 200 μM H2O2 for 0, 1, or 2 h as indicated. In some experiments, GCs were treated with pepstatin A (10 μg/ml), E64 (10 μg/ml), LY294002 (20 μM), perifosine (10 μM), Tiron (10 mM), Sirtinol (100 μM), SRT1720 (100 μM), 3-MA (10 mM), or Z-VAD-FMK (50 μM) 1 h before H2O2 exposure. For the suppression of melatonin-mediated antioxidative effects, a cocktail consists of inhibitors (hereinafter referred to as antioxidant inhibitors, or AOI) against glutathione/GSH (L-Buthionine sulfoximine; 1.5 mM), catalase/CAT (3-Amino-1,2,4-triazole; 10 mM), superoxide dismutase/SOD (diethyldithiocarbamic acid sodium salt trihydrate, 5 mM), glutathione peroxidase/GPx (mercaptosuccinic acid, 5 mM), and glutathione reductase/GR (carmustine, 5 μM) were added 1 h prior to H2O2 incubation. For RNA interference, GCs were transfected with Becn1 siRNA, Atg7 siRNA, Foxo1 siRNA, or scrambled control siRNA for 24 h, grown in medium with or without 10 μM melatonin for another 24 h, rinsed using PBS, and then exposed to 2 h of H2O2 incubation. For an overexpression experiment, the Flag-tagged FOXO1 plasmids or an empty control plasmid were individually transfected into GCs. 24 h later, cells were cultured with or without melatonin for an additional 24 h before they were used for the next assay.
2.4. Detection of ROS production
ROS (reactive oxygen species) levels were determined using the Reactive Oxygen Species Assay Kit (Beyotime Institute of Biotechnology, S0033) according to the manufacturer's instructions. This fluorescent strategy is based on the oxidative conversion of dichlorodihydrofluorescein (DCFH) to dichlorofluorescein (DCF), which emits green fluorescence upon excitation at 488 nm. The cells were imaged with a laser-scanning confocal microscope (Carl Zeiss, Zeiss LSM 710 META, Oberkochen, Germany). The results were calculated as fluorescence intensity in each GC by using the ImageJ 1.42q software (National Institutes of Health, Bethesda, MD, USA).
2.5. RNA interference
The siRNAs directed against Becn1 (sc-29798), Atg7 (sc-41448), Foxo1 (sc-35383) and the scrambled control siRNA (sc-37007) were obtained from Santa Cruz Biotechnology. siRNA transfection was performed using Lipofectamine 3000 reagent (Invitrogen, L3000015) according to the manufacturer's instructions.
2.6. Cell viability assay
Cell viability of GCs was measured using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, CK04), in which the tetrazolium salt (WST-8) is reduced by dehydrogenase activities in viable cells to generate a yellow water-soluble formazan dye. Intensity of colour is therefore directly proportional to the number of living cells in culture. The experimental procedures were carried out following the manufacturer's directions. Briefly, GCs were seeded in 96-well plates, and grown to 90% confluency for 4 days. After the indicated treatments, CCK-8 assay reagent (10 μL) was added to each well containing 100 μL medium, and incubated in the dark for 2 h at 37 ℃. The formation of formazan was assessed by determining the optical density (OD) at 450 nm under a microplate spectrophotometer (Thermo Fisher Scientific, Camarillo, CA, USA).
2.7. Measurement of ATP production
ATP levels in GCs were determined by a luciferin-luciferase system using the ATP Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's protocols. Briefly, cell lysates were centrifuged at 12,000 g at 4 ℃ for 10 min, and the supernatant was incubated in the ATP-detection buffer containing firefly luciferin and firefly luciferase enzyme reagent for 5 min. The RLU was then detected using a Glomax™ Luminometer (Promega, Madison, WI, USA). The ATP levels were normalized against protein concentrations determined by a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China).
2.8. Examination of total antioxidation capability
Total antioxidation capability (T-AOC) was determined using a Total Antioxidant Capability Assay Kit with FRAP method (Beyotime Institute of Biotechnology, Shanghai, China) as previously described [41]. Briefly, cellular homogenates were centrifuged at 12,000 g at 4 ℃ for 5 min, and the precipitates were lysed for determination of protein concentration using a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). 5 μL of the supernatants was incubated with 180 μL chromophoric substrate (Fe3+-TPTZ; Beyotime Institute of Biotechnology, Shanghai, China Beyotime) for 5 min at 37 ℃. The reduction of Fe3+-TPTZ was then detected at 593 nm using a microplate reader (Thermo Fisher Scientific, Camarillo, CA, USA) and the amount of Fe2+-TPTZ yielded per gram of total protein was calculated.
2.9. Acridine orange staining
Formation of acidic vesicular organelles (AVOs), a morphological characteristic of autophagy, is used to evaluate the abundance of autophagic vacuoles in the cells [42]. To detect AVOs formation, we performed vital staining with acridine orange in GCs as described previously [40]. Briefly, cells with indicated treatments were stained with 1 μg/ml acridine orange at 37 ℃ for 15 min. In acridine orange-stained cells, the cytoplasm and nucleus emits green fluorescence, whereas the acidic compartments shine bright red [43]. The green (510–530 nm) and red (650 nm) fluorescence emission illuminated with blue (488 nm) excitation light were visualized under a laser-scanning confocal microscope (Carl Zeiss, Zeiss LSM 710 META, Oberkochen, Germany).
2.10. Visualization of autophagosome formation
GCs were seeded in 24-well plates with 10 mm coverslips, and grown to 80% confluence prior to transient transfection with the GFP-MAP1LC3B expression vector, which is a kind gift from Prof. Jiyong Zhou (Zhejiang University). 24 h later, cells were cultured with or without 10 μM melatonin for another 24 h, rinsed using PBS, exposed to 2 h of H2O2 incubation, and the intracellular fluorescence of GFP-MAP1LC3B was then observed under a laser-scanning confocal microscope (Carl Zeiss, Zeiss LSM 710 META, Oberkochen, Germany). Experiments were performed in triplicate. The fields of each coverslip were divided into 9 component squares, and 3 of them were randomly selected for counting punctate GFP-MAP1LC3B at a magnification of 400×.
2.11. Quantitative RT-PCR (qRT-PCR)
The collection of total RNA and cDNA from GCs were performed as described previously [38]. The qRT-PCR was carried out using SYBR Premix Ex Taq (Takara, DRR420A) in a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primer sequences for the target genes are listed in Table S1. Data were normalized by the expression of Actb housekeeping gene. Specificity of each PCR amplification was verified by melting curve analysis.
2.12. Transmission electron microscopy
Cultured GCs were removed using a cell scrapper and collected by centrifugation at 1300 g for 15 min. Sample preparation for transmission electron microscopy was accomplished by Biological Electron Microscope Facility (BEMF) at Nanjing Agricultural University. Briefly, the cell pellets were immediately fixed in 2.5% glutaraldehyde (Sigma-Aldrich, 49626) for 24 h, and then postfixed with 1% osmic acid (Sigma-Aldrich, 75632) for 1.5 h, washed, dehydrated in graded concentrations of ethanol, and embedded in Araldite (Sigma-Aldrich, A3183). Samples were then cut into ultrathin sections (50 nm), stained with aqueous uranyl acetate (Polysciences, 6159-44-0) and lead citrate (Sigma-Aldrich, 15326). Representative fields were selected for imaging with a Hitachi H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
2.13. Immunofluorescence
After the desired treatments, GCs grown on coverslips were washed with PBS, and fixed using 4% paraformaldehyde (Sigma-Aldrich, P-6148) according to standard protocols. Cells were then permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, T8787) for 10 min at 4 ℃. After blocking with 1% BSA (Sigma-Aldrich, A3059) for 1 h at room temperature, the cell climbing sheets were incubated with rabbit anti-FOXO1 (1:100 dilution, Cell Signaling Technology, 2880) for 1 h at 37 ℃, followed by rinsing with PBS solution. Next, cells were stained for 1 h with a Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A-11008), and the nuclei were counterstained with DAPI (1:200 dilution, Sigma-Aldrich, D8417) for another 20 min. Fluorescent images were captured using a Zeiss LSM 710 META confocal microscope (Carl Zeiss, Oberkochen, Germany).
2.14. Immunohistochemical staining
Mice ovaries were fixed in buffered paraformaldehyde (4%), embedded in paraffin, sectioned to approximately 5 µm, and mounted on glass slides. The ovarian sections were deparafinized in xylene, rehydrated, and retrieved by microwave heating with buffer of citrate ([R&D Systems, 3161500G], 0.05% Tween-20 [Sigma-Aldrich, P1379], pH 6.0) for 0.5 h. Endogenous peroxidase activity was quenched by incubation with 3% H2O2 (Sigma-Aldrich, 216763-100 ml) for 10 min. After blocking in 1% BSA for 1 h, sections were immunostained with rabbit antibodies against MAP1LC3B (Sigma-Aldrich, L7543) or SQSTM1 (Abcam, ab101266), and corresponding secondary antibodies with biotin labeling. The immunoreactive signals were visualized using the 3, 3'-diaminobenzidine chromogen solution (Sigma-Aldrich, D8001). The nuclei were counterstained in hematoxylin (Sigma-Aldrich, H9627) prior to dehydration and coverslip placement.
2.15. Immunoblot analysis
GCs were lysed with ice-cold RIPA Lysis Buffer (Beyotime, P0013B) plus a complete protease inhibitor cocktail (Roche, 04693132001), and protein concentrations were determined using a BCA Protein Assay Kit (Beyotime, P0012) according to the manufacturer's instructions. Cell lysates were boiled for 5 min in SDS loading buffer (SunShineBio, SN336-2). Equal amount of sample proteins (15 μg/lane) were separated by electrophoresis through a 12% Express Plus™ PAGE gel (Genscript, M01210), and transferred to a PVDF membrane (Millipore, HATF09025) by electroblotting. Nonspecific binding sites were blocked with 5% BSA in TBST (Solarbio, T1085) for 1 h at room temperature, followed by overnight incubation at 4 ℃ with diluted primary antibodies (1:1000) as indicated. The blots were washed 3 times with TBST and incubated 2 h with a diluted (1:2000) secondary antibody against rabbit (Cell Signaling Technology, 7074) or mouse (Cell Signaling Technology, 7076). Immunoreactive bands were detected using a WesternBright ECL HRP substrate kit (Advansta, K-12045-C20) according to the manufacturer's instructions. The relative expression of target proteins was normalized to that of TUBA1A.
2.16. Co-immunoprecipitation
For co-immunoprecipitation experiments, PBS-washed GCs were lysed on ice with IP lysis buffer (Pierce, 26149) containing protease inhibitor cocktail (Roche, 04693132001). Whole-cell lysates (WCL) were subjected to immunoprecipitation with rabbit antibody against FOXO1 (Cell Signaling Technology, 2880). For each IP reaction, 2–4 μg of antibody were mixed with 1 ml of cell lysate. After an overnight incubation at 4 ℃, Protein A+G Agarose beads (Beyotime, P2012) were added. 1 h later, beads were washed with lysis buffer, and the immunoprecipitates were eluted using SDS loading buffer (SunShineBio, SN336-2). The supernatants were then processed for immunoblotting analysis with the indicated antibodies. The amounts of proteins co-immunoprecipitated with FOXO1 were normalized to TUBA1A levels of input samples.
2.17. Statistical analysis
All experiments were repeated at least three times, and all data were presented as means ± S.E. Statistical significance was analyzed by the SPSS version 16.0 software (SPSS, IL, USA). Differences between two groups were assessed using the Student t-test, and between multiple groups using one-way ANOVA. Values of P < 0.05 were considered significant.
3. Results
3.1. Inhibition of autophagy by melatonin attenuates oxidative damage in ovarian granulosa cells (GCs)
Using an established in vivo model for triggering oxidative stress in mouse ovarian GCs [38], we determined whether melatonin exerts any influence on GC autophagy upon oxidant stimulation. The results of immunohistochemical assay showed that the induction of autophagy was significantly enhanced in ovaries collected from mice subjected to 3-NP administration (Fig. 1A and B). In contrast, impaired autophghagic signals in follicular GCs were observed following melatonin injection. Notably, positive staining of the autophagy-related biomarkers, including MAP1LC3B and SQSTM1, was concentrated almost in the granulosa layers of antral follicles (Fig. 1A and B, lower panels), indicating that melatonin may regulate GCs-specific autophagy during oxidative stress. Accordingly, immunoblot analysis of total MAP1LC3B expression, MAP1LC3B-II accumulation and SQSTM1 degradation within ovarian GCs further confirmed that the oxidant 3-NP-induced autophagy was inhibited by melatonin (Fig. 1C–F). To better assess the inhibitory effects of melatonin on 3-NP induced autophagy, we treated mice with chloroquine, a lysosomal inhibitor that prevents autophagic degradation in the lysosomes, and investigated its influence on autophagic flux by determining MAP1LC3B protein abundance (Fig. S1). Markedly, chloroquine treatment increased MAP1LC3B-II accumulation in GCs of mice received 3-NP injection, but the increase was much smaller in the corresponding samples of mice injected with melatonin, indicating that melatonin blocked 3-NP-triggered autophagic flux in ovarian GCs. In addition, data obtained from CCK-8 assay showed a preventive effect of melatonin on GCs death in ovaries harvested from mice injected with 3-NP (Fig. 1G). These in vivo findings thus suggested the possibility that suppression of autophagy might be required for melatonin-mediated GC protection upon oxidative stress.
Fig. 1.

The inhibitory effects of MT on autophagy reduces oxidative damage in mouse follicular GCs. Mice were injected i.p. with melatonin (15 mg/kg) or 0.5% ethanol saline once daily at 8:00 a.m. for 7 days. From day 3 to day 7, mice received an additional i.p. injection of 3-NP (50 mg/kg) or 0.5% ethanol saline at 8:00 p.m. each day. Ovaries were collected 24 h after the final injection. Immunohistochemical staining of GCs in ovarian sections was detected using anti-MAP1LC3B (A) and anti-SQSTM1 (B). Bar, 100 µm. O, oocyte; GC, granulosa cells; B, basement membrane; T, theca cells. Areas outlined in red are enlarged in lower panels. (C) Immunoblotting analysis of MAP1LC3B and SQSTM1 in ovarian GCs harvested from mice subjected to the indicated treatments. (D–F) Quantification of total MAP1LC3B expression, MAP1LC3B-II accumulation, and SQSTM1 degradation. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3. **P < 0.01; NS, not significant, P > 0.05. (G) Viability of ovarian GCs retrieved from mice with indicated administration was assessed using CCK-8 assay. Data represent mean ± S.E; n = 3 in each group. **P < 0.01; NS, not significant, P > 0.05 (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.2. Melatonin counteracts oxidative damage in cultured GCs via inhibiting autophagic cell death
Given the detrimental effects of 3-NP on ATP generation [44], which is also involved in autophagy regulation, we doubted that the autophagic responses in ovarian GCs after 3-NP injection might not be solely induced by oxidative stress. On the other hand, a complex set of ovarian steroids and pituitary gonadotropins affected GC viability under physiological conditions [45]. To further clarify the correlation between autophagy and melatonin-mediated GC protection upon oxidative stimulation, as well as eliminate the nonspecific actions of ATP decrement and other endocrine factors on autophagy or GC survival, our subsequent experiments were performed with H2O2 treatment in primary cultured GCs collected from ovarian follicles. As expected, the H2O2-induced conversion of DCFH to DCF, which emits green fluorescence in ROS positive cells, was remarkably decreased by the treatment of Tiron (4,5-dihydroxy-1,3-benzene disulfonic acid-disodium salt), a powerful ROS scavenger (Fig. S2). To test whether oxidative stress activated autophagy in cultured GCs, cell lysates were harvested for immunoblot analysis of autophagy marker proteins, including MAP1LC3B and SQSTM1. As shown in Fig. S3, Tiron also significantly blocked the MAP1LC3B-II accumulation and SQSTM1 degradation in H2O2-treated GCs. Consistent with this, the level of H2O2-triggered cell death was markedly reduced in the presence of Tiron (Fig. S4). These data suggested that in vitro oxidative stress driven by H2O2 treatment promotes the induction of autophagy and GC injury. We also detected the possible regulation of H2O2 on ATP production in GCs. As shown in Fig. S5, no obvious reduction of ATP level was observed in cells treated with H2O2. Since H2O2 exhibited much stronger inhibitory effects on cell viability under the same conditions (Fig. S4), we believed that H2O2-induced GC death might not be attributed to ATP depletion. On the other hand, melatonin did not significantly change ATP level in GCs with or without H2O2 treatment, indicating that melatonin-mediated GC protection might not be achieved by influencing ATP production.
To determine whether melatonin regulates autophagy in cultured GCs upon oxidative stress, acridine orange staining was employed to visualize the production of acidic autolysosomes. As shown in Fig. 2A and B, GCs pretreated with melatonin displayed a marked reduction in the amount of AVOs after H2O2 incubation. Correspondingly, immunoblotting assay showed that the MAP1LC3B-II expression, conversion of MAP1LC3B-I to MAP1LC3B-II and SQSTM1 degradation following H2O2 exposure were significantly inhibited by melatonin (Fig. 2C–F). To obtain a better evaluation of melatonin-mediated suppression on autophagy, we next examined the autophagic flux in GCs treated with/without pepstatin A and E64 (P.E, both are lysosomal protease inhibitors) (Fig. 2G–J). By monitoring the GFP-MAP1LC3B puncta and MAP1LC3B blots, both of which were further accumulated by P.E adminstration in cells subjected to H2O2 exposure, our data indicated that the autophagic flux was enhanced during oxidative stress (Fig. 2G–J). In contrast, melatonin abrogated the generation of punctate GFP-MAP1LC3B and aggregated MAP1LC3B in H2O2-incubated cells despite P.E treatment, suggesting a potential role of melatonin in repressing autophagosome formation (Fig. 2G–J). Notably, the lysosomal inhibitor (pepstatin A and E64) alone did not significantly increase the accumulation of MAP1LC3B dots, indicating a low baseline autophagic flux in GCs under normal growth conditions (Fig. 2I). Correspondingly, no obvious effects of melatonin on basal autophagic flux were detected in GCs without H2O2 exposure (Fig. 2I). Moreover, the loss of GC viability caused by H2O2 stimulation was significantly reduced in the presence of melatonin (Fig. 2K), while Luzindole (a competitive antagonist of melatonin receptors) abolished melatonin-induced GC survival during oxidative stress (Fig. S6). The results further confirmed the in vivo observations, which implied that suppression of autophagy by melatonin might contribute to GC protection under oxidative stress conditions.
Fig. 2.

Melatonin represses H2O2-induced autophagy in cultured GCs. (A) GCs received 24 h of melatonin (10 μM) treatment were then rinsed in PBS, and incubated with 200 μM H2O2 for 2 h. The acidic vesicular organelles (AVOs, red) were detected using acridine orange staining. Bar, 10 µm. (B) The formation of autophagic vacuoles was quantified by calculating the amount of AVOs per cell. Experiments were repeated in triplicate, and 3 fields of each coverslip were selected at random for counting. Data represent mean ± S.E; n = 3 in each group. **P < 0.01. (C) GCs cultured with or without 10 μM melatonin for 24 h were then washed in PBS, and treated with H2O2 for 0–2 h. The expression of MAP1LC3B and SQSTM1 in GCs was determined by western blotting. (D–F) The MAP1LC3B-II accumulation, conversion of MAP1LC3B-I to MAP1LC3B-II and SQSTM1 degradation were quantified by densitometric analysis. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3. * *P < 0.01; NS, not significant, P > 0.05. (G) GCs transfected with GFP-MAP1LC3B plasmid for 24 h were cultured for another 24 h in the presence or absence of 10 μM melatonin before 2 h of H2O2 (200 μM) incubation. For the inhibition of autolysosome formation, pepstatin A (10 μg/ml) and E64 (10 μg/ml) were added 1 h prior to H2O2 (200 μM) exposure. Bar, 10 µm. (H) The formation of autophagosomes was assessed by quantifying the GFP-MAP1LC3B puncta per cell. Experiments were repeated in triplicate, and 3 fields of each coverslip were selected in random for counting. Data represent mean ± S.E; n = 3 in each group. *P < 0.05, **P < 0.01; N, not significant, P > 0.05. P.E., pepstatin A and E64. (I) GCs pretreated with 10 μM melatonin for 24 h were then rinsed in PBS, and exposed to 200 μM H2O2 for 2 h. To block the autophagic flux, pepstatin A (10 μg/ml) and E64 (10 μg/ml) were added 1 h before H2O2 exposure. Western blotting showed expression levels of MAP1LC3B and TUBA1A. (J) Quantification of immunoblot signals for MAP1LC3B-II accumulation. Data represent mean ± S.E; n = 3. *P < 0.05, **P < 0.01; N, not significant, P > 0.05. (K) GCs were grown in medium containing 10 μM melatonin for 24 h, washed using PBS, incubated with 200 μM H2O2 for 0–2 h, and then processed for determining cell viability using the CCK-8 assay. Data represent mean ± S.E; n = 3. *P < 0.05, **P < 0.01; NS, not significant, P > 0.05.
To verify this assumption, GCs were treated with melatonin or the autophagy inhibitor 3-methyladenine (3-MA) prior to H2O2 exposure. As shown in Fig. 3A, both melatonin and 3-MA markedly restored the viability of cells within 2 h after H2O2 incubation. Considering that apoptosis was also triggered in cells with continuing H2O2 stimulation [38], [46], we next tested whether melatonin-mediated autophagy correlated with apoptosis in GCs received short-term oxidative stress. However, compared with melatonin and/or 3-MA treatment, inhibition of apoptosis using Z-VAD-FMK (pancaspase inhibitor) failed to alleviate oxidative damage in GCs. In addition, no apparent activation of CASP3 was detected in the early stages of H2O2-induced GC death [40]. Our data thus suggested that melatonin preferentially inhibits apoptosis-independent autophagic death in GCs suffering acute oxidative stress. Since the autophagic process is controlled by several autophagy-related genes such as Becn1 and Atg7, we next tested whether antagonizing autophagy using small interfering RNA (siRNA) against Becn1 and Atg7 (Fig. 3B, C and Fig. S7) affected melatonin-mediated GC protection. As shown in Fig. S8 and Fig. 3D to F, the induction of autophagy and cell viability loss upon H2O2 stimulation were remarkably suppressed following siRNAs transfection. Consistently, melatonin displayed approximate inhibitory effects on oxidative stress-induced autophagy and GC death. Notably, melatonin did not significantly influence the autophagic activity or cell viability when the expression of Becn1 and Atg7 were silenced by RNAi. Based on these data, we proposed that melatonin protects GCs from oxidative injury via repressing autophagic death.
Fig. 3.

Melatonin preferentially inhibits the autophagic death to prevent oxidative stress-induced GC injury. (A) GCs with 24 h of melatonin (10 μM) treatment were rinsed in PBS, and then exposed to H2O2 (200 μM) for 2 h. The autophagy inhibitor 3-MA (10 mM), or the apoptosis inhibitor Z-VAD-FMK (50 μM) were added 1 h prior to H2O2 incubation. Cell viability was determined using the CCK-8 assay. Data represent mean ± S.E; n = 3 in each group. *P < 0.05 (**P < 0.01) vs. vehicle group at 0 h. # Represents P < 0.05 (## Represents P < 0.01) vs. H2O2-only-treated cells. & Represents P > 0.05 vs. H2O2-only-treated cells. N, not significant, P > 0.05. δ Represents P < 0.05 (δδ Represents P < 0.01) vs. Z-VAD-FMK-treated cells. (B and C) Primary cultured GCs remained as an untreated control or were transfected with Becn1 siRNA, Atg7 siRNA or scrambled control siRNA for 48 h. The protein levels of BECN1 and ATG7 were evaluated using western blotting. TUBA1A served as the control for loading. (D and E) GCs transfected with Becn1 siRNA, Atg7 siRNA or scrambled control siRNA for 24 h were cultured in media containing 10 μM melatonin for another 24 h before 2 h of H2O2 (200 μM) incubation. Cell lysates were then collected for western blotting assay. The MAP1LC3B-II accumulation and SQSTM1 degradation were quantified by densitometric analysis. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3 in each group. **P < 0.01; N, not significant, P > 0.05. (F) Cell viability was determined by CCK-8 assay in GCs with the indicated treatments as described above. ** Represents P < 0.01 compared to ‘Non siRNA’ condition. # Represents P > 0.05 compared to ‘Non siRNA’ condition. & Represents P > 0.05 compared to ‘H2O2 +melatonin’ condition.
3.3. The protective effect of Melatonin on GC survival via repressing autophagic death is independent of ROS scavenging
To test whether the antioxidant property of melatonin might influence autophagy by removing oxidative stress, the ROS production was measured in H2O2-incubated GCs following melatonin administration. As expected, the antioxidation capability of cells treated with melatonin was enhanced upon H2O2 exposure (Fig. 4A). The results of ROS detection also showed a reduction of oxidative stress in the presence of melatonin, but it was insufficient to exterminate excess intracellular ROS (Fig. 4B and C). Actually, compared with the control group, cells received melatonin treatment still exhibited significantly higher ROS levels following H2O2 incubation (Fig. 4B and C). Moreover, since melatonin provided much stronger inhibitory effects on autophagy induction under the same conditions (Fig. 2), we suspected that the suppression of autophagy might not be entirely attributed to ROS scavenging.
Fig. 4.

Melatonin decreases ROS generation in GCs with oxidative stimulation. (A) GCs pretreated with or without 10 μM melatonin for 24 h were then rinsed using PBS, and cultured in the presence or absence of H2O2 (200 μM). 2 h later, the total antioxidation capability (T-AOC) was evaluated by determining the reductive conversion of Fe3+-TPTZ to Fe2+-TPTZ. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 compared to control group. # Represents P < 0.05 compared to H2O2-only-treated cells. (B) The detection of ROS production in GCs with the indicated treatments as described above. Bar, 20 µm. (C) Quantification of intracellular ROS levels. The optical density was calculated in each GC with ImageJ 1.42q software. Experiments were repeated in triplicate, and three fields of each coverslip were selected in random for counting. Data represent mean ± S.E; n = 3. *P < 0.05, **P < 0.01; NS, not significant, P > 0.05.
Melatonin has been reported to act directly as an antioxidant or indirectly by activating several antioxidative components, including SOD, CAT, GPx, GR and GSH [47]. Our current experimental procedure excluded the direct antioxidative actions of melatonin, because GCs pretreated with this indoleamine were rinsed in PBS before H2O2 incubation. To further clarify whether melatonin-mediated autophagy suppression is correlated with ROS clearance, the antioxidation activity was blocked using specific antagonists (hereinafter referred to as antioxidant inhibitors, or AOI) against the downstream antioxidants of melatonin (Fig. 5A–C). As shown in Fig. 5D–F, AOI exerts no evident influence on melatonin-induced suppression of autophagy in cells with H2O2 exposure, implying an antioxidation-independent role of melatonin in autophagy regulation. This speculation was further confirmed by the observations from GCs treated with H2O2 in various concentrations as indicated for 2 h (Fig. 5G–K). It was found that the extent of autophagy and ROS production was increased in a dose-dependent manner when exposed to H2O2 (Fig. 5G–K). However, no additional induction of the autophagic biomarkers was detected at concentrations more than 150 μM (Fig. 5G–I). Notably, cells subjected to 150 μM of H2O2 incubation displayed similar levels of ROS generation compared with melatonin treatment upon oxidative stress (Fig. 5J and K). The results perfectly explained why melatonin could not inhibit autophagy via removing the intracellular ROS. Moreover, AOI failed to counteract the prosurvival effects of melatonin in H2O2-treated GCs (Fig. 5L). These data thus further supported a direct role of autophagy suppression in melatonin-mediated GCs protection without eliminating oxidative stress itself.
Fig. 5.

The inhibitory effect of melatonin on H2O2-triggered autophagic GC death is independent of ROS elimination. (A) Primary cultured GCs grown in medium containing 10 μM melatonin for 24 h were then rinsed in PBS, and exposed to 2 h of H2O2 (200 μM) incubation. The inhibitors of melatonin downstream antioxidants (AOI) were added 1 h prior to H2O2 treatment. The total antioxidation capability (T-AOC) was determined as described in Materials and Methods section. Data represent mean ± S.E; n = 3. *P < 0.05, **P < 0.01; NS, not significant, P > 0.05. (B) GCs were subjected to H2O2 exposure following melatonin and/or AOI treatment as mentioned above. The ROS levels were detected by dichlorofluorescein fluorescence (green), and nuclei were counterstained with DAPI (blue). Bar, 20 µm. (C) The optical density of intracellular ROS was quantified using ImageJ software. ** Represents P < 0.01 compared to control group; # Represents P < 0.05; N, not significant. (D) The immunoblotting detection of MAP1LC3B and SQSTM1 in GCs received the indicated treatments as described above. (E and F) Quantification of the MAP1LC3B-II accumulation and SQSTM1 degradation. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3 in each group. ** Represents P < 0.01 compared to control group. ## Represents P < 0.01 compared to H2O2-only-treated cells. N, not significant. (G) Primary cultured GCs treated with H2O2 at different concentrations as indicated for 2 h were collected for immunoblotting analysis of MAP1LC3B and SQSTM1. (H and I) The expression of MAP1LC3B-II and SQSTM1 were quantified by densitometric analysis. ** Represents P < 0.01 compared to control group; NS, not significant. (J) GCs pretreated with or without melatonin (10 μM) for 24 h were then rinsed in PBS, and incubated with H2O2 in various concentrations. 2 h later, the formation of ROS in GCs was observed under a laser confocal-scanning microscope. Bar, 20 µm. (K) Quantification of ROS levels by calculating the green fluorescence optical density in each GC. Experiments were repeated in triplicate, and three fields of each coverslip were selected at random for counting. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 compared to control group; NS, not significant. (L) GCs cultured with 10 μM melatonin for 24 h were then rinsed in PBS, and exposed to 2 h of H2O2 (200 μM) incubation. For the inhibition of melatonin-induced activation of downstream antioxidative components, cells were treated with AOI 1 h before H2O2 exposure. Cell viability was examined as described above. **P < 0.01; NS, not significant, P > 0.05.
3.4. Melatonin inhibits FOXO1-dependent autophagy in H2O2-incubated GCs
FOXO1, a major downstream effector of stress response signaling, has been suggested to induce autophagic cell death upon oxidative stimulation [37]. To test whether FOXO1 correlates with melatonin-mediated autophagy suppression during oxidative stress, the transcript encoding FOXO1 was silenced using RNA interference (Fig. 6A). Evidence from multiple types of mammalian cells indicates that stress-induced FOXO1 activation triggers the expression of several autophagy-related genes (Atgs), including Becn1, Rab7, Pik3c3, Atg7, Map1lc3b, Atg12 and Bnip3 [36], [48], [49]. As shown in Fig. 6B–E, both FOXO1 knockdown and melatonin treatment remarkably repressed Atgs transcription, MAP1LC3B-II accumulation and SQSTM1 degradation following oxidative stress. Notably, Foxo1 siRNA provided no additional inhibitory effects on autophagy when GCs were pretreated with melatonin (Fig. 6B–E), indicating that the suppression of FOXO1 by melatonin abrogated H2O2-induced autophagy. During oxidative stress, we also identified numerous vesicular structures with double membranes through the use of transmission electron microscopy (TEM), but such autophagic structures rarely appeared in GCs treated with Foxo1 siRNA and/or melatonin (Fig. 6F and G). Moreover, by determining cell viability under the same conditions (Fig. 6H), we further confirmed that FOXO1 inhibition is required for melatonin-mediated GC protection against oxidative damage. However, the results of qRT-PCR analysis and immunoblotting assay both showed that melatonin exerted no significant influence on FOXO1 expression in H2O2-treated cells despite siRNA transfection (Figs. 6I and 8A). Since FOXO1 functions primarily as a transcription factor in the nucleus, it raises the possibility that melatonin inhibits the activity of FOXO1 via regulating its subcellular localization.
Fig. 6.

Suppression of FOXO1-dependent autophagy by melatonin attenuates oxidative damage in GCs. (A) Primary cultured GCs were transfected with Foxo1 siRNA or scrambled control siRNA for 48 h. The expression of FOXO1 was determined by western blotting. (B) GCs transfected with Foxo1 siRNA or scrambled control siRNA were cultured with 10 μM melatonin for 24 h, washed in PBS, and then subjected to 2 h of H2O2 exposure (200 μM). qRT-PCR was performed to measure the mRNA levels of autophagy-related (Atg) genes in GCs. Expression data were normalized to that of Actb. Significances were marked as **P < 0.01 vs. SC siRNA group; ##P < 0.01 vs. SC siRNA+H2O2 group. N, not significant, P > 0.05. SC siRNA, scrambled control siRNA. (C) Immunoblot analysis of MAP1LC3B and SQSTM1 in GCs with the indicated treatments as described above. (D and E) The MAP1LC3B-II accumulation and SQSTM1 degradation were quantified by densitometric analysis. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 vs. control group; ## Represents P < 0.01 vs·H2O2 group. NS, not significant, P > 0.05. (F) GCs transfected with Foxo1 siRNA or scrambled control siRNA for 24 h were cultured for another 24 h in the presence or absence of 10 μM melatonin before 2 h of H2O2 (200 μM) incubation. Cells were then collected for TEM imaging of the autophagic structures. Bar, 1 µm. Enlarged images (below) show clearer autophagic vacuoles (red arrows). (G) Number of autophagic vacuoles per cell section in GCs. Bar graphs are mean ± S.E of results from 10 cell sections. **P < 0.01; NS, not significant, P > 0.05. (H) The detection of cell viability by CCK-8 assay. GCs were treated as above. Data represent mean ± S.E; n = 3 in each group. ** Represents P < 0.01 vs. scrambled siRNA-only-treated cells; ## Represents P < 0.01 vs. H2O2 group. NS, not significant, P > 0.05. (I) GCs transfected with Foxo1 siRNA or scrambled control siRNA for 24 h were exposed to various concentrations of melatonin (0, 5, 10, 20 μM). 24 h later, cells were washed in PBS and incubated with H2O2 (200 μM) for another 2 h. The expression of Foxo1 was determined by qRT-PCR. The relative expression level was normalized to that of Actb. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 vs. Foxo1 siRNA group; # Represents P < 0.01 vs. SC siRNA group. SC siRNA, scrambled control siRNA (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Fig. 8.

Inhibition of FOXO1 transcriptional activity through the melatonin-PI3K-AKT axis protects GCs from H2O2-induced autophagic PCD. (A) GCs transfected with Foxo1 siRNA or scrambled control siRNA for 24 h were cultured in media containing various concentrations of melatonin (0, 5, 10, 20 μM). 24 h later, cells were rinsed with PBS, and exposed to H2O2 (200 μM) incubation for another 2 h. The expression of phosphorylated FOXO1 (p-FOXO1) was determined by western blotting. (B) The phosphorylation level of FOXO1 was quantified by densitometric analysis. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 vs. Foxo1 siRNA group; # Represents P < 0.01 vs. SC siRNA group. SC siRNA, scrambled control siRNA. (C) GCs pretreated with or without 10 μM melatonin for 24 h were then rinsed in PBS, and subjected to 1 or 2 h of H2O2 (200 μM) incubation. For the inhibition of AKT, Perifosine (10 μM) was added 1 h before H2O2 exposure. Subcellular localization of FOXO1 was detected using anti-FOXO1 (green), and the nuclei were counterstained with DAPI (blue). Bar, 10 µm. (D) The percentage of cells with FOXO1 in the nucleus (green bars) and in the cytosol (orange bars). Experiments were repeated in triplicate, and 3 fields of each coverslip were selected in random for counting. Data represent mean ± S.E; n = 3. *P < 0.05, **P < 0.01. (E) A Flag-tagged FOXO1 (WT), FOXO1T24A,S253D,S316A (ADA), or an empty control plasmid (BP) was individually transfected into GCs. 24 h later, cells were cultured with or without melatonin (10 μM) for another 24 h. Immunofluorescence microscopy was performed to visualize subcellular localization of FOXO1 (green). The nuclei were counterstained with DAPI (blue). Bar, 5 µm. (F) The percentage of cells with FOXO1 in the nucleus or the cytosol under the indicated treatments. NPC, non-plasmid control; BP, blank plasmid; W, FOXO1-WT plasmid; ADA, FOXO1T24A,S253D,S316A plasmid. ** Represents P < 0.01 compared with the non-plasmid control; NS, not significant, P > 0.05; ##, P < 0.01. (G) qRT-PCR analysis of Atg genes transcription in GCs transfected with FOXO1-expressing vectors (FOXO1-WT, FOXO1T24A,S253D,S316A, and FOXO1N208A,H212R) in the presence or absence of melatonin (10 μM) as mentioned above. Expression data were normalized to that of Actb. Data represent mean ± S.E; n = 3 in each group. ** Represents P < 0.01 compared with nonplasmid control. # Represents P < 0.05 (## Represents P < 0.01) compared with FOXO1-WT (W) group. N Represents P > 0.05 compared with the FOXO1T24A,S253D,S316A (ADA) group. (H) Western blotting was performed to measure the protein levels of Flag-FOXO1, MAP1LC3B and SQSTM1 in GCs with the indicated treatments as described above. (I and J) Quantification of MAP1LC3B-II accumulation and SQSTM1 degradation. TUBA1A served as the control for loading. *P < 0.05 (**P < 0.01) vs. the non-plasmid control without melatonin treatment. ## Represents P < 0.01 vs. FOXO1-WT-transfected group without melatonin treatment. & Represents P > 0.05 vs. FOXO1T24A,S253D,S316A (ADA)-transfected group without melatonin treatment. δδ Represents P < 0.01 vs. FOXO1N208A,H212R (DBD)-transfected group without melatonin treatment. (K) Primary cultured GCs remained as an untreated control or were transfected with FOXO1-WT or FOXO1T24A,S253D,S316A for 24 h. Cells were then grown for another 24 h in the presence or absence of 10 μM melatonin, and collected for TEM imaging of the autophagic structures. Bar, 1 µm. Enlarged images (below) show clearer autophagic vacuoles (red arrows). (L) Number of autophagic vacuoles per cell section in GCs. Bar graphs are mean ± S.E of results from 10 cell sections. **P < 0.01; NS, not significant, P > 0.05. (M) Cell viability was measured by CCK-8 assay in GCs transfected with FOXO1 expression plasmids upon melatonin (10 μM) treatment. **P < 0.01; N, not significant, P > 0.05. BP, blank plasmid.
3.5. Melatonin protects GCs from H2O2-induced autophagic death through the PI3K-AKT pathway
Previous studies indicated that the PI3K-AKT pathway plays a key role in regulating nuclear/cytoplasmic shuttling of FOXO1 when GCs were exposed to oxidative stress [38], [39]. To further elucidate the antioxidation-independent mechanism of melatonin in GCs protection, we examined Ser473 phosphorylation of AKT (In this study, unless otherwise specified, p-AKT refers to phosphorylation of AKT at Ser473), which results in its activation. Western blot assays showed that melatonin remarkably inhibited H2O2-induced dephosphorylation of AKT without altering total AKT protein level (Fig. 7A–D). In contrast, the inhibition of PI3K with LY294002 abolished melatonin-induced AKT activation (Fig. 7E–G). These results were in agreement with an earlier report suggesting that the PI3K-AKT pathway acts downstream of melatonin signaling in mammalian cells [50].
Fig. 7.

Melatonin counteracts H2O2-induced autophagic GC death through the PI3K-AKT pathway. (A) After culturing with 10 μM melatonin for 24 h, GCs were washed in PBS, and then subjected to H2O2 (200 μM) incubation for 1 or 2 h. The expression of phosphorylated AKT (p-AKT) and total AKT was determined by western blotting. (B–D) The relative expression of total AKT, p-AKT, and the ratio of p-AKT to total AKT were quantified using densitometric analysis. TUBA1A served as the control for loading. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 compared to control group. # Represents P > 0.05 compared to control group. (E) GCs grown in medium containing 10 μM melatonin for 24 h were washed in PBS, and then incubated with 200 μM H2O2 for 2 h. For the inhibition of PI3K activity, LY294002 (20 μM) was added 1 h before H2O2 treatment. Western blotting was performed to measure the protein levels of p-AKT and total AKT. (F and G) Quantification of p-AKT expression and the ratio of p-AKT to total AKT. **P < 0.01; NS, not significant, P > 0.05. (H) GCs pretreated with or without 10 μM melatonin for 24 h were then rinsed in PBS, and cultured in the presence or absence of 200 μM H2O2 for 2 h. qRT-PCR was performed to measure the mRNA levels of autophagy-related (Atg) genes in GCs. Expression data were normalized to that of Actb. Data represent mean ± S.E; n = 3 in each group. * Represents P < 0.05 (** Represents P < 0.01) compared to control group. & Represents P > 0.05 compared to H2O2-only-treated cells. # Represents P < 0.05 (## Represents P < 0.01) compared to H2O2-only-treated cells. δ Represents P < 0.05 (δδ Represents P < 0.01) compared to melatonin+H2O2 group. (I) Immunoblotting analysis of MAP1LC3B, SQSTM1, AKT and p-AKT in GCs with the indicated treatments as described above. (J and K) Quantification of the MAP1LC3B-II accumulation and SQSTM1 degradation. Data represent mean ± S.E; n = 3. **P < 0.01; NS, not significant, P > 0.05. (L) TEM imaging of the autophagic structures in GCs with indicated treatments as mentioned above. Bar, 1 µm. Enlarged images (below) show clearer autophagic vacuoles (red arrows). (M) Number of autophagic vacuoles per cell section in GCs. Bar graphs are mean ± S.E of results from 10 cell sections. **P < 0.01; NS, not significant, P > 0.05. (N) Cell viability was determined by CCK-8 assay in GCs received the indicated treatments. Data represent mean ± S.E; n = 3. **P < 0.01; NS, not significant, P > 0.05 (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
We next investigated whether PI3K-AKT is required for melatonin-mediated autophagy suppression upon H2O2 stimulation. qRT-PCR analysis showed that perifosine (AKT inhibitor) abrogated melatonin-induced transcriptional downregulation of several FOXO1 target Atgs (Fig. 7H). In accordance with this, blocking PI3K-AKT pathway using perifosine led to AKT dephosphorylation, which was associated with MAP1LC3B-II accumulation, and SQSTM1 degradation despite melatonin treatment in GCs with H2O2 exposure (Fig. S9 and Fig. 7I–K). The inhibition of autophagy via the melatonin-PI3K-AKT axis was also demonstrated by monitoring the double membrane vesicles using transmission electron microscopy (TEM). As shown in Fig. 7L and M, treatment with perifosine restored the H2O2-triggered formation of autophagic vacuoles in the presence of melatonin. To further clarify whether the PI3K-AKT signaling affected the prosurvival action of melatonin upon oxidative stress, we then examined the cell viability of H2O2-treated GCs following melatonin and/or perifosine administration. As shown in Fig. 7N, the AKT inhibitor abolished melatonin-induced cell survival after H2O2 incubation, consistent with the observations from autophagy determination under the same conditions (Fig. 7H–K). Taken together, these data suggested that suppression of autophagy through the PI3K-AKT pathway is involved in melatonin-mediated GC protection against oxidative injury.
3.6. Suppression of FOXO1 transcriptional activity through the melatonin-PI3K-AKT axis antagonizes autophagic GC death upon oxidative stress
Given the role of PI3K-AKT signaling in repressing FOXO1 by promoting its phosphorylation and nuclear exportation, we next examined whether melatonin affected the subcellular localization of FOXO1 through this pathway. As shown in Fig. S10, Fig. 8A, and B, H2O2 incubation increased the expression of total FOXO1 and led to its dephosphorylation at Ser253 (In this study, p-FOXO1 refers to phosphorylation of FOXO1 at Ser253). Melatonin markedly inhibited H2O2-induced dephosphorylation of FOXO1 without affecting total FOXO1 protein level. Consistently, H2O2 exposure facilitated FOXO1 activation as suggested by increased nuclear localization, which was blocked after melatonin treatment (Fig. 8C and D). By contrast, the AKT inhibitor perifosine restored H2O2-induced nuclear transportation of FOXO1 in GCs despite melatonin administration (Fig. 8C and D). To further verify the inhibitory effects of melatonin-PI3K-AKT on FOXO1 activity, GCs were transfected with FOXO1-WT and a constitutively active FOXO1 mutant (FOXO1T24A,S253D,S316A/FOXO1-ADA; the 3 mutations were Threonine 24 to Alanine, and Serine 253 to Aspartate, and Serine 316 to Alanine). As shown in Fig. 8E and F, high levels of total FOXO1 protein were observed both in the cytoplasm and nucleus following enforced expression of FOXO1-WT or FOXO1T24A,S253D,S316A. However, GCs pretreated with melatonin for 24 h led to a marked redistribution of FOXO1 from the nucleus to the cytoplasm in the FOXO1-WT group. Conversely, the FOXO1 mutant without AKT phosphorylation sites (FOXO1T24A,S253D,S316A) abrogated melatonin-induced cytoplasmic localization of FOXO1, consistent with the results obtained from AKT inhibition as mentioned above (Fig. 8C and D). To investigate whether the nuclear/cytoplasmic shuttling of FOXO1 exerted any influence on melatonin-directed autophagy regulation, GCs were transfected with the expressing vectors of FOXO1, including FOXO1-WT (WT), FOXO1T24A,S253D,S316A, and FOXO1N208A,H212R/FOXO1-DBD (a FOXO1 mutant without DNA-binding capability). qRT-PCR analysis showed that all of the autophagic transcripts were remarkably increased after enforced expression of FOXO1-WT and FOXO1T24A,S253D,S316A, compared with levels in cells transfected with FOXO1N208A,H212R or blank control plasmid (Fig. 8G). Meanwhile, melatonin antagonized FOXO1-triggered transcriptional activation of Atgs in the FOXO1-WT group, but not in the FOXO1T24A,S253D,S316A group (Fig. 8G). Correspondingly, expression of FOXO1T24A,S253D,S316A restored MAP1LC3B-II expression, SQSTM1 degradation and autophagosome formation in cells treated with melatonin (Fig. 8H–L). Additionally, GCs transfected with FOXO1 plasmids displayed a remarkable decline in cell viability, which was significantly reversed by melatonin in the FOXO1-WT group. In contrast, the constitutively active form of FOXO1 (FOXO1T24A,S253D,S316A) counteracted melatonin-mediated GC protection (Fig. 8M). These findings were in agreement with the observations from autophagy determination under the same conditions (Fig. 8G–L), indicating that the phosphorylation and nuclear exclusion of FOXO1 might be an essential step in melatonin-mediated suppression of autophagic GC death through the PI3K-AKT signaling pathway.
3.7. Melatonin reduces oxidative injury in GCs by suppressing SIRT1-FOXO1-ATG7-dependent autophagy
The FOXO1 plasmid lacking DNA-binding capability (FOXO1N208A,H212R) was supposed to be a negative control for the stimulation of autophagy (Fig. 8G). However, the results of immunoblotting assay showed that the MAP1LC3B-II accumulation and SQSTM1 degradation was remarkably increased during enforced expression of this mutant (Fig. 8H and I). Notably, the induction of autophagy and cell viability loss in cells transfected with FOXO1N208A,H212R was prohibited after melatonin treatment, indicating that FOXO1-triggered autophagic GC death might be repressed by melatonin through an alternative mechanism which is independent of FOXO1 transcriptional activity. In fact, stress-induced acetylation of FOXO1 has been suggested to facilitate the autophagic process [37]. We thus examined the effects of melatonin on FOXO1 acetylation upon oxidative exposure. As shown in Fig. 9A–C, H2O2 incubation led to an elevated acetylation of FOXO1, which was maintained at significantly lower levels in cells pretreated with melatonin. Conversely, GCs received melatonin administration displayed enhanced expression of the deacetylase SIRT1, consistent with previous reports in several other tissues [51], [52]. To test whether FOXO1 acetylation is required for melatonin-mediated regulation of FOXO1 transcription independent autophagy, cells were treated with Sirtinol (SIRT1 inhibitor) or SRT1720 (SIRT1 activator) following FOXO1N208A,H212R transfection. Equal amounts of total FOXO1 proteins were detected in each group (Fig. 9D), enabling us to specify the effects of FOXO1 acetylation without differences in FOXO1 expression. As shown in Fig. 9D–G, inhibition of SIRT1 using Sirtinol not only induced FOXO1 acetylation, but also promoted MAP1LC3B-II accumulation and SQSTM1 degradation. In contrast, SRT1720 markedly attenuated the autophagic response in GCs, which was associated with compromised acetylation of FOXO1. Moreover, compared with melatonin treatment, SRT1720 exhibited similar level of suppression in FOXO1 acetylation and autophagy, but melatonin failed to further decrease the autophagic signals in GCs pretreated with the SIRT1 activator. Conversely, constitutively acetylated FOXO1 induced by Sirtinol treatment blocked the inhibitory effects of melatonin on autophagy. In accordance with this, the SIRT1 antagonist restored FOXO1N208A,H212R-induced autophagosome formation in the presence of melatonin (Fig. 9H and I). Collectively, these data demonstrated that deacetylation of FOXO1 through the melatonin-SIRT1 signaling represses autophagy in GCs.
Fig. 9.

Deacetylation of FOXO1 via the melatonin-SIRT1 signaling inhibits FOXO1-dependent autophagy in GCs. (A) GCs were cultured with 10 μM melatonin for 24 h, washed in PBS, and then exposed to H2O2 incubation for 1 or 2 h. The protein level of acetylated FOXO1 (Ac-FOXO1), total FOXO1 and the deacetylase SIRT1 was determined by western blotting. (B and C) The relative expression of Ac-FOXO1 and SIRT1 were quantified using densitometric analysis. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 compared to control group. # Represents P > 0.05 compared to control group. (D) GCs transfected with FOXO1N208A,H212R plasmid for 24 h were grown in medium containing 10 μM melatonin. 24 h later, cells were cultured for another 2 h in the presence or absence of Sirtinol (100 μM) or SRT1720 (100 μM). The expression of Ac-FOXO1, total FOXO1, MAP1LC3B and SQSTM1 was then detected by western blotting. (E–G) Quantification of FOXO1 acetylation, MAP1LC3B-II accumulation and SQSTM1 degradation. TUBA1A served as a loading control. Data represent mean ± S.E; n = 3. (H) GCs transfected with FOXO1N208A,H212R plasmid for 24 h were cultured for another 24 h in the presence or absence of 10 μM melatonin before 2 h of Sirtinol (100 μM) treatment. Cells were then collected for TEM imaging of the autophagic structures. Bar, 0.8 µm. Enlarged images (below) show clearer autophagic vacuoles (red arrows). (I) Number of autophagic vacuoles per cell section in GCs. Bar graphs are mean ± S.E of results from 10 cell sections. **P < 0.01 (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
We next investigated whether the acetylation of FOXO1 might influence its interaction with other autophagy-related (ATG) proteins, SIRT1 or the acetyltransferase EP300. GCs exposed to H2O2 incubation following melatonin treatment were lysed for coimmunoprecipitation with BECN1, MTOR, SQSTM1, ATG3, ATG5, ATG7, ATG12, SIRT1 and EP300. Immunoblotting analysis of the ATG proteins revealed that only ATG7 was coprecipitated by the FOXO1 antibody (Fig. 10A and B), implying a physical interaction between FOXO1 and ATG7 upon oxidative stimulation. In contrast, melatonin significantly reduced the binding affinity of ATG7 to FOXO1 during H2O2 exposure (Fig. 10A and B). Additionally, melatonin abrogated oxidative stress-induced acetylation of FOXO1 as indicated by enhanced interactions of FOXO1 and SIRT1 in contrast with EP300 (Fig. 10A, C and D), suggesting that the acetylation status of FOXO1 modified by SIRT1/EP300 counterbalance might be required for melatonin-regulated FOXO1-ATG7 combination upon oxidative stimulation. To verify this speculation, cells were treated with melatonin, Sirtinol or SRT1720 prior to H2O2 incubation. As shown in Fig. 10E–G, melatonin and the SIRT1 activator displayed a similar level of suppression in H2O2-triggered FOXO1 acetylation and FOXO1-ATG7 interaction, both of which could not be further inhibited by melatonin in GCs received SRT1720 administration. However, melatonin failed to block the acetylation of FOXO1 and its binding to ATG7 when SIRT1 was repressed using Sirtinol. Correspondingly, both melatonin and SRT1720 markedly restrained the decline in GC viability under oxidative stress, whereas constitutive acetylation of FOXO1 by Sirtinol abrogated the prosurvival effect of melatonin (Fig. 10H). Since H2O2-induced upregulation of ATG7 might also affect its interaction with FOXO1, cells were transfected with FOXO1N208A,H212R. Equal amounts of FOXO1 and/or ATG7 protein levels were observed in each treatment group (Fig. 10I), enabling us to specify the effects of FOXO1-ATG7 interaction without differences in their expression. Using coimmunoprecipitation analysis and CCK-8 assay (Fig. 10I–L), we further confirmed that the dissociation of FOXO1-ATG7 complex caused by FOXO1 deacetylation through the melatonin-SIRT1 pathway provides a preventive action on oxidative stress-induced autophagic GC death.
Fig. 10.

Melatonin prevents autophagic death by antagonizing the interaction of acetylated FOXO1 and ATG7 in H2O2-treated GCs. (A) GCs pretreated with 10 μM melatonin for 24 h were then washed in PBS, and grown in medium supplemented with H2O2 (200 μM). 2 h later, the cell lysates were processed for coimmunoprecipitation with anti-FOXO1, followed by probing with anti-BECN1, MTOR, SQSTM1, ATG3, ATG5, ATG7, ATG12, SIRT1 and EP300. WCL, whole-cell lysates. IP, immunoprecipitation. (B–D) The amount of coimmunoprecipitated ATG7, SIRT1 and EP300 for each IP reaction was normalized to TUBA1A content in the whole-cell lysates (input). Data represent mean ± S.E; n = 3. **P < 0.01. (E–H) GCs were cultured with 10 μM melatonin for 24 h, rinsed in PBS, and exposed to 200 μM H2O2 for 2 h. Sirtinol (100 μM) or SRT1720 (100 μM) was added 1 h prior to H2O2 incubation. After the indicated treatments, cells were collected for coimmunoprecipitation (E–G) or CCK-8 assay (H). For immunoprecipitation, the cell lysates were precipitated with anti-FOXO1, and probed with anti-Ac-FOXO1 or anti-ATG7. The protein levels of Ac-FOXO1 and ATG7 in the immunoprecipitates were quantified as described above. Data represent mean ± S.E; n = 3. ** Represents P < 0.01 vs. control group. ## Represents P < 0.01 vs. H2O2-only-treated cells. && Represents P < 0.01 vs. melatonin+H2O2 group. NS, not significant, P > 0.05. (I) GCs transfected with FOXO1N208A,H212R plasmid for 24 h were cultured for another 24 h in the presence or absence of 10 μM melatonin before 2 h of Sirtinol (100 μM) or SRT1720 (100 μM) treatment. Cell lysates were then extracted for co-immunoprecipitation with anti-FOXO1, followed by probing with anti-Ac-FOXO1 or anti-ATG7. (J and K) The amount of Ac-FOXO1 and ATG7 in the immunoprecipitates was quantified as mentioned above. **P < 0.01 vs. non treatment control. ##, P < 0.01; N, not significant, P > 0.05. (L) The detection of cell viability by CCK-8 assay. GCs were treated as above. Data represent mean ± S.E; n = 3 in each group. ** Represents P < 0.01 compared with the FOXO1N208A,H212R (DBD) group. ## Represents P < 0.01 compared with the FOXO1N208A,H212R (DBD)+melatonin group. NS, not significant, P > 0.05.
4. Discussion
The biosynthesis of sex steroids in ovarian GCs requires massive energy consumption and thus generates high levels of intracellular ROS [53]. Accelerated metabolic rates in the reproductive process also give rise to a large amount of oxygen free radicals [4]. Therefore, oxidative stress represents a cost of reproduction. On the other hand, environmental pollutants, ionizing radiation, malnutrition and unhealthy lifestyle factors including cigarette smoking, alcohol abuse, and drug use promote excess production of ROS in ovarian follicles [5]. ROS accumulation causes oxidative damage of GCs, which in turn triggering follicular atresia and relevant anovulatory disorders [5]. Melatonin, a major secretory product of the pineal gland, has been implicated in preserving normal functions of GCs and follicles during oxidative stress [26]. In this study, we proposed a novel role of melatonin in protecting ovarian GCs survival from oxidative damage by inhibiting autophagy. As reported, the cytoprotective effects of melatonin are achieved primarily through its antioxidant properties [47]. Interestingly, our data showed that melatonin-mediated suppression of autophagic GC death is independent of ROS clearance.
Based on our data obtained in vivo and in vitro, the current study demonstrated FOXO1 as a critical target of melatonin-mediated GC protection. Under oxidative stress, excessive activation of autophagy by FOXO1 induces autophagic GC death. The detrimental effects of autophagic response could be impaired by melatonin treatment, but it provided no additional protection when FOXO1 has been knocked down. In fact, melatonin promotes nuclear exclusion of FOXO1 via the PI3K-AKT pathway, which in turn counteracts the expression of several downstream proautophagic genes. Moreover, inhibition of FOXO1 acetylation and the resulting disassembly of FOXO1-ATG7 complex through the melatonin-SIRT1 signaling also attenuate autophagic death in GCs exposed to oxidative stimulation. Taken together, suppression of autophagy, downregulation of FOXO1 transcriptional activity, and deacetylation of FOXO1 by melatonin constitutes part of an adaptive mechanism for maintaining GC survival against oxidative injury (see Graphical abstract).
Although previous studies suggested a potential function for melatonin in preserving GC viability upon noxious stimuli [26], current knowledge regarding the defense mechanisms of melatonin is mostly confined to its regulation on apoptosis [54], which has traditionally been considered as the major cause of GC death and follicular atresia [55], [56]. However, recent literature showed that nonapoptotic forms of PCD (programmed cell death) such as autophagy also occurs within the atretic follicles [10]. Particularly, autophagic signals are detectable only in the granulosa layers of antral follicles [57]. Actually, growing evidence indicates that autophagy may serve primarily as a pro-death pathway that exacerbates GC damage under stressful conditions [12], [13]. Moreover, oxidative stress-activated autophagy has been reported to initiate PCD without apoptosis induction in multiple types of mammalian cells [46], [58]. Therefore, the present study investigated whether autophagy is correlated with the inhibitory effects of melatonin on oxidative GC injury. To our knowledge, this is the first evidence demonstrating a role of autophagy in melatonin-induced GC protection during oxidative stress.
It is still a matter of debate as to whether autophagy, not only facilitates cellular resistance to environmental stresses, but also promotes cell destruction through excessive self-digestion and degradation of vital components [8]. To clarify the interplay between autophagy and cell death in melatonin-treated GCs, we antagonized the autophagic process using 3-MA or siRNAs targeting Atg7 and Becn1. Remarkably, impaired autophagic signals correlated with elevated cell viability were observed in GCs subjected to oxidant incubation following melatonin administration. Consistently, the specific suppression of autophagy also relieved H2O2-induced GC injury. However, melatonin could not further decrease the rate of GC death when cellular autophagy was blocked. Thus, our data suggest that melatonin represses a detrimental form of autophagy in GCs suffering oxidative stimulation.
Melatonin serves as a robust antioxidant that scavenges ROS directly or indirectly by upregulating other antioxidative enzymes [59]. Recently, melatonin has been suggested to repress autophagy through redox-mediated elimination of free radicals [18], [19]. Consistent with previous reports [26], [54], we found that melatonin raised the antioxidation capability of GCs upon oxidative stress. However, the extent of ROS reduction in GCs pretreated with melatonin was significantly lower than its inhibitory effects on autophagy during H2O2 exposure. Correspondingly, the blocking of ROS clearance in melatonin-treated cells prior to oxidative stimulation did not significantly restore the autophagic activity that should have been recovered by antagonizing the downstream antioxidants of melatonin. In fact, cells received melatonin treatment before H2O2 incubation showed similar levels of ROS production induced by 150–200 μM H2O2, which tended to trigger a maximum autophagic response as suggested by our current experimental model. Collectively, this study first describes a specific regulation of GC autophagy by melatonin without removing oxidative stress.
As a conserved transcriptional regulator responsible for cell fate determination [28], [29], [31], [32], FOXO1 is selectively and highly expressed by GCs of growing follicles [60]. Immunohistochemical assay in rodent ovaries revealed intense nuclear staining of FOXO1 that was centralized within GCs during follicular atrsia [61]. Earlier reports demonstrated that FOXO1 has potential roles in modulating GC functions [62], [63], [64]. For example, enforced FOXO1 activation in GCs blocked gonadotrophin-induced upregulation of genes required for sterol/steroid and lipid production [62], [63]. Further investigations suggested the negative effects of FOXO1 on GC differentiation by downregulating the expression of NR5A1, INHA, EREG, CCND2 and CYP19A1 [64]. Our previous work also identified FOXO1 as a critical factor in promoting GC death upon oxidative stimulation [15], [38], [39]. These results thus indicate that the inhibition of FOXO1 might be essential for maintaining the well-being of GCs. However, it is still unknown whether FOXO1 contributes to melatonin-mediated regulation of GC protection during oxidative stress. Our current work showed that melatonin markedly inhibited autophagy and oxidative injury in mouse ovarian GCs, which was associated with FOXO1 suppression. Therefore, this might be the first evidence suggesting that melatonin restrains autophagic GC death through a FOXO1-dependent manner.
The potent functions of the forkhead transcription factors are tightly controlled by their subcellular localizaion and posttranslational modifications, including phosphorylation, acetylation, and ubiquitination [33], [34]. It remains unclear whether, among the various cellular responses to FOXO1, such as proliferation, apoptosis, cell cycle arrest and autophagy, there is a specific set of FOXO1 target genes, or the physiological actions varies depending on different cell types, tissues or environmental stimuli. Particularly, in neuron cells, the translocation of FOXO1 from the cytoplasm to the nucleus driven by oxidative stress triggered cell death [65]. In contrast, nuclear transportation of FOXO1 preserved cardiomyocyte survival by upregulating the transcription of autophagy-related genes (Atgs) during starvation [35]. However, the regulation modes of FOXO1 in the protective mechanisms of melatonin against oxidative injury (especially the autophagic death), has previously not been described in GCs. Here, we observed that melatonin inhibits FOXO1 activity via the PI3K-AKT pathway, which promotes phosphorylation and nuclear export of FOXO1, hence blocking the expression of several downstream Atg genes as reported [36], [48], [49], and thereby preventing autophagic GC death upon oxidative stimulation. These findings might provide the first evidence that melatonin modulates the autophagic response within GCs through a process involving suppression of FOXO1 transcriptional activity.
The transcriptional activator FOXO1 has been implicated in regulating transcription-independent autophagy [37], [66], [67]. Specifically, the acetylated forms of FOXO1 might be required for promoting autophagy in certain cancer cells [37]. Cellular stresses, including ROS accumulation and nutrient deprivation, not only decrease AKT-induced phosphorylation of FOXO1, but also facilitate its detachment from sirtuin proteins and consequent acetylation mediated by the acetyltransferase EP300 [68], [69]. SIRT1, the mammalian homolog of yeast silent information regulator 2 (Sir2) [70], catalyzes the deacetylation of FOXO1 under stressful conditions, since enforced SIRT1 expression efficiently attenuated FOXO1 acetylation upon oxidative stimulation [71]. However, few reports described whether the acetylation status of FOXO1 might be correlated with autophagy in response to melatonin signaling. According to our data, melatonin abrogated H2O2-induced FOXO1 acetylation as indicated by increased binding affinity of FOXO1 to SIRT1 in contrast with EP300, which was followed by impaired autophagic signals. In other words, these findings first demonstrate a role of melatonin in repressing autophagy via acetylation-dependent regulation of FOXO1 activity. As to the present study, this hypothesis perfectly explains why the FOXO1 mutant lacking DNA-binding capability also triggered a remarkable autophagic response, which should have been counteracted by melatonin-mediated transcriptional suppression of Atg genes.
Posttranslational modifications of FOXO1 affect its interactions with other proteins [34]. Here, we showed that the autophagy-related protein 7 (ATG7), which is required for MAP1LC3 conversion [72], acts as a FOXO1 binding partner in the regulation of FOXO1 acetylation-dependent autophagy by melatonin. In response to oxidative exposure that improved the autophagic induction, melatonin decreased the level of Atg7 transcripts via antagonizing FOXO1 transcriptional activity. Importantly, ATG7 formed complex with acetylated FOXO1, which was accompanied by intensified autophagic signals during oxidative stress. These results were in agreement with an earlier report suggesting that the interaction between ATG7 and acetylated FOXO1 facilitates the autophagic activity [37]. Conversely, melatonin markedly reduced the binding affinity of ATG7 to FOXO1 by promoting FOXO1 deacetylation, followed by dampened activation of autophagy, along with an enhancement of GC resistance to oxidative injury. To our knowledge, these findings might represent the first evidence demonstrating that melatonin-mediated deacetylation of FOXO1 and its detachment from ATG7 provides a protective effect on GC viability by suppressing oxidative stress-induced autophagic death.
Maintenance of redox balance is critical for GC survival in ovarian follicles [26]. The follicular antioxidants, including melatonin, SOD, GPx, and CAT would normally protect GCs from free radical damage and suppress atresia [73]. In contrast, reduced antioxidant enzyme levels are reported in the follicular fluid of women with unexplained infertility [74]. Moreover, persistent and excessive oxidative stimulation not only attenuates the effects of cellular antioxidative system [75], but also impairs the ROS scavenging ability of melatonin as suggested by our current study. In fact, we had previously assumed that a redox-dependent feedback pathway modulates autophagy when melatonin acts as an antioxidant to adjust ROS levels in GCs. However, our findings negate that speculation. It was found that the suppression of over-stimulated autophagic activity through the melatonin-FOXO1 signaling, which inhibits the self-destruction of cellular proteins and organelles, might provide a second level of defense when antioxidant capabilities are compromised. This process constitutes a selective survival advantage in conferring resistance to excessive oxidative stress.
When exposure to low dose of environmental stress, cells activate protective genes called vitagenes to maintain cellular homeostasis and survival in a process termed as “hormesis” [76]. However, cells appear poorly adapted for high dose of stimuli that induce cytotoxicity and cellular damage. Under this condition, pharmacological intervention might be essential for cell protection by upregulating vitagene activity through several defensive pathways, including SIRT1 and NRF2 [76]. Emerging evidence indicates a crosstalk between SIRT1 and NRF2 in cellular resistance to external stress upon drug treatment [77], [78]. Since the results of our present study suggests the involvement of SIRT1 signaling in melatonin-mediated GC protection, further researches might be conducted to investigate the interplay between SIRT1 and NRF2 signaling in the prosurvival mechanism of melatonin against oxidative damage. Moreover, it remains to be determined whether, among the diverse cellular responses to the SIRT1/NRF2 pathway, there are specific target sets of vitagenes upon melatonin treatment. These studies might lend support to develop a hormetic melatonin therapy to produce desirable clinical effects.
Melatonin has been suggested to inhibit the pathogenesis of anovulatory disorders by preserving GC survival, though the underlying mechanism remains largely undetermined [26]. The present study describes a novel role of melatonin in protecting against oxidative injury of GCs by suppressing FOXO1-dependent autophagy through the PI3K-AKT-FOXO1 axis and SIRT1-FOXO1-ATG7 pathway. Therefore, targeting melatonin-FOXO1 signaling may provide benefits to the clinical treatment for anovulatory infertility.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.31630072; No. 31601939), the Key Project of Chinese National Programs for Fundamental Research and Development (973 program no. 2014CB138502), Natural Science Foundation of Jiangsu Province (No. BK20150664), the Fundamental Research Funds for the Central Universities (No. KJQN201705), China Postdoctoral Science Special Foundation (No. 2016T90476), China Postdoctoral Science Foundation (No. 2015M581818), Jiangsu Planned Projects for Postdoctoral Research Funds (No. 1501047A). We thank Professor Shao-chen Sun at Nanjing Agricultural University for his help and assistance in manuscript preparation and submission
Acknowledgments
Conflict of interest
No potential conflicts of interest were disclosed.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.07.004.
Contributor Information
Ming Shen, Email: shenm2015@njau.edu.com.
Yan Cao, Email: 2015105014@njau.edu.cn.
Yi Jiang, Email: 2014105076@njau.edu.cn.
Yinghui Wei, Email: 309918676@qq.com.
Honglin Liu, Email: liuhonglin@njau.edu.cn.
Appendix A. Supplementary material
Supplementary material
References
- 1.Baker T.G. A quantitative and cytological study of germ cells in human ovaries. Proc. R. Soc. Lond. B Biol. Sci. 1963;158:417–433. doi: 10.1098/rspb.1963.0055. [DOI] [PubMed] [Google Scholar]
- 2.Alonso-Pozos I., Rosales-Torres A.M., Avalos-Rodriguez A., Vergara-Onofre M., Rosado-Garcia A. Mechanism of granulosa cell death during follicular atresia depends on follicular size. Theriogenology. 2003;60:1071–1081. doi: 10.1016/s0093-691x(03)00123-7. [DOI] [PubMed] [Google Scholar]
- 3.Uttara B., Singh A.V., Zamboni P., Mahajan R.T. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agarwal A., Gupta S., Sharma R.K. Role of oxidative stress in female reproduction. Reprod. Biol. Endocrinol. 2005;3:28. doi: 10.1186/1477-7827-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agarwal A., Aponte-Mellado A., Premkumar B.J., Shaman A., Gupta S. The effects of oxidative stress on female reproduction: a review. Reprod. Biol. Endocrinol. 2012;10:49. doi: 10.1186/1477-7827-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B., Klionsky D.J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 7.Kaur J., Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 8.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dadakhujaev S., Jung E.J., Noh H.S., Hah Y.S., Kim C.J., Kim D.R. Interplay between autophagy and apoptosis in TrkA-induced cell death. Autophagy. 2009;5:103–105. doi: 10.4161/auto.5.1.7276. [DOI] [PubMed] [Google Scholar]
- 10.Choi J., Jo M., Lee E., Choi D. Induction of apoptotic cell death via accumulation of autophagosomes in rat granulosa cells. Fertil. Steril. 2011;95:1482–1486. doi: 10.1016/j.fertnstert.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Hulas-Stasiak M., Gawron A. Follicular atresia in the prepubertal spiny mouse (Acomys cahirinus) ovary. Apoptosis. 2011;16:967–975. doi: 10.1007/s10495-011-0626-9. [DOI] [PubMed] [Google Scholar]
- 12.Duerrschmidt N., Zabirnyk O., Nowicki M., Ricken A., Hmeidan F.A., Blumenauer V. Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology. 2006;147:3851–3860. doi: 10.1210/en.2006-0088. [DOI] [PubMed] [Google Scholar]
- 13.Serke H., Vilser C., Nowicki M., Hmeidan F.A., Blumenauer V., Hummitzsch K. Granulosa cell subtypes respond by autophagy or cell death to oxLDL-dependent activation of the oxidized lipoprotein receptor 1 and toll-like 4 receptor. Autophagy. 2009;5:991–1003. doi: 10.4161/auto.5.7.9507. [DOI] [PubMed] [Google Scholar]
- 14.Vilser C., Hueller H., Nowicki M., Hmeidan F.A., Blumenauer V., Spanel-Borowski K. The variable expression of lectin-like oxidized low-density lipoprotein receptor (LOX-1) and signs of autophagy and apoptosis in freshly harvested human granulosa cells depend on gonadotropin dose, age, and body weight. Fertil. Steril. 2010;93:2706–2715. doi: 10.1016/j.fertnstert.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 15.Shen M., Jiang Y., Guan Z., Cao Y., Li L., Liu H. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy. 2017;13:1364–1385. doi: 10.1080/15548627.2017.1327941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manchester L.C., Coto-Montes A., Boga J.A., Andersen L.P., Zhou Z., Galano A. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015;59:403–419. doi: 10.1111/jpi.12267. [DOI] [PubMed] [Google Scholar]
- 17.Reiter R.J., Mayo J.C., Tan D.X., Sainz R.M., Alatorre-Jimenez M., Qin L. Melatonin as an antioxidant: under promises but over delivers. J. Pineal Res. 2016;61:253–278. doi: 10.1111/jpi.12360. [DOI] [PubMed] [Google Scholar]
- 18.Vega-Naredo I., Caballero B., Sierra V., Garcia-Macia M., de Gonzalo-Calvo D., Oliveira P.J. Melatonin modulates autophagy through a redox-mediated action in female Syrian hamster Harderian gland controlling cell types and gland activity. J. Pineal Res. 2012;52:80–92. doi: 10.1111/j.1600-079X.2011.00922.x. [DOI] [PubMed] [Google Scholar]
- 19.Khaldy H., Escames G., Leon J., Bikjdaouene L., Acuna-Castroviejo D. Synergistic effects of melatonin and deprenyl against MPTP-induced mitochondrial damage and DA depletion. Neurobiol. Aging. 2003;24:491–500. doi: 10.1016/s0197-4580(02)00133-1. [DOI] [PubMed] [Google Scholar]
- 20.Chang C.F., Huang H.J., Lee H.C., Hung K.C., Wu R.T., Lin A.M. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and alpha-synuclein aggregation. J. Pineal Res. 2012;52:312–321. doi: 10.1111/j.1600-079X.2011.00945.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H., Chen J., Lu X., Shen C., Zeng J., Chen L. Melatonin protects against rotenone-induced cell injury via inhibition of Omi and Bax-mediated autophagy in Hela cells. J. Pineal Res. 2012;52:120–127. doi: 10.1111/j.1600-079X.2011.00926.x. [DOI] [PubMed] [Google Scholar]
- 22.Brzezinski A., Seibel M.M., Lynch H.J., Deng M.H., Wurtman R.J. Melatonin in human preovulatory follicular fluid. J. Clin. Endocrinol. Metab. 1987;64:865–867. doi: 10.1210/jcem-64-4-865. [DOI] [PubMed] [Google Scholar]
- 23.Sundaresan N.R., Marcus Leo M.D., Subramani J., Anish D., Sudhagar M., Ahmed K.A. Expression analysis of melatonin receptor subtypes in the ovary of domestic chicken. Vet. Res. Commun. 2009;33:49–56. doi: 10.1007/s11259-008-9071-9. [DOI] [PubMed] [Google Scholar]
- 24.Tian X., Wang F., Zhang L., He C., Ji P., Wang J. Beneficial effects of melatonin on the in vitro maturation of sheep oocytes and its relation to melatonin receptors. Int. J. Mol. Sci. 2017:18. doi: 10.3390/ijms18040834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yie S.M., Niles L.P., Younglai E.V. Melatonin receptors on human granulosa cell membranes. J. Clin. Endocrinol. Metab. 1995;80:1747–1749. doi: 10.1210/jcem.80.5.7745030. [DOI] [PubMed] [Google Scholar]
- 26.Tamura H., Nakamura Y., Korkmaz A., Manchester L.C., Tan D.X., Sugino N. Melatonin and the ovary: physiological and pathophysiological implications. Fertil. Steril. 2009;92:328–343. doi: 10.1016/j.fertnstert.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Soares J.M., Jr., Simoes M.J., Oshima C.T., Mora O.A., De Lima G.R., Baracat E.C. Pinealectomy changes rat ovarian interstitial cell morphology and decreases progesterone receptor expression. Gynecol. Endocrinol. 2003;17:115–123. [PubMed] [Google Scholar]
- 28.Nemoto S., Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 29.Accili D., Arden K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 30.Barthel A., Schmoll D., Unterman T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Greer E.L., Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 32.Salih D.A., Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogt P.K., Jiang H., Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. 2005;4:908–913. doi: 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- 34.Calnan D.R., Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 35.Sengupta A., Molkentin J.D., Yutzey K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009;284:28319–28331. doi: 10.1074/jbc.M109.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hariharan N., Maejima Y., Nakae J., Paik J., Depinho R.A., Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Y., Yang J., Liao W., Liu X., Zhang H., Wang S. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 38.Shen M., Lin F., Zhang J., Tang Y., Chen W.K., Liu H. Involvement of the up-regulated FoxO1 expression in follicular granulosa cell apoptosis induced by oxidative stress. J. Biol. Chem. 2012;287:25727–25740. doi: 10.1074/jbc.M112.349902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen M., Liu Z., Li B., Teng Y., Zhang J., Tang Y. Involvement of FoxO1 in the effects of follicle-stimulating hormone on inhibition of apoptosis in mouse granulosa cells. Cell Death Dis. 2014;5:e1475. doi: 10.1038/cddis.2014.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen M., Jiang Y., Guan Z., Cao Y., Sun S.C., Liu H. FSH protects mouse granulosa cells from oxidative damage by repressing mitophagy. Sci. Rep. 2016;6:38090. doi: 10.1038/srep38090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L., Hao J., Hu J., Pu J., Lu Z., Zhao L. Protective effects of ginsenosides against Bisphenol A-induced cytotoxicity in 15P-1 Sertoli cells via extracellular signal-regulated kinase 1/2 signalling and antioxidant mechanisms. Basic Clin. Pharmacol. Toxicol. 2012;111:42–49. doi: 10.1111/j.1742-7843.2012.00857.x. [DOI] [PubMed] [Google Scholar]
- 42.Paglin S., Hollister T., Delohery T., Hackett N., McMahill M., Sphicas E. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 43.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liot G., Bossy B., Lubitz S., Kushnareva Y., Sejbuk N., Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X.N., Greenwald G.S. Synergistic effects of steroids with FSH on folliculogenesis, steroidogenesis and FSH- and hCG-receptors in hypophysectomized mice. J. Reprod. Fertil. 1993;99:403–413. doi: 10.1530/jrf.0.0990403. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y., McMillan-Ward E., Kong J., Israels S.J., Gibson S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–182. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H.M., Zhang Y. Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J. Pineal Res. 2014;57:131–146. doi: 10.1111/jpi.12162. [DOI] [PubMed] [Google Scholar]
- 48.Webb A.E., Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 2014;39:159–169. doi: 10.1016/j.tibs.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Vos K.E., Coffer P.J. The extending network of FOXO transcriptional target genes. Antioxid. Redox Signal. 2011;14:579–592. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 50.Picinato M.C., Hirata A.E., Cipolla-Neto J., Curi R., Carvalho C.R., Anhe G.F. Activation of insulin and IGF-1 signaling pathways by melatonin through MT1 receptor in isolated rat pancreatic islets. J. Pineal Res. 2008;44:88–94. doi: 10.1111/j.1600-079X.2007.00493.x. [DOI] [PubMed] [Google Scholar]
- 51.Yu L., Sun Y., Cheng L., Jin Z., Yang Y., Zhai M. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J. Pineal Res. 2014;57:228–238. doi: 10.1111/jpi.12161. [DOI] [PubMed] [Google Scholar]
- 52.Zhao L., An R., Yang Y., Yang X., Liu H., Yue L. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J. Pineal Res. 2015;59:230–239. doi: 10.1111/jpi.12254. [DOI] [PubMed] [Google Scholar]
- 53.Rapoport R., Sklan D., Hanukoglu I. Electron leakage from the adrenal cortex mitochondrial P450scc and P450c11 systems: nadph and steroid dependence. Arch. Biochem. Biophys. 1995;317:412–416. doi: 10.1006/abbi.1995.1182. [DOI] [PubMed] [Google Scholar]
- 54.Song C., Peng W., Yin S., Zhao J., Fu B., Zhang J. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci. Rep. 2016;6:35165. doi: 10.1038/srep35165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsuda-Minehata F., Inoue N., Goto Y., Manabe N. The regulation of ovarian granulosa cell death by pro- and anti-apoptotic molecules. J. Reprod. Dev. 2006;52:695–705. doi: 10.1262/jrd.18069. [DOI] [PubMed] [Google Scholar]
- 56.Tilly J.L., Tilly K.I., Kenton M.L., Johnson A.L. Expression of members of the bcl-2 gene family in the immature rat ovary: equine chorionic gonadotropin-mediated inhibition of granulosa cell apoptosis is associated with decreased bax and constitutive bcl-2 and bcl-xlong messenger ribonucleic acid levels. Endocrinology. 1995;136:232–241. doi: 10.1210/endo.136.1.7828536. [DOI] [PubMed] [Google Scholar]
- 57.Choi J.Y., Jo M.W., Lee E.Y., Yoon B.K., Choi D.S. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil. Steril. 2010;93:2532–2537. doi: 10.1016/j.fertnstert.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 58.Pan X., Liu D., Wang J., Zhang X., Yan M., Zhang D. Peneciraistin C induces caspase-independent autophagic cell death through mitochondrial-derived reactive oxygen species production in lung cancer cells. Cancer Sci. 2013;104:1476–1482. doi: 10.1111/cas.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Genc A., Ucok K., Sener U., Koyuncu T., Akar O., Celik S. Association analyses of oxidative stress, aerobic capacity, daily physical activity, and body composition parameters in patients with mild to moderate COPD. Turk. J. Med. Sci. 2014;44:972–979. doi: 10.3906/sag-1308-65. [DOI] [PubMed] [Google Scholar]
- 60.Richards J.S., Sharma S.C., Falender A.E., Lo Y.H. Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-I, estrogen, and the gonadotropins. Mol. Endocrinol. 2002;16:580–599. doi: 10.1210/mend.16.3.0806. [DOI] [PubMed] [Google Scholar]
- 61.Shi F., LaPolt P.S. Relationship between FoxO1 protein levels and follicular development, atresia, and luteinization in the rat ovary. J. Endocrinol. 2003;179:195–203. doi: 10.1677/joe.0.1790195. [DOI] [PubMed] [Google Scholar]
- 62.Liu Z., Rudd M.D., Hernandez-Gonzalez I., Gonzalez-Robayna I., Fan H.Y., Zeleznik A.J. FSH and FOXO1 regulate genes in the sterol/steroid and lipid biosynthetic pathways in granulosa cells. Mol. Endocrinol. 2009;23:649–661. doi: 10.1210/me.2008-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan H.Y., O'Connor A., Shitanaka M., Shimada M., Liu Z., Richards J.S. Beta-catenin (CTNNB1) promotes preovulatory follicular development but represses LH-mediated ovulation and luteinization. Mol. Endocrinol. 2010;24:1529–1542. doi: 10.1210/me.2010-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park Y., Maizels E.T., Feiger Z.J., Alam H., Peters C.A., Woodruff T.K. Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J. Biol. Chem. 2005;280:9135–9148. doi: 10.1074/jbc.M409486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng W.H., Kar S., Quirion R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J. Biol. Chem. 2000;275:39152–39158. doi: 10.1074/jbc.M002417200. [DOI] [PubMed] [Google Scholar]
- 66.Zhao Y., Wang L., Yang J., Zhang P., Ma K., Zhou J. Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy. 2010;6:988–990. doi: 10.4161/auto.6.7.13289. [DOI] [PubMed] [Google Scholar]
- 67.Medema R.H., Jaattela M. Cytosolic FoxO1: alive and killing. Nat. Cell Biol. 2010;12:642–643. doi: 10.1038/ncb0710-642. [DOI] [PubMed] [Google Scholar]
- 68.Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 69.Daitoku H., Sakamaki J., Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta. 2011;1813:1954–1960. doi: 10.1016/j.bbamcr.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 70.Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 71.Salminen A., Kaarniranta K., Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int. J. Mol. Sci. 2013;14:3834–3859. doi: 10.3390/ijms14023834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kabeya Y., Mizushima N., Yamamoto A., Oshitani-Okamoto S., Ohsumi Y., Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 73.Tamura H., Takasaki A., Taketani T., Tanabe M., Kizuka F., Lee L. Melatonin as a free radical scavenger in the ovarian follicle. Endocr. J. 2013;60:1–13. doi: 10.1507/endocrj.ej12-0263. [DOI] [PubMed] [Google Scholar]
- 74.Paszkowski T., Traub A.I., Robinson S.Y., McMaster D. Selenium dependent glutathione peroxidase activity in human follicular fluid. Clin. Chim. Acta. 1995;236:173–180. doi: 10.1016/0009-8981(95)98130-9. [DOI] [PubMed] [Google Scholar]
- 75.Kirkham P.A., Barnes P.J. Oxidative stress in COPD. Chest. 2013;144:266–273. doi: 10.1378/chest.12-2664. [DOI] [PubMed] [Google Scholar]
- 76.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010;13:1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang K., Gao X., Wei W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-beta1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017;361:63–72. doi: 10.1016/j.yexcr.2017.09.042. [DOI] [PubMed] [Google Scholar]
- 78.Shah S.A., Khan M., Jo M.H., Jo M.G., Amin F.U., Kim M.O. Melatonin stimulates the SIRT1/Nrf2 signaling pathway counteracting lipopolysaccharide (LPS)-induced oxidative stress to rescue postnatal rat brain. CNS Neurosci. Ther. 2017;23:33–44. doi: 10.1111/cns.12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material