

Abstract
Key points
We studied the role of the large‐conductance Ca2+‐activated K+ channel (BK) and voltage‐dependent K+ channels (Kv) on [Ca2+]i responses to a wide range of hypoxia at different resting cell membrane potential (E m).
BK/Kv were mostly closed at rest in normoxia. BK/Kv became basally active when cells were depolarized by elevated [KCl]o (>12 mm). Regardless of whether BK/Kv were closed or basally open, hypoxia‐induced elevation of [Ca2+]i was enhanced 2‐ to 3‐fold by inhibitors of BK/Kv.
Hypoxia‐induced elevation of [Ca2+]i was enhanced ∼2‐fold by an inhibitor of Kv2, a major Kv in rat glomus cells.
Hypoxia did not inhibit BK in inside‐out patches.
Our study supports a scheme in which activation of BK/Kv strongly limits the magnitude of hypoxia‐induced [Ca2+]i rise, with Kv having a much greater effect than BK.
Abstract
Large‐conductance KCa (BK) and other voltage‐dependent K+ channels (Kv) are highly expressed in carotid body (CB) glomus cells, but their role in hypoxia‐induced excitation is still not well defined and remains controversial. We addressed this issue by studying the effects of inhibitors of BK (IBTX) and BK/Kv (TEA/4‐AP) on [Ca2+]i responses to a wide range of hypoxia at different levels of resting cell membrane potential (E m). IBTX and TEA/4‐AP did not affect the basal [Ca2+]i in isolated glomus cells bathed in 5 mm KClo, but elicited transient increases in [Ca2+]i in cells that were moderately depolarized (11–20 mV) by elevation of [KCl]o (12–20 mm). Thus, BK and Kv were mostly closed at rest and activated by depolarization. Four different levels of hypoxia (mild, moderate, severe, anoxia) were used to produce a wide range of [Ca2+]i elevation (0–700 nm). IBTX did not affect the rise in [Ca2+]i, but TEA/4‐AP strongly (∼3‐fold) enhanced [Ca2+]i rise by moderate and severe levels of hypoxia. Guangxitoxin, a Kv2 blocker, inhibited the whole‐cell current by ∼50%, and enhanced 2‐fold the [Ca2+]i rise elicited by moderate and severe levels of hypoxia. Anoxia did not directly affect BK, but activated BK via depolarization. Our findings do not support the view that hypoxia inhibits BK/Kv to initiate or maintain the hypoxic response. Rather, our results show that BK/Kv are activated as glomus cells depolarize in response to hypoxia, which then limits the rise in [Ca2+]i. Inhibition of Kv may provide a mechanism to enhance the chemosensory activity of the CB and ventilation.
Keywords: hypoxia, carotid body, voltage‐dependent K+ channel, BK, TASK
Key points
We studied the role of the large‐conductance Ca2+‐activated K+ channel (BK) and voltage‐dependent K+ channels (Kv) on [Ca2+]i responses to a wide range of hypoxia at different resting cell membrane potential (E m).
BK/Kv were mostly closed at rest in normoxia. BK/Kv became basally active when cells were depolarized by elevated [KCl]o (>12 mm). Regardless of whether BK/Kv were closed or basally open, hypoxia‐induced elevation of [Ca2+]i was enhanced 2‐ to 3‐fold by inhibitors of BK/Kv.
Hypoxia‐induced elevation of [Ca2+]i was enhanced ∼2‐fold by an inhibitor of Kv2, a major Kv in rat glomus cells.
Hypoxia did not inhibit BK in inside‐out patches.
Our study supports a scheme in which activation of BK/Kv strongly limits the magnitude of hypoxia‐induced [Ca2+]i rise, with Kv having a much greater effect than BK.
Introduction
Carotid body (CB) glomus cells continuously monitor changes in arterial and adjust the degree of ventilation to help maintain a constant level of blood . The signalling process begins with the altered activity of O2 sensor(s) within the CB. This results in the generation of a signal that modulates the activity of O2‐sensitive ion channels, and thereby the membrane potential and excitability of glomus cells (Buckler & Vaughan‐Jones, 1994; Kumar & Prabhakar, 2012; Lopez‐Barneo et al. 2016). It is widely accepted that hypoxia depolarizes glomus cells by inhibiting the outward K+ current, which results in opening of the voltage‐dependent Ca2+ channel (Cav) and elevation of [Ca2+]i (Urena et al. 1994; Kumar, 2007; Lopez‐Barneo et al. 2008; Buckler, 2015). The rise in [Ca2+]i stimulates the secretion of transmitters such as acetylcholine and ATP that bind to their receptors at the terminals of carotid sinus afferent nerve that has inputs to the cardiorespiratory brainstem nuclei (Pardal et al. 2000; Zhang et al. 2000; Lopez‐Barneo et al. 2001).
The idea that hypoxia inhibits the K+ current to elicit the hypoxic response (elevation of [Ca2+]i and secretion of transmitters) is now well accepted. However, the precise roles of different types of K+ channels in the initiation and maintenance of the hypoxic response are still not well defined and reported findings are contradictory. TASK (mainly TASK‐1/3), large‐conductance Ca2+‐activated K+ (BK) and voltage‐dependent K+ (Kv) are three major K+ channel types expressed in rat glomus cells, where we refer to Kv as a group of voltage‐dependent and tetraethylammonium (TEA)/ 4‐aminopyridine (4‐AP)‐sensitive K+ channels that does not include BK. If a K+ channel is active at rest and is inhibited by hypoxia, then closure of the K+ channel is expected to cause depolarization and help sustain the depolarized state, provided that the resting background Na+ conductance remains unaffected. A clear case for such a mechanism is demonstrated by the inhibition of TASK, the background K+ channel in glomus cells (Buckler et al. 2000; Kim et al. 2009), and this signalling mechanism is not disputed for rat and mouse glomus cells. The role of TASK in hypoxia‐induced excitation of glomus cells in other species such as rabbit and human remains uncertain, as TASK current and its sensitivity to hypoxia has not yet been reported in these species.
BK and Kv as potential hypoxia‐sensitive K+ channels have been the focus of many studies since some time. Unfortunately, conflicting findings have been reported on the roles of BK and Kv in the regulation of the hypoxic response. Species differences may exist, but even in the same species such as the rat, opposite results have been observed. In rat glomus cells, a group of studies showed evidence that BK/Kv were active at rest and that hypoxia inhibited them to depolarize the cells and produce the hypoxic response (Wyatt & Peers, 1995; Pardal et al. 2000; Peers & Wyatt, 2007). In mouse glomus cells, hypoxia also inhibited BK/Kv, and this was proposed to underlie the hypoxic response (Perez‐Garcia et al. 2004; Otsubo et al. 2011). In contrast, another group of studies presented evidence that BK/Kv were closed at rest and therefore had no effect on the hypoxic response (Buckler, 1997; Gomez‐Nino et al. 2009; Donnelly et al. 2011) or reduced the hypoxic response (Pardal et al. 2000; Wasicko et al. 2006). Thus, the contradictory findings and different proposed roles for BK/Kv have limited our understanding of the ionic mechanisms responsible for basal glomus cell excitability and the hypoxic response.
In excitable cells, activation of BK and/or Kv following depolarization provides the repolarizing force to limit further depolarization as well as to terminate the excitation. Thus, regardless of the state of BK/Kv in glomus cells at rest, the depolarization elicited by hypoxia (via inhibition of TASK, for example) is predicted to activate BK/Kv that then limits the hypoxic response. Because several studies have reported that hypoxia inhibits BK/Kv (Wyatt & Peers, 1995; Riesco‐Fagundo et al. 2001; Williams et al. 2004), one proposed idea was that the reduced repolarizing force (due to inhibition of BK/Kv) allowed the hypoxic response to occur (Gomez‐Nino et al. 2009). Such a mechanism may explain the failure of K+ channel inhibitors to enhance the hypoxic response observed in some studies (Gomez‐Nino et al. 2009; Donnelly et al. 2011), but fails to account for the strong potentiating effect of K+ channel inhibitors on the hypoxic response observed in other studies (Buckler, 1997; Pardal et al. 2000; Wasicko et al. 2006).
To address the question on the role of BK and Kv in the hypoxic response, we performed a more detailed study of the effects of hypoxia on [Ca2+]i using many coverslips of cells derived from multiple cell isolations, using cells at different levels of resting cell membrane potential (E m), and using cells exposed to four different levels of hypoxia (mild hypoxia, moderate hypoxia, severe hypoxia and anoxia) that elicit a wide range of elevations in [Ca2+]i. One of our goals was to test the possibility that different resting cell E m and associated changes in basal [Ca2+]i alter the basal levels of BK/Kv and thus the sensitivity of glomus cells to K+ channel inhibitors and hypoxia. Using iberiotoxin (IBTX) to inhibit BK, and TEA/4‐AP to inhibit BK/Kv, we studied whether and how much BK and Kv modulate the hypoxic response. We also assessed the direct and indirect effect of hypoxia on BK/Kv in inside‐out and cell‐attached patches. Although hypoxia has been reported to inhibit BK in perforated and inside‐out patches (Wyatt & Peers, 1995; Riesco‐Fagundo et al. 2001; Williams et al. 2004), the effects of hypoxia on BK and Kv without the experimental control of cell E m and [Ca2+]i are not clearly known.
Our results showed that TEA/4‐AP had no effect on basal [Ca2+]i, showing that BK and Kv were mostly closed at rest in normoxia. TEA/4‐AP caused an increase in [Ca2+]i in glomus cells that were mildly depolarized using elevated [KCl]o, indicating that BK and Kv were basally open in these cells. Therefore, differences in resting cell E m levels may possibly explain why K+ channel inhibitors increased [Ca2+]i and secretory activity in some studies, but not in others. Moderate and severe levels of hypoxia elicited a rise in [Ca2+]i that was strongly enhanced by TEA/4‐AP, moderately enhanced by guangxitoxin‐1E (GxTX) (an inhibitor of Kv2) and very weakly enhanced by IBTX. Thus, the contribution of BK alone in limiting the rise in [Ca2+]i in response to hypoxia was very small, that by Kv2 was moderate, and that by BK and Kv together was strong. Our findings in rat glomus cells show that activation of BK/Kv strongly limits the hypoxic response, and therefore do not support the scheme in which hypoxia inhibits BK/Kv to initiate or potentiate the hypoxic response.
Methods
Ethical approval
All experiments were carried out according to the guidelines set by the Animal Care and Use Committee of Rosalind Franklin University under Institutional Animal Care and Use Committee (IACUC) protocol #B14‐11, and conform to the principles and regulations as described by Grundy (2015). The animal care and use programme is AAALAC‐accredited and the approved Assurance with Office of Laboratory Animal Welfare number is A3279‐01. There are no ethical concerns.
Cell isolation
Rats (Sprague‐Dawley; postnatal days 16–24) purchased from Envigo (Indianapolis, IN, USA) were acclimatized in house for ∼1 week. Standard laboratory diet (Teklad Global 19% Protein Extruded Rodent Diet, Harlan #2019S) and drinking water were provided. Rats were deeply anaesthetized by inhalation of isoflurane until cessation of breathing, and decapitated with a sharp razor blade. CBs were removed and placed in ice‐cold, low Ca2+, low Mg2+ phosphate‐buffered saline (low Ca2+/Mg2+ PBS: 137 mm NaCl, 2.8 mm KCl, 2 mm KH2PO4, 0.07 mm CaCl2, 0.05 mm MgCl2, pH 7.4). Each CB (six to eight in total) was cut into several pieces and placed in a solution containing trypsin (0.4 mg ml−1) and collagenase (0.4 mg ml−1) in low Ca2+/Mg2+ PBS and incubated at 37°C for about 25 min. Following trituration, the dispersed cells were separated by centrifugation at low speed (1200 × g for 5 min) and suspended in CB growth medium [Ham's F‐12, 10% fetal bovine serum, 23 mm glucose, 4 mm Glutamax‐1 (l‐alanyl glutamine), 10 kU penicillin, 10 kU streptomycin and 300 μg ml−1 insulin]. Cells were plated on to polylysine‐pretreated glass coverslips and incubated at 37°C for ∼3 h in a humidified atmosphere of 95% air/5% CO2. Cells were used the same day within 6 h after plating.
Electrophysiology
Electrophysiological recording was performed using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA, USA). Cell‐attached patches were formed with gentle suction with sylgard‐coated borosilicate glass pipettes. Channel current was filtered at 2 kHz using an eight‐pole Bessel filter (–3 dB; Frequency Devices, Ottawa, IL, USA) and transferred to a computer using the Digidata 1320 interface at a sampling rate of 20 kHz. Single‐channel currents were analysed with the pCLAMP program (version 10). Channel openings were analysed to obtain channel activity (NPo, where N is the number of channels in the patch, and Po is the probability of a channel being open). Minimum open time duration was set at 0.1 ms. NPo was determined from ∼30 s of current recordings. Experiments were performed on cells prepared from different days. A maximum of two coverslips of cells was used on the day of the experiment so that each experiment was done on at least three separate cell preparations.
For whole‐cell studies, the pipette solution contained (mm): 140 KCl, 1 MgCl2, 5 EGTA, 10 Hepes, 11 glucose and 2 MgATP (pH 7.3). The bath (physiological) solution contained (mm): 117 NaCl, 5 KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (adjusted to pH 7.3 with NaOH). In experiments using cell‐attached and inside‐out patches, pipette solution contained (mm): 140 KCl (or 117 NaCl, 23 NaHCO3 and 5 KCl), 1 MgCl2, 5 EGTA, 10 Hepes and 11 glucose (pH 7.3). Extracellular bath (physiological) solution contained (mm): 117 NaCl (or 135 KCl), 5 KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (adjusted to pH 7.3 with NaOH). In all electrophysiological experiments, cells were perfused by gravity with solution preheated just before entry to the perfusion chamber using an in‐line solution heater and a temperature controller (Warner Instruments, Hamden, CT, USA). The temperature of the bath perfusion solution cells was maintained constant at 34–35°C.
[Ca2+]i measurement
Isolated cells plated on glass coverslips were incubated with 2 μm fura‐2 acetoxymethyl ester (fura‐2 AM) for 30 min at 37°C in culture medium. A coverslip with attached cells was placed in a recording chamber positioned on the stage of an inverted microscope (IX71; Olympus America Inc., Centre Valley, PA, USA). Fura‐2 was alternately excited at 340 and 380 nm and the emitted fluorescence was filtered at 510 nm and recorded using a charge‐coupled device (CCD)‐based imaging system running SimplePCI software (Hamamatsu, Welwyn Garden City, UK). The cells in the recording chamber were continuously perfused with a solution containing (mm): 117 NaCl, 5 KCl, 23 NaHCO3, 1 MgCl2, 1 CaCl2, 10 Hepes and 11 glucose (pH 7.3). Ratiometric data were calibrated by applying experimentally determined constants to the equation: [Ca2+] = K d × β × (R–R min)/(R max–R) (Grynkiewicz et al. 1985). Values for R max (12.0), R min (0.1) and β (11.6) were determined in vitro, and a K d value of 300 nm for fura‐2 was assumed. Each experiment was performed using coverslips of cells prepared on different days and no more than two coverslips were used for each day. Basal and peak [Ca2+]i values were each determined by averaging over 8–10 s (four or five frames) of data.
High K+ perfusion solutions with desired levels of free [Ca2+] were prepared according to the WEBMAXC STANDARD procotol (https://web.stanford.edu/~cpatton/webmaxcS.htm) using 1 mm BAPTA (in mm: 117 KCl, 23 KHCO3, 1 MgCl2, 10 Hepes, 20 mannitol and 11 glucose; pH 7.3). We prepared 200 nm, 500 nm and 1 μM Ca2+ solutions using this protocol. Free [Ca2+] was then determined by in vitro calibration using fura‐2 (1 μm) with a K d value (285 nm) for Ca2+ at 37°C and β value (9.8) that was determined in vitro. [Ca2+] values were 163 ± 11, 550 ± 18 and 948 ± 21 nm (mean ± SE of three determinations). We used these solutions to study BK in glomus cells. Ionomycin (2 μm) was used in some cell‐attached patch experiments.
Hypoxia studies
In our experiments that used a recording chamber that was open to the atmosphere, we generated four hypoxic solutions that produced increases in [Ca2+]i that ranged from ∼0% (mild hypoxia) to ∼200% (anoxia) of that produced by 20 mm KClo. These levels of hypoxia were achieved by bubbling solutions with 5% CO2/95% N2 with 2 units ml−1 of glucose oxidase/100 units ml−1 of catalase added to the solution (anoxia: AN; Baumann et al. 2008; Kim et al. 2011), 5% CO2/95% N2 with 0.4 units ml−1 of glucose oxidase/20 units ml−1 of catalase added to the solution (severe hypoxia: SH), 5% CO2/1% O2/94% N2 (moderate hypoxia: MoH), or 5% CO2/2% O2/90% N2 (mild hypoxia: MiH). Control solution was gassed with 5% CO2/95% air. A reproducible level of [O2] in the perfusion chamber was produced for each level of hypoxia, as judged by %O2 measurements using an O2 meter (ISO2, World Precision Instruments, Sarasota, FL, USA). After a steady‐state basal [Ca2+]i was obtained with normoxic solution, the perfusion solution was switched to a desired hypoxic solution. The temperature of the perfusion solutions was 35.0 ± 0.5°C. In three trials, the mean O2 contents in hypoxic solutions in the recording chamber were ∼0.5% (3.5 mmHg) for anoxia, ∼1.0% (7.6 mmHg) for severe hypoxia, ∼1.6% (12.2 mmHg) for moderate hypoxia and ∼2.6% (20 mmHg) for mild hypoxia. The O2 meter was calibrated to 0% with solution bubbled with pure nitrogen for 60 min and to 21% with solution gassed with air for 30 min at 37°C.
Materials
TEA, IBTX and 4‐AP were purchased from Tocris (Ellisville, MO, USA). Fura‐2 and fura‐2‐AM was purchased from TEF Labs (Austin, TX, USA). Catalase and glucose oxidase were purchased from Sigma‐Aldrich (St Louis, MO, USA). GxTX was purchased from Alomone Labs (Jerusalem, Israel). Gas tanks were from Linde Gas (USA). All other chemicals were purchased from Sigma‐Aldrich.
Statistical analysis
Student's t test (for paired comparison of two sets of data) and one‐way ANOVA (comparison of three or more sets of data) were used for data analysis using PRISM software. For fura‐2 experiments, a mean value of [Ca2+]i was determined from each coverslip of O2‐responding cells, and used to calculate the final mean ± SEM from the total number of coverslips used. Post hoc testing following ANOVA was based on Bonferroni correction. P < 0.05 was considered significant. Individual P values are indicated as necessary.
Results
Whole‐cell and single‐channel currents in isolated rat glomus cells
We first assessed the magnitude of whole‐cell BK and Kv currents activated by depolarization. As reported previously, whole‐cell currents showed a biphasic response to a voltage ramp applied from −100 to +20 mV. Currents averaged from 12 cells with similar levels of control whole‐cell currents (i.e. ∼2 nA at +20 mV) are shown in Fig. 1 A. A very small current (∼10–40 pA) was activated at potentials between −100 and −30 mV, and this was followed by a large current activation (1–2 nA) when cells were depolarized beyond −30 mV. Application of 100 nm IBTX produced a very small inhibition of the flat part of the current response, but inhibited the current more strongly as the cell depolarized, as shown more clearly in Fig. 1 B. Application of TEA/4‐AP (3 mm each) that block Kv as well as BK produced a strong reduction of the whole‐cell current. Figure 1 C shows subtracted currents for BK (IBTX‐sensitive), Kv (TEA/4‐AP‐sensitive current minus IBTX‐sensitive current), and the sum of BK and Kv between −80 and −20 mV. On average, the magnitude of Kv activated by depolarization beyond −20 mV was ∼2.5‐fold greater than that of BK (Fig. 1 C).
Figure 1. Effects of IBTX and TEA/4‐AP on the whole‐cell current in isolated glomus cells.
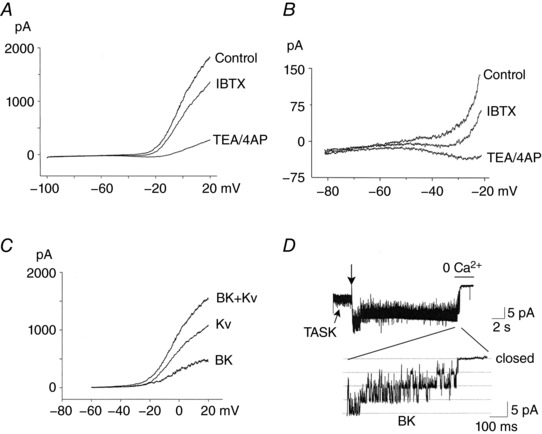
A, whole‐cell currents were recorded from isolated glomus cells using a ramp protocol. Cells were held at −100 mV and voltage ramps applied (500 ms duration) to +20 mV and then back to −100 mV. Cells were perfused with solution containing 100 nm IBTX first until steady state current was obtained and then with TEA (3 mm)/4‐AP (3 mm), and whole‐cell current recorded again. Currents were averaged from 12 cells with relatively similar control current sizes. B, current tracings between −80 and −20 mV shown at an expanded scale. C, IBTX‐sensitive (BK), TEA/4‐AP‐sensitive (BK + Kv) and Kv currents between −60 and +20 mV are shown (average of 12 cells). D, cell‐attached patch showing TASK openings. At the downward arrow, inside‐out patch is formed into a Ca2+‐containing bath solution, which activates BK. Removal of bath Ca2+ closes BK. Expanded tracing showing closing of BK upon removal of Ca2+. Similar responses were observed in 18 cells.
In cell‐attached patches with 140 mm KCl in the pipette and perfused with a physiological bath solution (5 mm KClo), and with the pipette potential (V p) set at 0 mV, TASK was active and no BK was observed (Fig. 1 D). When inside‐out patches were formed (indicated by arrow) and thus exposed to the bath solution that contained 1 mm Ca2+, several BK channels opened in most patches, and closed when Ca2+ was removed from the perfusion solution (Fig. 1 D). The expanded current tracing shows the closing of BK upon washout of Ca2+ from the bath solution. Such channels were not observed when 100 nm IBTX was present in the pipette and bath solutions in all 12 patches. We assessed the expression of BK by counting the number of channels in inside‐out patches at high (1 mm) and low (0 Ca2+ with 1 mm EGTA) cytosolic [Ca2+]. On average, three BK channels were present in each patch (range = 0–6 channels; n = 18). We were unable to identify Kv channels, probably because they were closed and single channel conductance levels are too small to be clearly resolved under these experimental conditions.
Effects of IBTX and TEA/4‐AP on [Ca2+]i in isolated glomus cells
In the next series of experiments, we assessed the role of BK and BK/Kv in the regulation of basal [Ca2+]i using IBTX and TEA/4‐AP. If BK or Kv is sufficiently active at rest, then its inhibition is predicted to depolarize the cells, open Cav and elevate [Ca2+]i. In glomus cells perfused with a bath solution containing 5 mm KCl, IBTX (100 nm) produced no significant effect on mean basal [Ca2+]i, whereas anoxia and 20 mm KCl elicited strong increases in [Ca2+]i, showing that BK was mostly closed at rest (Fig. 2 A).
Figure 2. Effect of IBTX on glomus cell [Ca2+]i at different [KCl]o .
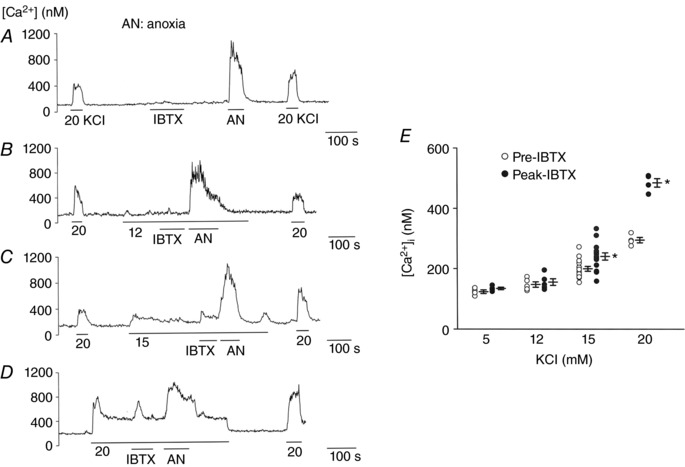
A, glomus cells were perfused with normoxic bath perfusion solution containing 5 mm KCl, and changes in [Ca2+]i were recorded in response to 20 mm KCl, 100 nm IBTX and anoxia (total n = 34 cells from 4 coverslips). Numbers without units are in mm. B–D, same as in A except that the bath perfusion solution was switched to that containing 12 mm (B; n = 28 cells), 15 mm (C; n = 156) or 20 mm (D; n = 69) KCl, and change in [Ca2+]i was recorded in response to 100 nm IBTX and anoxia. Four to 15 coverslips of cells were used for each level of KClo. E, pre‐IBTX and peak‐IBTX [Ca2+]i responses were determined and plotted at different [KCl]o. Each circle is the averaged [Ca2+]i value from one coverslip containing multiple cells. Vertical lines beside the circles represent mean ± SE for each level of [KCl]o. *Significantly different from the respective control value (P < 0.05). P = 0.16 for 12 mm, P < 0.001 for 15 mm and P < 0.001 for 20 mm KCl.
We then studied would happen to BK if glomus cells were depolarized moderately. Based on our whole‐cell current recordings, we predicted BK would become active as cells begin to depolarize, and IBTX would cause additional depolarization and elevation of [Ca2+]i. To test this prediction, glomus cells were perfused with a solution containing 12, 15 or 20 mm KCl. If one assumes a value of 0.05 for P Na/P K, 140 mm for [K+]i and [Na+]o, and 15 mm for [Na+]i, a resting E m of −64 mV is calculated based on the Goldman‐Hodgkin‐Katz (GHK) constant field equation when [K+]o is 5 mm. At [K+]o of 12, 15, 20 and 30 mm, the cell E m is calculated to be −53, −49, −44 and −36 mV, producing cell depolarization of 11, 15, 20 and 28 mV, respectively. Anoxia elevated [Ca2+]i to a level similar to that elicited by 30 mm KCl, indicating that anoxia depolarized the cells by ∼28 mV. The levels of depolarization would still be the same even if [K+]i was lower or higher, because of the strict dependence of changes in E m on [K+]o.
Application of 12 mm KClo produced a small transient increase in [Ca2+]i such that the steady state basal [Ca2+]i remained only slightly elevated or unchanged (Fig. 2 B and E). Higher [KCl]o (15 and 20 mm) induced a larger transient rise in [Ca2+]i that decreased to a steady state level below the peak level but above the basal level (Fig. 2 C and D). IBTX did not produce a significant increase in peak [Ca2+]i at 12 mm KClo, but elicited significant elevations of peak [Ca2+]i at 15 and 20 mm KClo, as shown in the scatter plots of pre‐IBTX and peak‐IBTX levels in Fig. 2 E, where each data point represents one coverslip of cells. The IBTX‐induced increase in [Ca2+]i was generally short‐lasting, suggesting that the rise in [Ca2+]i was followed by Ca2+ extrusion from the cell and/or uptake by intracellular stores. All cells whose [Ca2+]i was increased by 20 mm KClo responded strongly to anoxia, indicating that they were O2‐sensitive glomus cells. These results show that, when cells are depolarized by 15 mV or more, BK becomes sufficiently active such that its inhibition by IBTX is able to elicit a transient increase in [Ca2+]i even at elevated [KCl]o.
To assess the role of BK and Kv together, we measured [Ca2+]i in response to TEA (3 mm)/4‐AP (3 mm) that block not only BK but also all subtypes of Kv. At 5 mm KClo, TEA/4‐AP did not affect the basal [Ca2+]i, indicating that BK/Kv were mostly closed at rest (Fig. 3 A). In cells moderately depolarized with 12, 15 and 20 mm KClo, TEA/4‐AP elicited transient increases in peak [Ca2+]i in a manner that was qualitatively similar to those produced by IBTX alone (Fig. 3 B and D). Figure 3 E plots [Ca2+]i determined before addition of TEA/4‐AP (pre TEA/4‐AP) and [Ca2+]i at the peak observed following application of TEA/4‐AP (peak TEA/4‐AP). The magnitude of [Ca2+]i rise produced by TEA/4‐AP was significantly greater than that produced by IBTX for each level of KClo (Fig. 3 F), indicating that inhibition of Kv contributed strongly to the rise in [Ca2+]i.
Figure 3. Effect of TEA/4‐AP on glomus cell [Ca2+]i at different [KCl]o .
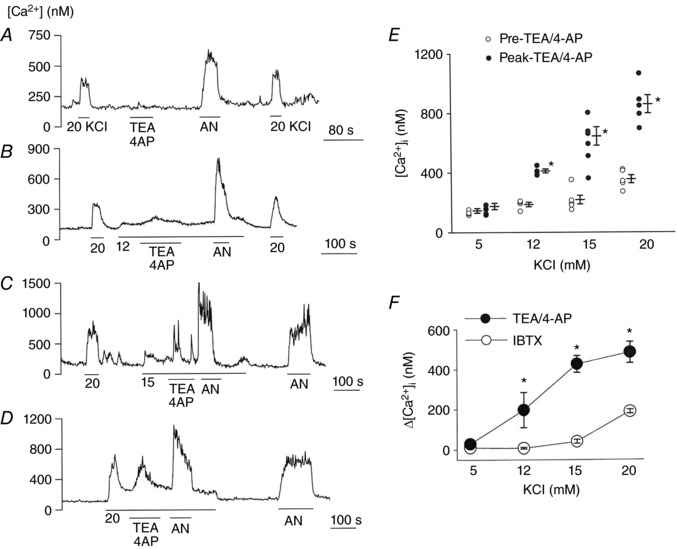
A, glomus cells perfused with normoxic bath perfusion solution containing 5 mm KCl, and changes in [Ca2+]i were recorded in response to 20 mm KCl, TEA/4‐AP (3 mm each) and anoxia (four coverslips of cells: total of 44 cells). B–D, same as in A except that the bath perfusion solution was switched to that containing 12 mm (B; n = 26 cells), 15 mm (C; n = 37 cells) or 20 mm (D; n = 64 cells) KClo, and change in [Ca2+]i was recorded in response to TEA/4‐AP and anoxia (4–6 coverslips of cells were used for each level of KClo.). E, pre‐TEA/4‐AP and peak‐TEA/4‐AP [Ca2+]i responses were determined and plotted at different [KCl]o. Each circle is the average [Ca2+]i value from one coverslip of cells. Vertical lines are mean ± SEM of 4–6 coverslips of cells. *Significantly different from the respective control (P < 0.01 for 12, 15 and 20 mm KCl). F, changes in [Ca2+]i (Δ[Ca2+]i) produced by IBTX and TEA/4‐AP were determined and plotted for each level of [KCl]o. Each point is the mean ± SEM. *Significantly different from the respective control value (P < 0.01).
These effects of IBTX and TEA/4‐AP on [Ca2+]i at elevated levels of KClo may be different from those observed in similarly depolarized cells bathed in 5 mm KClo. Nevertheless, comparison of the effects produced by IBTX and TEA/4‐AP shows that if the resting E m of glomus cells is shifted toward more positive levels for some reason, BK and Kv can become basally open. The K+ channel inhibitors are then able to cause further depolarization and elevation of [Ca2+]i in these cells, and this may possibly explain why IBTX and TEA were able to elevate [Ca2+]i in resting glomus cells in some studies.
TEA/4‐AP potentiate hypoxia‐induced rise in [Ca2+]i
The above results show that depolarization of glomus cells with high [KCl]o activates BK/Kv, as expected of voltage‐dependent K+ channels. Thus, we predicted that depolarization produced by hypoxia also activates BK/Kv. To determine whether and how much BK/Kv affect the increase in [Ca2+]i elicited by hypoxia, we tested the effect of TEA/4‐AP on [Ca2+]i rise. As before, anoxia elicited a strong elevation of [Ca2+]i at 5 mm [KCl]o. The rise in [Ca2+]i elicited by anoxia was significantly increased in the presence of TEA/4‐AP (Fig. 4 A and E), regardless of whether the inhibitors were added before (Fig. 4 A: left) or after (Fig. 4 A: right) the control anoxia test. These results showed that activation of BK/Kv reduced the anoxic [Ca2+]i response. We then repeated the same experiment using mild, moderate and severe levels of hypoxia. Under our experimental conditions, the basal [Ca2+]i in normoxia was 141 ± 7 nm (n = 24 coverslips of cells), and [Ca2+]i rose to 739 ± 41 nm (anoxia; n = 11), 285 ± 34 nm (severe hypoxia; n = 16), 178 ± 6 nm (moderate hypoxia; n = 16) and 146 ± 10 nm (mild hypoxia; n = 5) in response to different levels of hypoxia. Under identical perfusion conditions, 20 mm KClo increased [Ca2+]i to 418 ± 12 nm (n = 24) in normoxia. Thus, the four levels of hypoxia we used in these experiments covered a wide range of [Ca2+]i rise.
Figure 4. TEA/4‐AP enhance hypoxia‐induced elevation of [Ca2+]i .
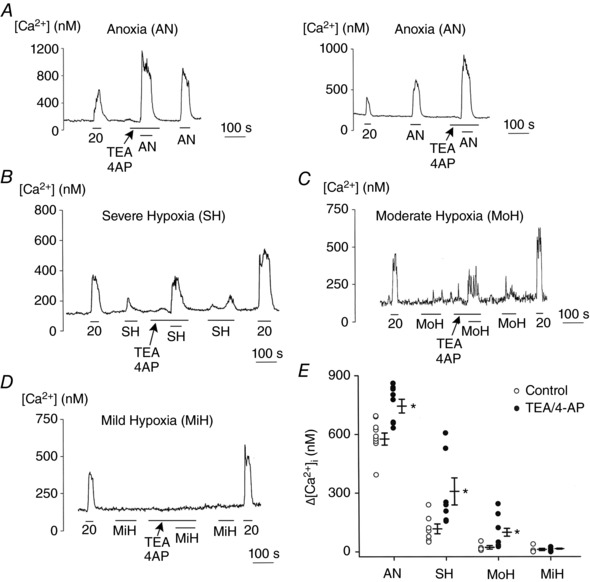
A, (Left): glomus cells are perfused with normoxic bath perfusion solution containing 5 mm KCl, and change in [Ca2+]i recorded in response to anoxia with and without TEA/4‐AP (3 mm each). (Right): the order of application of anoxia and TEA/4‐AP is switched. Together, nine coverslips of cells are used (total of 82 cells). B–D, same experiments as in A, except that changes in [Ca2+]i were recorded in response to severe hypoxia (SH: B), moderate hypoxia (MoH: C) and mild hypoxia (MiH: D). Changes in [Ca2+]i in response to hypoxia were recorded with and without TEA/4‐AP. Each of the 5–7 coverslips contains multiple cells; a total of 53 cells (SH), 136 cells (MoH) and 144 cells (MiH) are used. E, scatter plot showing change in [Ca2+]i (Δ[Ca2+]i) produced by anoxia and hypoxia. Vertical lines are mean ± SEM of 5–9 coverslips of cells, where each circle is the average value from a coverslip containing multiple glomus cells. *Significantly different from the respective control value (P < 0.001 for AN, P = 0.007 for SH, P = 0.023 for MoH and P = 0.42 for MiH).
The increase in [Ca2+]i (Δ[Ca2+]i) elicited by severe hypoxia was strongly enhanced (∼3‐fold) by TEA/4‐AP (Fig. 4 B and E). Similarly, the increase in [Ca2+]i produced by moderate hypoxia was also markedly enhanced (∼3.5‐fold) by TEA/4‐AP, although the absolute levels of [Ca2+]i were much lower than those observed with severe hypoxia (Fig. 4 C and E). A mild level of hypoxia obtained by bubbling the perfusion solution with 2% O2 did not produce a significant increase in basal [Ca2+]i with or without TEA/4‐AP (Fig. 4 D and E). This indicated that a mild level of hypoxia did not depolarize glomus cells enough to activate BK/Kv under our experimental conditions. In some of these experiments, the basal [Ca2+]i rose very slowly over time, and the responses to 20 mm KCl tended to be slightly higher at the end than those at the beginning of cell perfusion. Despite this caveat, the recovery of the hypoxic responses after washout of TEA/4‐AP assured us that the potentiating effects of K+ channel inhibitors were real. Together, our results show that as glomus cells depolarize in response to moderate/severe hypoxia and anoxia, BK/Kv become active and help limit the degree of depolarization and the rise in [Ca2+]i. The strong limiting effect of BK/Kv on [Ca2+]i is consistent with a net activation of BK/Kv by hypoxia, but is not consistent with the idea that hypoxia inhibits BK/Kv to elicit or sustain the hypoxic response.
IBTX has little or no effect on hypoxia‐induced rise in [Ca2+]i
To determine the contribution of BK alone to the suppression of hypoxia‐induced rise in [Ca2+]i, we studied the effect of IBTX on the rise in [Ca2+]i elicited by anoxia and three different levels of hypoxia at 5 mm [KCl]o. Anoxia elicited a strong increase in [Ca2+]i, and this increase was not affected by IBTX (Fig. 5 A and D). IBTX also failed to produce a significant effect on the mean [Ca2+]i rise elicited by severe hypoxia (Fig. 5 B). These results suggest that BK activated by severe hypoxia and anoxia is not sufficiently high in magnitude to suppress the [Ca2+]i rise. Further supporting the general lack of effect of BK on the hypoxic response was the finding that IBTX produced no significant enhancement of [Ca2+]i rise by a moderate level of hypoxia (Fig. 5 C and D), compared to a 3.5‐fold increase by TEA/4‐AP. These results together with those observed with TEA/4‐AP show that BK alone has a weak or no effect on hypoxia‐induced rise in [Ca2+]i, whereas BK/Kv together produce a strong limiting effect. This is probably because the magnitude of BK/Kv together is much larger than that of BK alone.
Figure 5. Lack of effect of IBTX on anoxia‐ and hypoxia‐induced elevation of [Ca2+]i .
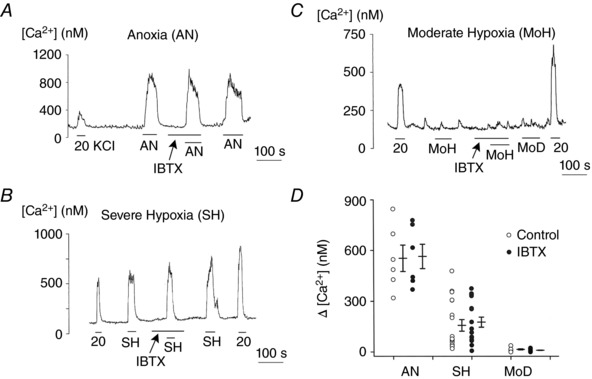
A, glomus cells were perfused with normoxic bath perfusion solution containing 5 mm KCl, and change in [Ca2+]i was recorded in response to anoxia with and without 100 nm IBTX. Five coverslips of cells were used (total of 106 cells). B and C, same experiments as in A, except that changes in [Ca2+]i were recorded in response to severe hypoxia (B; 18 coverslips: n = 158 cells) and moderate hypoxia (C; 9 coverslips: n = 106 cells). Changes in [Ca2+]i in response to hypoxia were recorded with and without IBTX. D, scatter plot showing change in [Ca2+]i (Δ[Ca2+]i) produced by anoxia and hypoxia. Vertical lines are mean ± SEM of 5–18 coverslips of cells, where each circle is the mean value from a coverslip containing multiple glomus cells. No significance was present between control and IBTX groups (P = 0.92 for AN, P = 0.85 for SH and P = 0.81 for MoH).
Guangxitoxin enhances hypoxia‐induced rise in [Ca2+]i
Studies above with IBTX and TEA/4‐AP show that activation of both BK and Kv are necessary to limit the rise in [Ca2+]i in response to hypoxia. As BK alone has a weak or no effect on [Ca2+]i rise, could Kv alone also have a weak effect? To address this, we tested the effect of GxTX (100 nm), an inhibitor of Kv2, on the whole‐cell current. GxTX reduced the whole‐cell current by 52% at +0 mV, indicating that Kv2 is a major Kv whose contribution to the outward current is more than twice that by BK (Fig. 6 A). As predicted, TEA produced an additional inhibition of the whole‐cell current, indicating the presence of other types of Kv in rat glomus cells. We tested the effect of GxTX on the elevation of [Ca2+]i produced by severe and moderate hypoxia. GxTX significantly enhanced the increase in [Ca2+]i produced by severe hypoxia in all nine coverslips of cells tested (Fig. 6 B). GxTX also enhanced the [Ca2+]i rise induced by moderate hypoxia (Fig. 6 C). These results are summarized in Fig. 6 D and show that Kv2 alone is able to significantly limit the hypoxia‐induced rise in [Ca2+]i. This is consistent with the idea that the magnitude of the outward K+ current activated by moderate or severe hypoxia needs to be larger than that provided by BK alone to be effective in limiting the rise in [Ca2+]i.
Figure 6. GxTX enhances hypoxia‐induced elevation of [Ca2+]i .
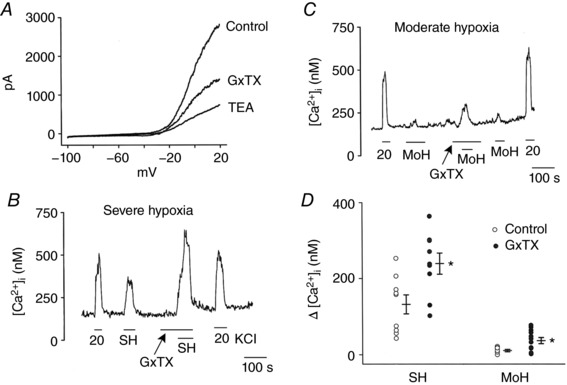
A, whole‐cell currents recorded from isolated glomus cells using a ramp protocol. Cells were held at −100 mV and voltage ramps applied (500 ms duration) to +20 mV and then back to −100 mV. Cells were perfused with solution containing 100 nm GxTX first until steady state current was observed, and then with 3 mm TEA, and whole‐cell currents recorded again. Currents were averaged from sic cells with relatively similar control current sizes. B and C, glomus cells were perfused with normoxic bath perfusion solution containing 5 mm KCl, and changes in [Ca2+]i were recorded in response to severe hypoxia (B; 9 coverslips: n = 121 cells) or moderate hypoxia (C; 12 coverslips: n = 235 cells) with and without GxTX. D, summary scatter plot showing the enhancing effect of GxTX on [Ca2+]i rise induced by severe and moderate hypoxia. Each circle is the mean value from a coverslip. Vertical lines are mean ± SEM obtained from 9–12 coverslips of cells. *Significantly different from the respective control value (P < 0.001 for SH and P = 0.008 for MoH).
Effects of TEA/4‐AP and IBTX on hypoxia‐induced rise in [Ca2+]i in partially depolarized cells
What would be the role of BK/Kv in the hypoxic response if these K+ channels were basally active? To address this, we induced depolarization of glomus cells by elevating [KCl]o and then studied the effect of TEA/4‐AP on [Ca2+]i response to hypoxia. At 12 mm KClo, which depolarizes the cells by ∼11 mV, TEA/4‐AP significantly enhanced the elevation of [Ca2+]i produced by anoxia as well as by severe hypoxia (Fig. 7 A–C). In the two tracings shown in Fig. 7 A, similar effects of TEA/4‐AP were obtained whether TEA/4‐AP was applied before or after the control anoxia test. The enhancing effect of TEA/4‐AP at 12 mm KClo was greater with severe hypoxia than anoxia, similar to that observed at 5 mm KClo (see Fig. 4). These results show that in glomus cells already mildly depolarized by ∼11 mV, hypoxia also causes activation of BK/Kv to limit the [Ca2+]i response.
Figure 7. Effects of TEA/4‐AP and IBTX on anoxia‐ and hypoxia‐induced elevation of [Ca2+]i at 12 mm KClo .
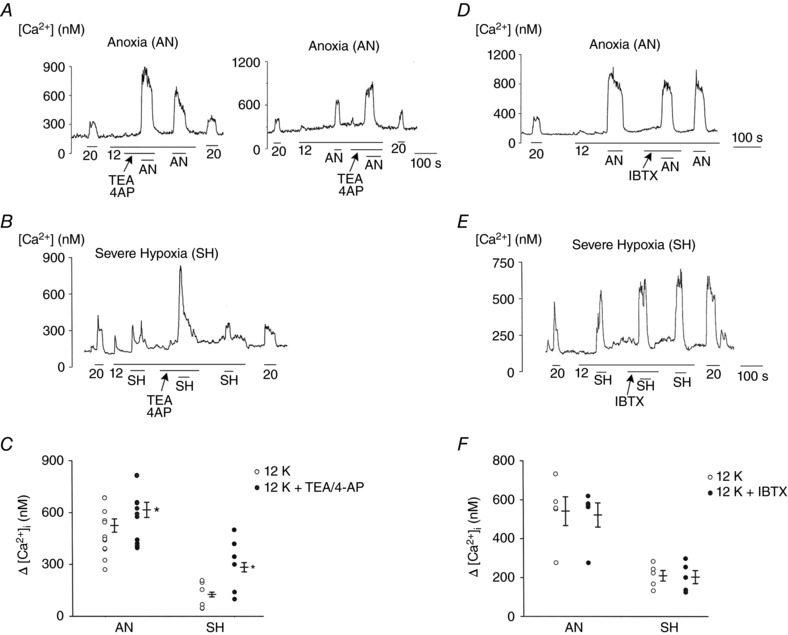
A, glomus cells are perfused with normoxic bath perfusion solution containing 12 mm KCl, and changes in [Ca2+]i were recorded in response to anoxia with and without TEA/4‐AP (3 mm each). Two tracings are shown where TEA/4‐AP was added before or after the control anoxia test. TEA/4‐AP enhanced anoxia (AN)‐induced increase in [Ca2+]i (12 coverslips: n = 136 cells). B, changes in [Ca2+]i in response to severe hypoxia recorded with and without TEA/4‐AP at 12 mm KClo (6 coverslips: n = 60 cells). C, scatter plot showing the enhancing effect of TEA/4‐AP on [Ca2+]i rise induced by anoxia and severe hypoxia. Each circle is the mean value from a coverslip. Verticals bars are mean ± SEM obtained from 6–12 coverslips of cells. *Significantly different from the respective control value (P = 0.002 for AN and P = 0.005 for SH). D, changes in [Ca2+]i in response to anoxia recorded with and without IBTX at 12 mm KClo (5 coverslips: n = 45 cells). E, changes in [Ca2+]i in response to severe hypoxia recorded with and without IBTX at 12 mm KClo (5 coverslips: n = 32 cells). F, scatter plot showing no significant effect of IBTX on averaged increase in [Ca2+]i (Δ[Ca2+]i) produced by anoxia and hypoxia (P > 0.05). Each circle is the mean value from a coverslip. Verticals bars are mean ± SEM obtained from five coverslips of cells. No significance difference was found between control and IBTX groups (P = 0.58 for AN and P = 0.74 for SH).
IBTX, however, failed to produce a significant change in [Ca2+]i produced by anoxia and severe hypoxia at 12 mm KClo (Fig. 7 D–F). Thus, in glomus cells depolarized by ∼11 mV, BK alone was not sufficient to suppress the rise in [Ca2+]i elicited by hypoxia. These results obtained with IBTX and TEA/4‐AP are similar to our findings observed at 5 mm KClo (see Fig. 5), and indicate that the size of the K+ current activated by hypoxia needs to be larger than that provided by BK alone to be effective in reducing the rise in [Ca2+]i regardless of whether the resting E m is ∼ −60 or ∼ −49 mV.
Hypoxia causes activation of BK in cell‐attached patches
Our findings above indicate that hypoxia causes activation of BK/Kv, which helps limit the rise in [Ca2+]i. To show activation of BK/Kv current, we studied the effect of anoxia on K+ channels in cell‐attached patches perfused with physiological solution containing 5 mm KCl in the pipette and bath solutions. When the pipette potential was set at 0 mV, however, the baseline current noise was relatively high and we were unable to identify clear channel openings in normoxia and anoxia. This is probably because of small single channel conductance levels of K+ channels under the ionic conditions used (Fig. 8 A). Thus, whether anoxia activates K+ channels could not be determined under physiological ionic conditions.
Figure 8. Activation of BK by hypoxia in cell‐attached patches at different [KCl]o and pipette potential.
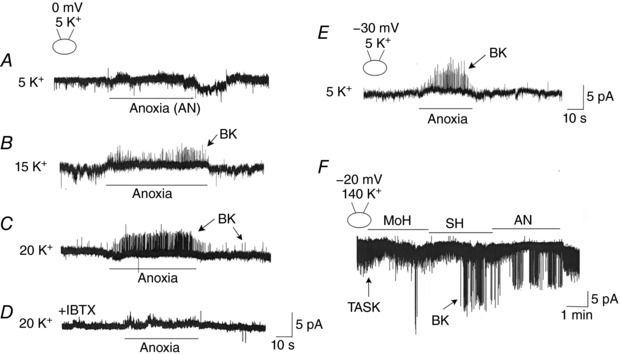
A, cell‐attached patch is formed with pipette and bath solutions containing 5 mm KCl and pipette potential held at 0 mV. The perfusion solution was switched from normoxic to anoxic solution. No channel activity was identified in 20 patches. B and C, cell‐attached patches were perfused with high [KCl]o (15 or 20 mm) to depolarize the cells. Anoxia reversibly activated BK in 5 of 14 patches (15 mm K+) and in 6 of 12 patches (20 mm K+). D, same experiment as C, except that IBTX (100 nm) was added to the pipette and bath solutions. No BK was detected in 12 patches. E, same as in A except that the pipette potential was set at −30 mV to depolarize the patch membrane. Anoxia reversibly activated BK in 7 of 8 patches. F, cell‐attached patch is formed and pipette potential set at −20 or −30 mV. Bath and pipette solutions contained 140 mm KCl. High basal TASK activity is present in cell‐attached patches. Hypoxia and anoxia activated BK and inhibited TASK reversibly in 4 of 7 cells. In the three patches, no BK was observed despite the inhibition of TASK by hypoxia. The reduced amplitude of BK caused by hypoxia is due to depolarization of the cell.
To be able to test the effect of hypoxia on BK, we needed a condition that allowed a basal level of BK opening to be detected. This was accomplished by perfusing cells with elevated [KCl]o to produce mild levels of depolarization. When glomus cells were perfused with solution containing 15 mm KCl that depolarizes the cells by ∼15 mV (based on our estimation described above), we were still unable to identify basal BK openings. However, opening of BK could be observed in anoxia, probably due to additional depolarization elicited by anoxia (see arrow; Fig. 8 B). When cells were perfused in solution containing 20 mm KCl that depolarizes glomus cells by ∼20 mV, a low level of basal BK activity was detected in some patches in normoxia. In such patches, anoxia quickly and reversibly activated BK (see arrows; Fig. 8 C). The anoxia‐activated channel was not detected in all 12 patches when IBTX was added to the pipette and bath solutions (Fig. 8 D), indicating that the observed channels were BK.
Anoxia also reversibly activated BK when the bath [KCl]o was 5 mm and the pipette potential was set at −30 mV to depolarize the membrane in cell‐attached patches (Fig. 8 E). In other cell‐attached patches in which the pipette potential was set at −20 or −30 mV, and the pipette solution contained 140 mm KCl and the bath solution contained 5 mm K+, we observed robust TASK openings in the inward current direction (Fig. 8 F). Hypoxia caused inhibition of TASK and activation of BK in a reversible and [O2]‐dependent manner in 4 of 7 patches tested. In Ca2+‐free bath and pipette solutions, the high‐conductance BK channels were not observed in all 9 patches. The decrease in BK single channel amplitude caused by hypoxia and anoxia is expected, because of depolarization of the cell and accompanying reduction of the transmembrane potential gradient. Our results show that in glomus cells that are mildly depolarized with 15–20 mm KClo to elicit basal BK openings, anoxia activates BK presumably via additional depolarization. We were unable to identify Kv channels under our ionic conditions, probably because of their very small single channel conductance levels (<0.5 pS). The single channel openings of such K+ channels would be hidden within the noise of the baseline membrane current.
Hypoxia does not directly affect BK
Our results above show that hypoxia causes activation of BK in glomus cells that are partially depolarized by elevated [KCl]o. Although activation of BK was observed, it is quite possible that hypoxia caused a partial direct inhibition of BK, which was not sufficient to override the stimulatory effect of depolarization. A possible direct effect of hypoxia on BK was studied using inside‐out patches at symmetrical 140 mm KCl. In the presence of ∼1 μm Ca2+ in the bath solution, the patch membrane was depolarized to elicit a mild level of BK activity (Fig. 9 A). When a relatively steady state level of BK activity was observed, anoxic solution was applied for ∼1 min. No significant effect of anoxia on BK was present in these inside‐out patches, as illustrated in the scatter plot in Fig. 9 A. In the presence of 0.3 mm Ca2+ in the bath solution, BK was highly active in inside‐out patches as predicted. In these patches, anoxia also showed no significant effect on BK, regardless of the initial level of channel activity (Fig. 9 B).
Figure 9. Hypoxia does not directly affect BK.
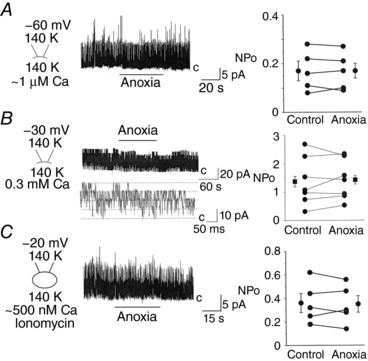
A, inside‐out patches were perfused with normoxic solution containing ∼1 μm Ca2+ to activate outward BK current. The pipette and bath solutions contained 140 mm KCl and the pipette potential was held at −60 mV. BK activities in normoxia and anoxia in 5 patches from 3 cell preparations shown along with the mean ± SEM. The closed state is indicated by ‘c’. Anoxia produced no significant effect on BK activity (P = 0.70). B, inside‐out patches perfused with normoxic solution containing 0.3 mm Ca2+ to activate outward BK current. The pipette and bath solutions contained 140 mm KCl and the pipette potential was held at −30 mV. BK activities in normoxia and anoxia in 7 patches are shown along with the mean ± SEM. Anoxia produced no significant effect on BK activity (P = 0.58). C, cell‐attached patches perfused with normoxic solution containing ∼500 nm Ca2+ and BK is activated using ionomycin (2 μm). The pipette and bath solutions contained 140 mm KCl and the pipette potential was held at −20 mV (bottom three data points) or −30 mV. BK activities in normoxia and anoxia in 5 patches are shown along with the mean ± SEM. Anoxia produced no significant effect on BK activity (P = 0.46).
We also tested the effect of anoxia on BK in cell‐attached patches perfused with solution containing 140 mm K+ and ∼500 nm free Ca2+. Under this condition, cell E m is held at ∼0 mV and therefore no additional depolarization can occur in response to hypoxia. Ionomycin (2 μm) was added to the perfusion solution to elevate [Ca2+]i. BK openings were observed when the patch membrane was mildly depolarized, but anoxia again produced no significant effect on BK (Fig. 9 C). These results obtained from inside‐out and cell‐attached patches show that hypoxia does not directly affect BK in our glomus cell preparations at ∼35°C. Thus, the stimulatory effect of hypoxia on BK in cell‐attached patches perfused with solution containing 5 mm KClo, as shown in Fig. 8, is probably entirely due to cell depolarization and elevation of [Ca2+]i. We were unable to study the effect of hypoxia on Kv in cell‐attached patches, as they could not be detected clearly under these experimental conditions because of opening of other higher conductance channels such as TASK and BK.
Discussion
This study was instigated by the contradictory role of BK/Kv in glomus cell function described in the literature. Our goal was to re‐examine the role of BK and Kv in basal glomus cell excitability and in the generation of the hypoxic response using a more comprehensive approach. To achieve this goal, we used four different levels of hypoxia in glomus cells maintained at several different levels of resting cell E m, and studied direct and indirect effects of anoxia on BK in inside and cell‐attached patches. For each experiment, we used many coverslips of cells prepared from multiple cell isolations to obtain a large sample size. Our study in isolated rat glomus cells showed that BK/Kv were closed at rest, indicating that BK/Kv did not participate in the initiation of the hypoxic response (i.e. the rise in [Ca2+]i). BK/Kv became active when cells depolarized in response to hypoxia, and strongly limited the rise in [Ca2+]i. Our findings support a scheme in which hypoxia‐induced depolarization activates BK/Kv to provide a hyperpolarizing force that limits the magnitude of the hypoxic response, but do not support the scheme in which hypoxia inhibits BK/Kv to elicit and sustain the hypoxic response.
Function of BK and Kv in isolated rat glomus cells in normoxia
The lack of effect of IBTX and TEA/4‐AP on basal [Ca2+]i at 5 mm KClo shows that BK and Kv are not functionally active for modulating the resting cell E m and Cav in isolated rat glomus cells. Our findings are in agreement with earlier studies in which TEA and IBTX produced no effect on [Ca2+]i and the secretory response in isolated rat glomus cells (Buckler, 1997; Gomez‐Nino et al. 2009; Donnelly et al. 2011), but not with studies in which K+ channel inhibitors depolarized and elicited a secretory response (Wyatt & Peers, 1995; Pardal et al. 2000). In our own experiments (Fig. 1 B), IBTX and TEA/4‐AP produced a very small inhibition of the whole‐cell current in the flat part of the current response, suggesting that very low levels of BK and Kv may be present at these negative potentials. However, inhibition of BK/Kv was not enough to affect Cav function and cause an increase in [Ca2+]i.
The transient rise in [Ca2+]i produced by IBTX and TEA/4‐AP in cells perfused with >12 mm KClo indicated that BK and Kv were basally open when the resting cell E m was shifted to positive levels. This makes sense, as depolarization is predicted to open BK and Kv. This could explain the basal opening of BK observed in perforated patches and CB slices where IBTX and TEA caused cell depolarization and secretory response, respectively (Wyatt & Peers, 1995; Pardal et al. 2000). Within intact CB or CB slices, glomus cells may undergo spontaneous depolarization to elicit a spontaneous transient rise in [Ca2+]i that is often observed even in normoxia. In such an event, BK/Kv may open whenever depolarization occurs, and this could also explain the [Ca2+]i response of glomus cells to BK/Kv inhibitors. Such mechanisms need to be explored further to help understand the behaviour of these ion channels in vivo.
BK/Kv limit the elevation of [Ca2+]i during hypoxia
The notion that hypoxia inhibits the K+ conductance to elicit the hypoxic response is now widely accepted. As discussed above, the inhibition of BK/Kv has been commonly described as a mechanism underlying the hypoxic response. In rat/mouse glomus cells, the reduction of the whole‐cell current by hypoxia is rather small at ∼20% (Hatton et al. 1997; Lopez‐Lopez et al. 1997; Perez‐Garcia et al. 2004; Peers & Wyatt, 2007). A significant portion of the inhibition is probably due to inhibition of TASK, suggesting that the true inhibition of BK/Kv by hypoxia is probably much less than 20%. These considerations further suggest that inhibition of BK/Kv is unlikely to be the cause of the hypoxic response in rat and mouse glomus cells. Although we have not directly tested the effect of hypoxia on Kv, the strong potentiation of the hypoxic [Ca2+]i response by TEA/4‐AP shows that activation of BK/Kv, and not inhibition, determines the magnitude of the observed hypoxic response.
Our findings support the earlier observations that TEA/4‐AP increase the hypoxia‐induced elevation of [Ca2+]i (Buckler, 1997; Wasicko et al. 2006). The enhanced secretory response to hypoxia by TEA observed in CB slices also shows that hypoxia caused activation of BK/Kv to limit and thus optimize the secretory response (Pardal et al. 2000). However, a lack of effect of TEA (without 4‐AP) has also been reported on hypoxia‐induced elevation of [Ca2+]i and catecholamine secretion (Gomez‐Nino et al. 2009). The interpretation of these findings was that inhibition of BK/Kv by hypoxia opposed the depolarization‐induced activation of BK/Kv, and TEA was therefore unable to potentiate the hypoxic response. This explanation is based on earlier reports that hypoxia causes inhibition of BK/Kv (Peers, 1990; Wyatt & Peers, 1995; Riesco‐Fagundo et al. 2001). The reason for different reported effects of K+ channel inhibitors on the hypoxic response is difficult to explain. It seems unlikely that the use of only TEA, rather than TEA/4‐AP, was the cause of different findings, as the 4‐AP‐sensitive Kv is small in rat glomus cells. It is possible that the level of hypoxia was too severe (∼10 mmHg O2) to observe a significant effect of TEA in a particular glomus cell preparation used (Gomez‐Nino et al. 2009). In our study, the enhancing effect of TEA/4‐AP on [Ca2+]i was relatively mild with anoxia, but strong with severe and moderate levels of hypoxia. Our results also indicate that the level of K+ current that needs to be activated to suppress the [Ca2+]i rise depends on the severity of hypoxia, with progressively larger current activation required as hypoxia becomes more severe.
Kv2 as the primary Kv subtype that limits the hypoxic response
In rat glomus cells, the size of the GxTX‐sensitive K+ current was about twice that of the IBTX‐sensitive current. Our finding that GxTX, but not IBTX, can significantly augment the hypoxia‐induced rise in [Ca2+]i is in keeping with the idea that the size of the outward K+ current activated needs to be larger than that of BK to produce a significant suppressive effect on [Ca2+]i rise. As Kv2 is smaller than Kv (Fig. 6 A), it also makes sense that the enhancing effect of GxTX on [Ca2+]i rise produced by hypoxia is less than that produced by TEA/4‐AP. For severe hypoxia, for example, the increase in [Ca2+]i rise produced by TEA/4‐AP was ∼3‐fold, compared to ∼2‐fold by GxTX. Therefore, the relatively small size of BK compared to Kv and Kv2 would explain why blocking BK alone has little or no effect on the hypoxia‐induced increase in [Ca2+]i. Although BK alone has a minor effect on the [Ca2+]i rise produced by hypoxia, it contributes significantly to limiting the depolarization and [Ca2+]i rise when BK works together with Kv to produce a larger outward K+ current.
Hypoxia activates BK via cell depolarization
Several studies have reported that hypoxia inhibits BK (Wyatt & Peers, 1995; Hatton et al. 1997; Riesco‐Fagundo et al. 2001; Williams et al. 2004). However, a lack of effect of hypoxia on BK was observed in rabbit glomus cells (Ganfornina & Lopez‐Barneo, 1992). Our own studies showed that anoxia did not significantly affect BK in inside‐out patches, and actually activated BK in cell‐attached patches under physiological conditions (5 mm KClo). Furthermore, hypoxia had no effect on BK in cell‐attached patches of fully depolarized cells with a resting E m of ∼0 mV, showing that activation of BK observed under normal conditions (5 mm KClo) was due to cell depolarization. We are unable to offer a satisfactory explanation for the conflicting findings on the effect of hypoxia on BK. The Wistar rat strain was used in early studies (Hatton et al. 1997; Riesco‐Fagundo et al. 2001), and we have used the Sprague‐Dawley strain. It is possible that BK expressed in glomus cells from these two rat strains are modulated differently by hypoxia, possibly due to expression of different BK isoforms or splice variants. Our current recording was performed at ∼35°C, whereas those in other studies using rat glomus cells were done at room temperature (20–24°C). It is possible that hypoxia inhibits BK at 24°C but not at 35°C in rat glomus cells, although this seems somewhat unlikely at least in inside‐out patches.
Depolarization, spontaneous action potentials and BK/Kv
We have not recorded changes in cell E m during hypoxia, and therefore we do not know whether the depolarization produced by hypoxia elicited action potentials in our cell preparations. Earlier studies in rat glomus cells have shown that severe hypoxia (<14 mmHg) and anoxia caused cell depolarization (∼10–20 mV) and elicited action potentials that were 30–40 mV in amplitude from the depolarized level (Buckler & Vaughan‐Jones, 1994; Buckler, 1997, 1999). Spontaneous action potentials have also been observed in rabbit glomus cells (Lopez‐Lopez et al. 1989; Urena et al. 1989). The action potentials that are elicited during hypoxia should strongly activate BK/Kv, as the cell E m reaches levels more positive than ∼ −20 mV. The magnitude of BK/Kv activation is therefore expected to be much higher than that produced by sustained depolarization of 10–20 mV. The strong hyperpolarizing action of BK/Kv should help rapidly repolarize the cell during each action potential, reduce the level of depolarization and the action potential frequency, and limit the rise in [Ca2+]i during hypoxia.
As BK and Kv are also highly expressed in rabbit glomus cells, we speculate that these K+ channels serve a role similar to that in rat glomus cells. The resting E m of rabbit glomus cells was found to lie between −60 and −50 mV (Rocher et al. 2005), indicating that Kv is probably mostly inactive at rest. In one study, however, blocking the inactivating Kv with 4‐AP was found to cause depolarization in rabbit glomus cells, suggesting that Kv4 was basally active at rest (Perez‐Garcia et al. 2000). Whether such depolarization was associated with a rise in [Ca2+]i or an enhanced secretory response was not determined. In rabbit glomus cells, the inactivating Kv4 was found to be the primary O2‐sensitive K+ channel (Sanchez et al. 2002; Lopez‐Lopez & Perez‐Garcia, 2007). Thus, hypoxia‐induced inhibition of Kv4 and depolarization‐induced activation of BK/Kv may determine the magnitude of the repolarizing current in rabbit glomus cells during hypoxia. A comprehensive study that correlates changes in cell E m and [Ca2+]i in response to a range of hypoxia and specific K+ channel blockers is necessary to further clarify the role of Kv4 and BK/Kv in hypoxia sensing in rabbit glomus cells. In mouse glomus cells, we predict the role of BK/Kv to be similar to that in the rat, because BK, Kv and TASK functions are similar in the two species (Perez‐Garcia et al. 2004; Ortega‐Saenz et al. 2010; Turner & Buckler, 2013; Kang et al. 2014). However, a study of the effects of IBTX and TEA/4‐AP on [Ca2+]i changes in normoxia and hypoxia is also needed to confirm such a role for BK/Kv in mouse glomus cells. In all experiments using isolated cells, we need to keep in mind that the hypoxic sensitivity of isolated glomus cells and the frequency of spontaneous depolarization and action potentials elicited by hypoxia may be different in cells in vivo.
Important role of BK/Kv in the regulation of the hypoxic response
Based on our findings in isolated rat glomus cells, we propose a scheme in which the hypoxic response is reduced by the hyperpolarizing action of BK/Kv that are activated during depolarization produced by hypoxia. This would explain the enhancing effect of TEA on hypoxia‐induced secretory activity observed in CB slices (Pardal et al. 2000). Our study shows that Kv is the major outward K+ current that suppresses the hypoxia‐induced elevation of [Ca2+]i. Although BK alone is not strong enough to affect the hypoxic response, it contributes significantly to the limiting effect when Kv is also activated. Our recent study showed that hypoxia‐induced rise in [Ca2+]i activates a Na+‐permeable, non‐selective cation channel (NSC), which is predicted to enhance depolarization and rise in [Ca2+]i (Kang et al. 2014). Therefore, activation of NSC and BK/Kv at different levels of cell E m may determine the magnitude of hypoxia‐induced rise in [Ca2+]i, as illustrated in Fig. 10 A. These mechanisms are probably important for producing the optimal elevation of [Ca2+]i and secretory response for a given level of hypoxia. Further studies are needed to determine the contributions of NSC and BK/Kv to the hypoxic response at different levels of hypoxia.
Figure 10. Scheme illustrating the mechanisms that regulate the rise in [Ca2+]i produced by hypoxia, and the contribution by BK/Kv.
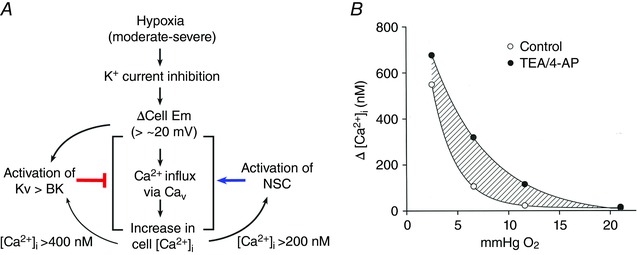
A, moderate and severe levels of hypoxia cause cell depolarization and activate Cav and elevate [Ca2+]i. A non‐selective cation channel (NSC) is activated by the rise in [Ca2+]i and is predicted to enhance the influx of Na+ and depolarization. Depolarization also activates BK and Kv that provide the repolarizing force to limit the rise in [Ca2+]i. Based on our studies, we propose that activation of NSC occurs first followed by activation of BK, as [Ca2+] rises. The contribution of NSC and BK/Kv should depend on the degree of depolarization and rise in [Ca2+]. Thus, two opposing mechanisms (NSC and BK/Kv) regulate the degree of elevation of [Ca2+]i in response to hypoxia. B, the relationship between O2 pressure and elevation of [Ca2+]i in the presence and absence of TEA/4‐AP. Per cent O2 was converted to pressure (mmHg) based on measurement of O2 content using an O2 meter. The shaded area between the two curves represents the contribution by BK/Kv to the [Ca2+]i rise at different levels of hypoxia. Thus, BK/Kv produce a strong limiting effect on hypoxia‐induced rise in [Ca2+]i. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 10 B is a re‐plot of Fig. 4 E and illustrates the contribution of BK/Kv in hypoxia‐induced elevation of [Ca2+]i. The suppressive effect of BK/Kv on hypoxia‐induced elevation of [Ca2+]i is present over a wide range of hypoxia. No suppressive effect of BK/Kv is observed with mild hypoxia that does not elevate mean basal [Ca2+]i. Thus, the strong shift of the curve to the right by TEA/4‐AP reveals how much BK/Kv reduce the hypoxic response. Inhibition of Kv should enhance the hypoxic chemosensory sensitivity of glomus cells and increase ventilation, similar to that observed with TASK inhibition (Cotten, 2013).
Additional information
Competing interests
The author have no competing interests.
Author contributions
This work was performed in the Department of Physiology at Chicago Medical School, Rosalind Franklin University located in North Chicago, IL, USA. DK conceived and designed the work. JW and DK acquired, analysed and interpreted the data, and drafted the manuscript. All authors have approved the final version of the manuscript and have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by an NIH grant to DK (HL111497).
Translational perspective.
Dysfunction of the carotid body is associated with pathophysiological conditions such as sleep apnoea, hypertension, heart failure, diabetes and respiratory distress. The carotid body is therefore an important potential target for the treatment of these conditions. Ion channels are central players in chemoreceptor function as they regulate cell excitability, Ca2+ signalling and transmitter secretion. Therefore, a full understanding of how ion channels regulate chemoreceptor activity is of fundamental importance. Our study on BK and Kv, two well‐expressed K+ channels, shed light on and clarify the mechanisms by which these two K+ channels modulate chemoreceptor sensitivity in normoxia and hypoxia. Based on the results, Kv can be a potential therapeutic target to either enhance or diminish ventilation and autonomic function. For example, activators of Kv are predicted to be of benefit for treating hypertension and heart failure, whereas inhibitors of Kv are predicted to help with respiratory distress syndromes.
Acknowledgements
We thank Mr James O. Hogan for technical assistance in the isolation and culture of CB cells. We thank Dr Carl White for providing the resources for Ca2+ imaging studies and giving us advice on the analysis of fura‐2 fluorescence.
Edited by: Harold Schultz & Gregory Funk
Linked articles This article is highlighted by a Perspective by López‐Barneo. To read this Perspective, visit https://doi.org/10.1113/JP275591.
References
- Baumann RP, Penketh PG, Seow HA, Shyam K & Sartorelli AC (2008). Generation of oxygen deficiency in cell culture using a two‐enzyme system to evaluate agents targeting hypoxic tumor cells. Radiat Res 170, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ (1997). A novel oxygen‐sensitive potassium current in rat carotid body type‐1 cells. J Physiol 498, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ (1999). Background leak K+‐currents and oxygen sensing in carotid body type 1 cells. Respir Physiol 115, 179–187. [DOI] [PubMed] [Google Scholar]
- Buckler KJ (2015). TASK channels in arterial chemoreceptors and their role in oxygen and acid sensing. Pflugers Arch 467, 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ & Vaughan‐Jones RD (1994). Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol 476, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA & Honore E (2000). An oxygen‐, acid‐ and anaesthetic‐sensitive TASK‐like background potassium channel in rat arterial chemoreceptor cells. J Physiol 525, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten JF (2013). TASK‐1 (KCNK3) and TASK‐3 (KCNK9) tandem pore potassium channel antagonists stimulate breathing in isoflurane‐anesthetized rats. Anesth Analg 116, 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DF, Kim I, Yang D & Carroll JL (2011). Role of MaxiK‐type calcium dependent K+ channels in rat carotid body hypoxia transduction during postnatal development. Respir Physiol Neurobiol 177, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD & Lopez‐Barneo J (1992). Potassium channel types in arterial chemoreceptor cells and their selective modulation by oxygen. J Gen Physiol 100, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Nino A, Obeso A, Baranda JA, Santo‐Domingo J, Lopez‐Lopez JR & Gonzalez C (2009). MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am J Physiol Cell Physiol 297, C715–722. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M & Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260, 3440–3450. [PubMed] [Google Scholar]
- Hatton CJ, Carpenter E, Pepper DR, Kumar P & Peers C (1997). Developmental changes in isolated rat type I carotid body cell K+ currents and their modulation by hypoxia. J Physiol 501, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Wang J, Hogan JO, Vennekens R, Freichel M, White C & Kim D (2014). Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non‐selective cation channel in chemoreceptor cells of rat carotid body. J Physiol 592, 1975–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I & Carroll JL (2009). Heteromeric TASK‐1/TASK‐3 is the major oxygen‐sensitive background K+ channel in rat carotid body glomus cells. J Physiol 587, 2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Papreck JR, Kim I, Donnelly DF & Carroll JL (2011). Changes in oxygen sensitivity of TASK in carotid body glomus cells during early postnatal development. Respir Physiol Neurobiol 177, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P ( 2007). Sensing hypoxia in the carotid body: from stimulus to response. Essays Biochem 43, 43–60. [DOI] [PubMed] [Google Scholar]
- Kumar P & Prabhakar NR (2012). Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2, 141–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Ortega‐Saenz P, Gonzalez‐Rodriguez P, Fernandez‐Aguera MC, Macias D, Pardal R & Gao L (2016). Oxygen‐sensing by arterial chemoreceptors: mechanisms and medical translation. Mol Aspects Med 47‐48, 90–108. [DOI] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Ortega‐Saenz P, Pardal R, Pascual A & Piruat JI (2008). Carotid body oxygen sensing. Eur Respir J 32, 1386–1398. [DOI] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Pardal R & Ortega‐Saenz P (2001). Cellular mechanism of oxygen sensing. Annu Rev Physiol 63, 259–287. [DOI] [PubMed] [Google Scholar]
- Lopez‐Lopez J, Gonzalez C, Urena J & Lopez‐Barneo J (1989). Low selectively inhibits K channel activity in chemoreceptor cells of the mammalian carotid body. J Gen Physiol 93, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Lopez JR, Gonzalez C & Perez‐Garcia MT (1997). Properties of ionic currents from isolated adult rat carotid body chemoreceptor cells: effect of hypoxia. J Physiol 499, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Lopez JR & Perez‐Garcia MT (2007). Oxygen sensitive Kv channels in the carotid body. Respir Physiol Neurobiol 157, 65–74. [DOI] [PubMed] [Google Scholar]
- Ortega‐Saenz P, Levitsky KL, Marcos‐Almaraz MT, Bonilla‐Henao V, Pascual A & Lopez‐Barneo J (2010). Carotid body chemosensory responses in mice deficient of TASK channels. J Gen Physiol 135, 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsubo T, Kostuk EW, Balbir A, Fujii K & Shirahata M (2011). Differential expression of large‐conductance Ca‐activated K channels in the carotid body between DBA/2J and A/J strains of mice. Front Cell Neurosci 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardal R, Ludewig U, Garcia‐Hirschfeld J & Lopez‐Barneo J (2000). Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci USA 97, 2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C ( 1990). Hypoxic suppression of K+ currents in type 1 carotid body cells: selective effect on the Ca2+‐activated K+ current. Neurosci Lett 119, 253–256. [DOI] [PubMed] [Google Scholar]
- Peers C & Wyatt CN (2007). The role of maxiK channels in carotid body chemotransduction. Respir Physiol Neurobiol 157, 75–82. [DOI] [PubMed] [Google Scholar]
- Perez‐Garcia MT, Colinas O, Miguel‐Velado E, Moreno‐Dominguez A & Lopez‐Lopez JR (2004). Characterization of the Kv channels of mouse carotid body chemoreceptor cells and their role in oxygen sensing. J Physiol 557, 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Garcia MT, Lopez‐Lopez JR, Riesco AM, Hoppe UC, Marban E, Gonzalez C & Johns DC (2000). Viral gene transfer of dominant‐negative Kv4 construct suppresses an O2‐sensitive K+ current in chemoreceptor cells. J Neurosci 20, 5689–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesco‐Fagundo AM, Perez‐Garcia MT, Gonzalez C & Lopez‐Lopez JR (2001). O2 modulates large‐conductance Ca2+‐dependent K+ channels of rat chemoreceptor cells by a membrane‐restricted and CO‐sensitive mechanism. Circ Res 89, 430–436. [DOI] [PubMed] [Google Scholar]
- Rocher A, Geijo‐Barrientos E, Caceres AI, Rigual R, Gonzalez C & Almaraz L (2005). Role of voltage‐dependent calcium channels in stimulus‐secretion coupling in rabbit carotid body chemoreceptor cells. J Physiol 562, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez D, Lopez‐Lopez JR, Perez‐Garcia MT, Sanz‐Alfayate G, Obeso A, Ganfornina MD & Gonzalez C (2002). Molecular identification of Kvα subunits that contribute to the oxygen‐sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J Physiol 542, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PJ & Buckler KJ (2013). Oxygen and mitochondrial inhibitors modulate both monomeric and heteromeric TASK‐1 and TASK‐3 channels in mouse carotid body type‐1 cells. J Physiol 591, 5977–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urena J, Fernandez‐Chacon R, Benot AR, Alvarez de Toledo GA & Lopez‐Barneo J (1994). Hypoxia induces voltage‐dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA 91, 10208–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urena J, Lopez‐Lopez J, Gonzalez C & Lopez‐Barneo J (1989). Ionic currents in dispersed chemoreceptor cells of the mammalian carotid body. J Gen Physiol 93, 979–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasicko MJ, Breitwieser GE, Kim I & Carroll JL (2006). Postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol 154, 356–371. [DOI] [PubMed] [Google Scholar]
- Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C & Kemp PJ (2004). Hemoxygenase‐2 is an oxygen sensor for a calcium‐sensitive potassium channel. Science 306, 2093–2097. [DOI] [PubMed] [Google Scholar]
- Wyatt CN & Peers C (1995). Ca2+‐ activated K+ channels in isolated type‐1 cells of the neonatal rat carotid body. J Physiol 483, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhong H, Vollmer C & Nurse CA (2000). Co‐release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol 525, 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]