

Abstract
Key points
The reason(s) for the increased central arterial stiffness in chronic obstructive pulmonary disease (COPD) are not well understood.
In this study, we inhibited the carotid chemoreceptor with both low‐dose dopamine and hyperoxia, and observed a decrease in central arterial stiffness and muscle sympathetic nervous activity in COPD patients, while no change was observed in age‐ and risk‐matched controls.
Carotid chemoreceptor inhibition increased vascular conductance, secondary to reduced arterial blood pressure in COPD patients.
Findings from the current study suggest that elevated carotid chemoreceptor activity may contribute to the increased arterial stiffness typically observed in COPD patients.
Abstract
Chronic obstructive pulmonary disease (COPD) patients have increased central arterial stiffness and muscle sympathetic nervous activity (MSNA), both of which contribute to cardiovascular (CV) dysfunction and increased CV risk. Previous work suggests that COPD patients have elevated carotid chemoreceptor (CC) activity/sensitivity, which may contribute to the elevated MSNA and arterial stiffness. Accordingly, the effect of CC inhibition on central arterial stiffness, MSNA and CV function at rest in COPD patients was examined in a randomized placebo‐controlled study. Thirteen mild–moderate COPD patients (forced expired volume in 1 s (FEV1) predicted ± SD: 83 ± 18%) and 13 age‐ and risk‐matched controls completed resting CV function measurements with either i.v. saline or i.v. dopamine (2 μg kg−1 min−1) while breathing normoxic or hyperoxic air (100% O2). On a separate day, a subset of COPD patients and controls completed MSNA measurements while breathing normoxic or hyperoxic air. Arterial stiffness was determined by pulse‐wave velocity (PWV) and MSNA was measured by microneurography. Brachial blood flow was determined using Doppler ultrasound, cardiac output was estimated by impedance cardiography, and vascular conductance was calculated as flow/mean arterial pressure (MAP). CC inhibition with dopamine decreased central and peripheral PWV, and MAP (P < 0.05) while increasing vascular conductance in COPD. No change in CV function was observed with dopamine in controls. CC inhibition with hyperoxia decreased peripheral PWV and MSNA (P < 0.05) in COPD, while no change was observed in controls. CC inhibition decreased PWV and MSNA, and improved vascular conductance in COPD, suggesting that tonic CC activity is elevated at rest and contributes to the elevated arterial stiffness in COPD.
Keywords: COPD, arterial Stiffness, carotid chemoreceptor
Key points
The reason(s) for the increased central arterial stiffness in chronic obstructive pulmonary disease (COPD) are not well understood.
In this study, we inhibited the carotid chemoreceptor with both low‐dose dopamine and hyperoxia, and observed a decrease in central arterial stiffness and muscle sympathetic nervous activity in COPD patients, while no change was observed in age‐ and risk‐matched controls.
Carotid chemoreceptor inhibition increased vascular conductance, secondary to reduced arterial blood pressure in COPD patients.
Findings from the current study suggest that elevated carotid chemoreceptor activity may contribute to the increased arterial stiffness typically observed in COPD patients.
Introduction
Chronic obstructive pulmonary disease (COPD) is a respiratory disorder characterized by progressive, partially reversible airway obstruction, dyspnoea and marked exercise intolerance. Although COPD is considered a disease of the lung, there is evidence of substantial systemic manifestations, such as cardiovascular (CV) disease, that develop as a consequence of COPD (Wouters, 2002). Previous work has shown elevated sympathetic nervous activity (SNA) and central arterial stiffness in both hypoxaemic and non‐hypoxaemic COPD patients, both of which would contribute to CV deterioration, and are linked to increased CV events/mortality (Sabit et al. 2007; Vlachopoulos et al. 2010; Andreas et al. 2014).
The mechanism(s) causing elevated SNA and central arterial stiffness in COPD are poorly understood; however, recent work suggests that greater activity of the carotid chemoreceptor (CC) may play a role (Stickland et al. 2016). The CCs are located within the carotid body at the level of the carotid sinus and are sensitive to circulating stimuli including O2, CO2, inflammation (IL‐6, TNF‐α) and reactive oxygen species, and are key mediators of ventilatory control and sympathetic vasoconstrictor outflow (Guyenet, 2000; Ding et al. 2011; Porzionato et al. 2013). Importantly, the CC has been shown to be tonically active and sensitized in conditions such as chronic heart failure (CHF), leading to elevations in SNA (Sun et al. 1999; Marcus et al. 2014). Recently we have shown enhanced resting CC activity and sensitivity in non‐hypoxaemic non‐hypercapnic COPD patients (Stickland et al. 2016). CC sensitivity was related to peripheral pulse‐wave velocity (PWV), and CC inhibition by breathing 100% O2 normalized PWV in COPD patients (Stickland et al. 2016). These findings are consistent with previous work in hypoxaemic COPD patients with severe respiratory failure demonstrating that breathing 100% O2 reduced resting SNA (Heindl et al. 2001). Combined with previous work in CHF, these data suggest that the CC may play an important role in CV regulation and may help explain the increased sympathetic outflow and central arterial stiffness even in stable non‐hypoxaemic COPD patients. Therefore, the aim of the present study was to examine the effect of CC inhibition on central arterial stiffness and CV function at rest in COPD patients. It was hypothesized that CC inhibition with either 100% inspired O2, or intravenous (i.v.) dopamine would reduce central arterial stiffness, secondary to reduced sympathetic vasoconstrictor outflow in COPD patients, while no change would be observed in controls.
Methods
Ethical approval
The present study was a randomized, single‐blind, placebo controlled design and was approved by the University of Alberta Health Research Ethics Board (Biomedical Panel Protocol no. 00043106). Participant written informed consent was obtained prior to any research procedures. The study conformed to the standards set by the latest revision of the Declaration of Helsinki.
Participants and procedure
Thirteen non‐hypoxaemic mild–moderate COPD patients (GOLD Stage I and II, post bronchodilator (forced expired volume in 1 s (FEV1)/forced vital capacity (FVC) ratio < 0.7, FEV1 ≥ 50% predicted) with a smoking history > 10 pack‐years and 13 age‐ and risk‐matched controls with minimal smoking history (<10 pack‐years) were recruited. Participants were carefully screened to exclude supplemental oxygen therapy, diabetes, CV disease, body mass index ≥ 35, severe inflammatory disorders (such as connective tissue disease), severe sleep apnoea (STOP‐Bang score < 3, apnoea–hypopnoea index < 30 as evaluated by overnight sleep monitoring with ApneaLink Plus, ResMed Ltd, Bella Vista, Australia) and recent respiratory exacerbations (<6 months). Medication use (not including respiratory medication) was similar between the control and COPD groups (Table 1). The lack of CV disease was also confirmed by a normal resting blood pressure (systolic < 140 mmHg) and 12‐lead ECG, as well as normal blood pressure and ECG responses to maximal exercise.
Table 1.
Participant characteristics
| Control | COPD | P value | |
|---|---|---|---|
| Participants | 13 | 13 | — |
| Male/female | 8/5 | 10/3 | — |
| Age (years) | 64 ± 14 | 61 ± 14 | 0.67 |
| Height (cm) | 168.7 ± 9.3 | 170.2 ± 5.5 | 0.63 |
| Mass (kg) | 78.0 ± 15.1 | 74.4 ± 14.7 | 0.55 |
| BMI (kg m−2) | 27.2 ± 3.7 | 25.6 ± 3.8 | 0.26 |
| Smoking history (pack‐years) | 3.9 ± 4.5 | 41.8 ± 29.7 | <0.001 |
| Current smoker (n) | 0 | 9 | — |
| Modified MRC dyspnoea (scale 0–4) | 0.0 ± 0.0 | 1.3 ± 0.5 | <0.001 |
| Medication use (n) | |||
| SABA | 0 | 8 | — |
| ICS | 0 | 8 | — |
| Combined ICS/LABA | 0 | 6 | — |
| β‐blocker | 1 | 2 | — |
| ACE–ARB | 0 | 1 | — |
| Diuretics | 1 | 0 | — |
| Statins | 4 | 3 | — |
| Pulmonary function | |||
| FEV1 (L) | 3.0 ± 0.8 | 2.4 ± 0.6 | <0.001 |
| FEV1 (% predicted) | 106.9 ± 13.9 | 83.1 ± 18.0 | <0.001 |
| FEV1 (Z‐score) | 0.13 ± 0.86 | −1.41 ± 1.26 | <0.001 |
| FEV1/FVC (% predicted) | 97.2 ± 5.7 | 79.9 ± 10.1 | <0.001 |
| FEV1/FVC (Z‐score) | −0.75 ± 0.47 | −2.28 ± 1.17 | <0.001 |
| FRC (% predicted) | 98.4 ± 17.6 | 118.1 ± 20.5 | 0.015 |
| RV (% predicted) | 87.8 ± 18.9 | 113.9 ± 21.4 | 0.004 |
| TLC (% predicted) | 100.3 ± 15.3 | 104.5 ± 15.0 | 0.555 |
| D LCO (% predicted) | 93.4 ± 10.5 | 65.1 ± 21.6 | <0.001 |
| Cardiopulmonary peak exercise responses | |||
| (mL kg−1 min−1) | 33.3 ± 9.3 | 24.9 ± 8.9 | 0.028 |
| (% predicted) | 130 ± 25 | 88 ± 26 | 0.002 |
| (L min−1) | 2.83 ± 0.86 | 1.98 ± 0.71 | 0.011 |
| (L min−1) | 92.6 ± 29.3 | 68.2 ± 17.3 | 0.017 |
| (mmHg) | 34.1 ± 4.8 | 32.8 ± 4.9 | 0.489 |
| HR (beats min−1) | 162 ± 20 | 144 ± 22 | 0.065 |
| (%) | 95.8 ± 4.2 | 97.0 ± 1.5 | 0.412 |
| (%) | 95.6 ± 3.2 | 95.4 ± 2.5 | 0.864 |
| Nadir / | 29.2 ± 2.2 | 34.9 ± 5.5 | <0.001 |
| Change in IC baseline to peak exercise (L) | 0.1 ± 0.4 | −0.4 ± 0.4 | 0.007 |
Data are presented as n or means ± SD. ACE: angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; BMI: body mass index; COPD: chronic obstructive pulmonary disease; D LCO: diffusing capacity of the lung for carbon monoxide; FEV1: forced expiratory volume in 1 s; FRC: functional residual capacity; FVC: forced vital capacity; HR: heart rate; IC: inspiratory capacity; ICS: inhaled corticosteroid; LABA: long‐acting β2‐agonist; MRC: Medical Research Council; : partial pressure of carbon dioxide; RV: residual volume; SABA: short‐acting β‐agonist; S pO 2: oxygen saturation measured by pulse oximeter; TLC: total lung capacity; : carbon dioxide production; : minute ventilation; : oxygen uptake. Z‐score calculated using Global Lung Initiative 2012 Equations (Quanjer et al. 2012).
Three sessions were completed over a 3 week period in the following order: (1) participant enrolment and medical history, standard pulmonary function and cardiopulmonary exercise test as previously described (Edgell et al. 2015; Stickland et al. 2016), (2) basal chemoreceptor reflex assessment, and (3) the experimental trial. Prior to all study procedures, participants were asked to abstain from caffeine, smoking, vigorous exercise, alcohol and respiratory medications for at least 6 h preceding testing.
Basal chemoreceptor assessment
Prior to data collection, participants rested quietly in the supine position while breathing normoxic air on a mouthpiece for 10 min to obtain baseline data. Basal CC activity was then evaluated by examining the transient reduction in minute ventilation in response to 2 min of hyperoxia (inspired O2 fraction, = 1.0). The difference between the baseline and nadir (15 s) ventilation was used as the index of basal CC activity (Dejours, 1962; Stickland et al. 2016). Following a minimum 10 min break, a new baseline ventilation was obtained, and participants then completed a standard hyperoxic hypercapnic rebreathe test to determine central medullary chemoreceptor sensitivity (Read, 1967). A 4 L rebreathing bag was filled with a specialized gas mixture (7% CO2, 50% O2 and 43% N2). Following a brief bout of hyperoxic breathing ( = 0.5), participants then began rebreathing until the end‐tidal CO2 values reached 55 mmHg or participants requested to stop. After another minimum 10 min break, baseline ventilation was obtained again and CC sensitivity was evaluated by measuring the hypoxic ventilatory response using a stepwise method, as previously described (Edgell et al. 2015; Stickland et al. 2016). Briefly, participants breathed normoxic air for 3 min followed by 3 min at a target arterial oxygen saturation (S po 2) of 90% and then 3 min at a target S pO 2 of 85%. Arterial saturation was analysed as opposed to end‐tidal O2 due to the uncertainty regarding the accuracy of estimating arterial blood gas from end‐tidal expired gas in patients with lung disease (Jones et al. 1979). Sufficient rest was given between trials to ensure that ventilation and arterial blood pressure returned to the initial baseline before continuing to the next test.
Experimental trial
Following instrumentation, participants breathed freely on the mouthpiece for 15 min to obtain baseline data. Interventions were then conducted in random order: (1) saline and normoxia, (2) saline and hyperoxia ( = 1.0), (3) dopamine and normoxia, and (4) dopamine and hyperoxia. Each intervention was separated by 10 min. For the dopamine trials, one minute steady‐state mean data were recorded following a 10 min wash‐in. Two minute hyperoxia interventions were conducted with either saline or dopamine infusion and measurements were analysed in the second minute (Stickland et al. 2011). On a separate day, a subset of seven COPD patients and seven controls completed resting muscle SNA (MSNA) measurements while breathing normoxic or hyperoxic air.
Dopamine
Prior to the experimental trial, an i.v. catheter was inserted into the antecubital vein of the left arm. Participants received either isotonic i.v. saline or i.v. dopamine HCl (2 μg kg−1 min−1; Hospira, Lake Forest, IL, USA) via an automated constant‐infusion pump (Alaris, San Diego, CA, USA). Dopamine does not cross the blood–brain barrier and would not affect the central chemoreceptors (Zlokovic, 2008). Previous research has shown that this specific dose effectively inhibits the carotid chemoreceptors in humans (Lahiri et al. 1980; Stickland et al. 2011; Edgell et al. 2015).
Hyperoxia
During both the chemoreceptor assessment and the experimental trials, 2 min hyperoxia interventions ( = 1.0) were conducted. Transient hyperoxia has been previously shown to rapidly inhibit the CC (Nye et al. 1981). Prolonged bouts of hyperoxia were avoided as long‐term hyperoxia can act as a central stimulant, which would confound results from the current study (Dean et al. 2004).
Cardiorespiratory measures
For both the chemoreceptor reflex assessment and the experimental trial, all data were recorded and integrated with a data acquisition system (Powerlab 16/30; ADInstruments, New South Wales, Australia) and stored for subsequent analysis using associated software (LabChart 8.0 Pro; ADInstruments). Minute ventilation was measured by a pneumotachometer (3700 series, Hans Rudolph, Kansas City, MO, USA) and expired CO2 and O2 were measured (CD‐3A and S‐3A; AEI Technologies, Naperville, IL, USA) continuously from a small sample port off the mouthpiece to obtain end‐tidal CO2 and O2. Inspired gas was humidified (HC150; Fisher and Paykel Healthcare, Auckland, New Zealand) and delivered continuously using a flow‐through system to prevent rebreathing of expired gas. Inspired gas was modulated using air–oxygen (normoxia to hyperoxia) or air–nitrogen (normoxia to hypoxia) blender systems. Heart rate was determined from the R–R interval with a single lead ECG (lead II, Dual Bio Amp; ADInstruments). Cardiac output was determined with impedance cardiography (Physioflow, Manatec, Paris, France), which has been previously validated (Edmunds et al. 1982). Arterial oxygen saturation was estimated with pulse oximetry (N‐595; Nellcor Oximax, Boulder, CO, USA) using a left ear‐lobe sensor. Blood pressure was determined using finger plethysmography (Finometer Midi, Amsterdam, the Netherlands) and was calibrated to brachial artery pressure at regular intervals using manual auscultation.
Doppler ultrasound
Similar to previous work (Edgell et al. 2015), brachial arterial mean blood velocity was obtained from the right arm using pulse‐wave Doppler ultrasound (GE, Vivid‐7, 4–5 MHz, <60 deg angle of insonation). Briefly, the average velocity was multiplied by the cross‐sectional area of the brachial artery and then multiplied by 60 to determine brachial flow (mL min−1) (Stickland et al. 2011; Edgell et al. 2015). Brachial conductance was calculated as brachial flow/mean arterial pressure.
Arterial stiffness
Arterial stiffness was determined by PWV. Pulse waves were gathered simultaneously using applanation tonometry (Mikro‐tip Catheter Transducers model SPT‐301, Millar Instruments, Inc., Houston, TX, USA) from the carotid, radial and femoral arteries. PWV was calculated as PWV = D × Δt −1, where D was the distance (m) between sites and Δt was the time difference (s) between pulse waves using the foot‐to‐foot method (Laurent et al. 2006). Carotid–femoral (cfPWV) and carotid–radial (crPWV) pulse‐wave velocity were used as indices of central and peripheral arterial stiffness, respectively (Andreas et al. 2014; Stickland et al. 2016). Distance was measured on the skin using a tape measure beginning at the sternal notch and extending to the recording sites of the cfPWV and crPWV. At least 10 consecutive beats were averaged to represent PWV over a complete respiratory cycle (Laurent et al. 2006).
Muscle sympathetic nerve activity
MSNA was recorded via peroneal microneurography as previously described (Charkoudian et al. 2017). Briefly, the raw sympathetic signal was rectified and integrated to produce a neurogram with a characteristic bursting pattern. MSNA was then analysed using a semi‐automated peak detection algorithm (Chart 8.1.3; ADInstruments) and expressed as the change in burst frequency (bursts min−1) and incidence (bursts (100 heart beats)−1) from baseline to hyperoxia.
Statistical analysis
Data are presented as means ± standard error of the mean (SEM) unless otherwise stated. For all inferential analysis, statistical significance was set a priori at P < 0.05. Primary endpoints were carotid–femoral pulse‐wave velocity and brachial artery conductance. A multi‐factorial ANOVA was used to evaluate the effect of CC inhibition with either dopamine or hyperoxia on key dependent variables in COPD and controls. If a main effect or interaction effect was found, a Bonferroni multiple comparisons test was used to locate the differences. Unpaired t analysis was used to evaluate participant characteristics and chemoreceptor reflexes between groups. All statistical analyses were performed using Sigma Plot Software version 13 (Systat Software Inc., Chicago, IL, USA).
Results
Participants
Descriptive participant characteristics for the COPD and control groups are displayed in Table 1. There were no between‐group differences in age, height, body mass or body mass index (BMI). As expected, the COPD group had a significantly greater smoking history, increased airway obstruction, functional residual capacity and residual volume compared to controls (Table 1). COPD patients had significantly lower peak oxygen uptake but there was no evidence of arterial desaturation at rest or peak exercise in either group (Table 1).
Chemoreceptor reflex
The transient reduction in minute ventilation during hyperoxia and the slope of the ventilatory response to hypoxia were greater in COPD patients when compared to controls (Table 2). These data suggest that COPD patients have greater CC activity and sensitivity. There was no difference in the hyperoxic hypercapnic ventilatory responses between groups indicating that central chemoreceptor sensitivity was similar between COPD and controls (Table 2).
Table 2.
Central and carotid chemoreceptor reflex responses
| Control | COPD | |
|---|---|---|
| Transient hyperoxia | ||
| Baseline (L min−1) | 7.88 ± 0.51 | 8.41 ± 0.83 |
| Nadir (15 s) (L min−1) | 7.76 ± 0.61 | 6.16 ± 0.61 |
| Nadir (15 s) change in from baseline (L min−1) | 0.12 ± 0.46 | 2.25 ± 0.59* |
| Hyperoxic hypercapnic ventilatory response | ||
| Δ/Δ slope (L min−1 mmHg−1) | 1.22 ± 0.27 | 0.93 ± 0.18 |
| Hypoxic ventilatory response | ||
| Δ/ΔS pO 2 slope (L min−1 %−1) | 0.04 ± 0.03 | 0.23 ± 0.08* |
Data are presented as means ± SEM. : partial pressure of carbon dioxide; S pO 2: oxygen saturation measured by pulse oximeter; : minute ventilation. * P < 0.05 between groups
Respiratory responses to carotid chemoreceptor inhibition
See Table 3 for grouped cardiorespiratory data. Minute ventilation was similar between groups during saline infusion while breathing room air (Table 3). CC inhibition with hyperoxia and low‐dose dopamine significantly reduced minute ventilation in COPD but had no effect in controls. When given both dopamine and hyperoxia, there was no further reduction in minute ventilation, suggesting that the interventions were not additive and the dopamine was likely a stronger CC inhibitor than inhaled O2.
Table 3.
Respiratory and cardiovascular data with either saline or dopamine while breathing room air or 2 min of hyperoxia (100% O2) in controls and COPD
| Group | Saline normoxia | Saline hyperoxia | Dopamine normoxia | Dopamine hyperoxia | |
|---|---|---|---|---|---|
| (L min−1) | Control | 7.3 ± 0.4 | 7.9 ± 0.4 | 8.0 ± 0.7 | 8.2 ± 0.8 |
| COPD | 8.5 ± 0.8 | 7.7 ± 0.7* | 7.0 ± 0.6*ǂ | 7.3 ± 0.5*ǂ | |
| (mmHg) | Control | 37.1 ± 1.3 | 34.8 ± 1.3 | 37.9 ± 1.6 | 34.9 ± 1.5 |
| COPD | 34.0 ± 2.2 | 32.0 ± 2.1 | 36.2 ± 1.4 | 33.9 ± 1.7 | |
| (mmHg) | Control | 103.5 ± 1.9 | 576.8 ± 11.2# | 103.5 ± 2.1 | 562.6 ± 18.6# |
| COPD | 110.5 ± 3.2 | 553.3 ± 19.2* | 105.5 ± 2.2 | 567.7 ± 21.4* | |
| S pO 2 (%) | Control | 95.5 ± 0.7 | 98.7 ± 0.5# | 95.5 ± 0.6 | 99.0 ± 0.3# |
| COPD | 94.3 ± 0.5 | 97.2 ± 0.7* | 93.4 ± 0.6 | 97.6 ± 0.6* | |
| (L min−1) | Control | 4.8 ± 0.3 | 5.0 ± 0.4 | 5.0 ± 0.4 | 5.1 ± 0.5 |
| COPD | 4.5 ± 0.3 | 4.7 ± 0.3 | 4.6 ± 0.3 | 4.9 ± 0.3 | |
| HR (beats min−1) | Control | 62.3 ± 2.4 | 62.1 ± 2.4 | 67.3 ± 2.8 | 65.1 ± 2.7 |
| COPD | 62.2 ± 2.7 | 58.9 ± 2.7 | 64.8 ± 2.5 | 64.0 ± 2.7 | |
| SV (mL beat−1) | Control | 78.8 ± 6.7 | 80.9 ± 6.4 | 75.1 ± 6.1 | 79.3 ± 6.5 |
| COPD | 73.5 ± 5.0 | 80.6 ± 5.9 | 72.6 ± 6.3 | 83.3 ± 8.8 | |
| MAP (mmHg) | Control | 91.0 ± 2.3 | 93.7 ± 2.5 | 91.9 ± 2.8 | 92.4 ± 2.9 |
| COPD | 93.2 ± 2.1 | 92.7 ± 2.2 | 87.0 ± 2.9*ǂ | 85.7 ± 3.3*ǂ | |
| PP (mmHg) | Control | 45.6 ± 3.3 | 45.5 ± 3.0 | 47.8 ± 3.3 | 47.5 ± 2.4 |
| COPD | 48.9 ± 3.5 | 46.5 ± 2.7 | 42.4 ± 2.4*ǂ | 43.6 ± 3.0*ǂ | |
| /MAP (mL min−1 mmHg−1) | Control | 53.9 ± 4.7 | 53.7 ± 4.9 | 54.9 ± 4.6 | 56.0 ± 5.2 |
| COPD | 47.7 ± 3.1 | 49.5 ± 3.0 | 53.8 ± 3.8* | 57.6 ± 4.1* | |
| Brachial artery diameter (mm) | Control | 3.79 ± 0.18 | 3.78 ± 0.19 | 3.85 ± 0.17 | 3.83 ± 0.16 |
| COPD | 3.97 ± 0.19 | 4.03 ± 0.18 | 4.08 ± 0.17*ǂ | 4.10 ± 0.19*ǂ |
Data are presented as means ± SEM. HR: heart rate; MAP: mean brachial arterial pressure; : partial pressure of carbon dioxide; : partial pressure of oxygen; PP: brachial artery pulse pressure; : cardiac output; /MAP: vascular conductance; S pO 2: oxygen saturation measured by pulse oximeter; SV: stroke volume;: minute ventilation. # P < 0.05 vs. control saline normoxia, * P < 0.05 vs. COPD saline normoxia, ǂ P < 0.05 vs. control within condition.
Cardiovascular responses to carotid chemoreceptor inhibition
See Table 3, and Figs 1 and 2 for grouped cardiovascular data. Both central (cfPWV) and peripheral (crPWV) pulse‐wave velocity were elevated in COPD during saline infusion while breathing room air when compared to controls (Fig. 1). Carotid–radial PWV was reduced with breathing 2 min of 100% O2 in COPD but not in controls. Dopamine reduced both cfPWV and crPWV in COPD but not in controls (Fig. 1). MAP and pulse pressure were significantly reduced with dopamine in COPD but not with hyperoxia (Table 3). No change in mean arterial pressure was observed in controls between conditions. Cardiac output, stroke volume, heart rate and brachial artery blood flow were unaffected by either hyperoxia or dopamine in both groups (Table 3, Fig. 2). Both brachial and total vascular conductance were significantly increased with dopamine in COPD but unchanged in controls, while conductance was unaffected with hyperoxia in either group (Table 3, Fig. 2). Brachial artery diameter was similar between groups with saline (Table 3). Dopamine increased brachial artery diameter in COPD, while no change was observed in controls (Table 3).
Figure 1. Carotid–femoral (cfPWV; A) and carotid–radial (crPWV; B) pulse‐wave velocity with either saline or dopamine while breathing room air or 2 min of hyperoxia (100% O2) in controls and COPD.
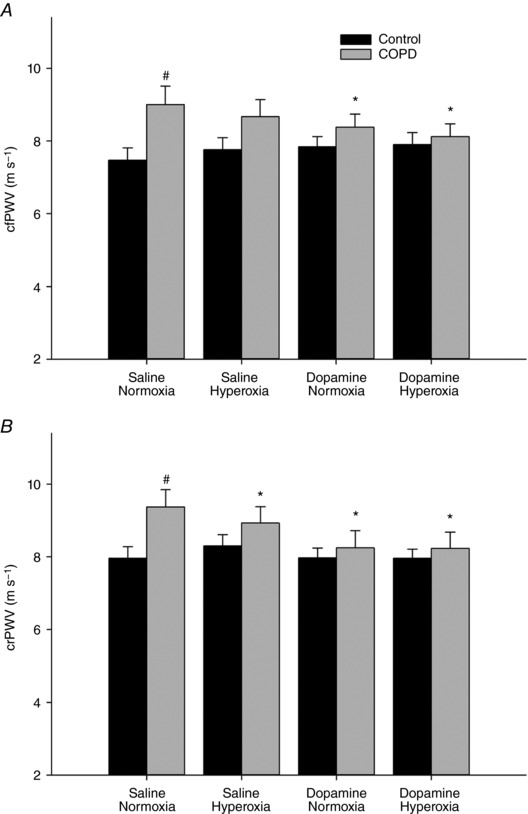
Both cfPWV and crPWV were elevated in COPD compared to control, in the saline and normoxia condition. Dopamine reduced both cfPWV and crPWV in COPD, while hyperoxia reduced crPWV in COPD. # P < 0.05 vs. control saline normoxia, * P < 0.05 vs. COPD saline normoxia.
Figure 2. Brachial blood flow (A) and conductance (B) with either saline or dopamine while breathing room air or 2 min of hyperoxia (100% O2) in controls and COPD.
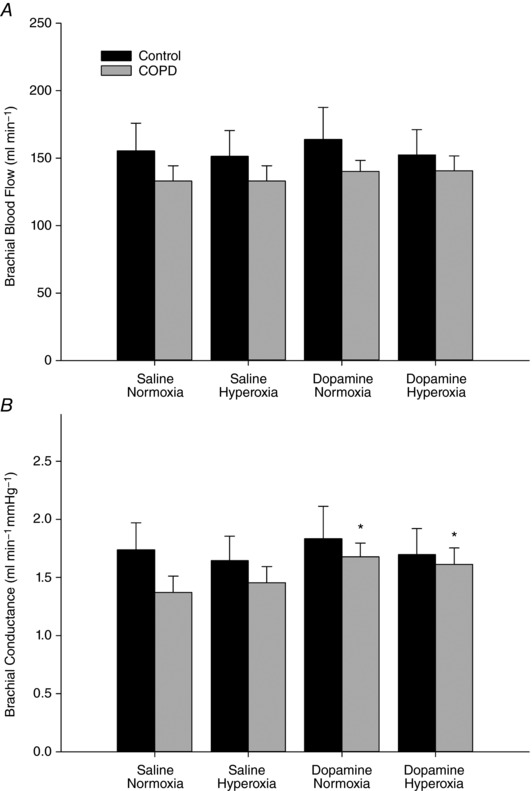
Dopamine increased conductance in COPD. * P < 0.05 vs. COPD saline normoxia.
In the subset of patients with MSNA recordings, there were no differences in resting burst frequency (42 ± 4 vs. 43 ± 8 bursts min−1, P = 0.61) or burst incidence (64 ± 9 vs. 65 ± 13 bursts (100 heart beats)−1, P = 0.79) between COPD patients and controls, respectively. CC inhibition with hyperoxia significantly decreased MSNA in COPD but not controls (Fig. 3).
Figure 3. Change in muscle sympathetic nerve activity (MSNA) burst frequency (A) and burst incidence (B) from normoxia to hyperoxia in controls and COPD.
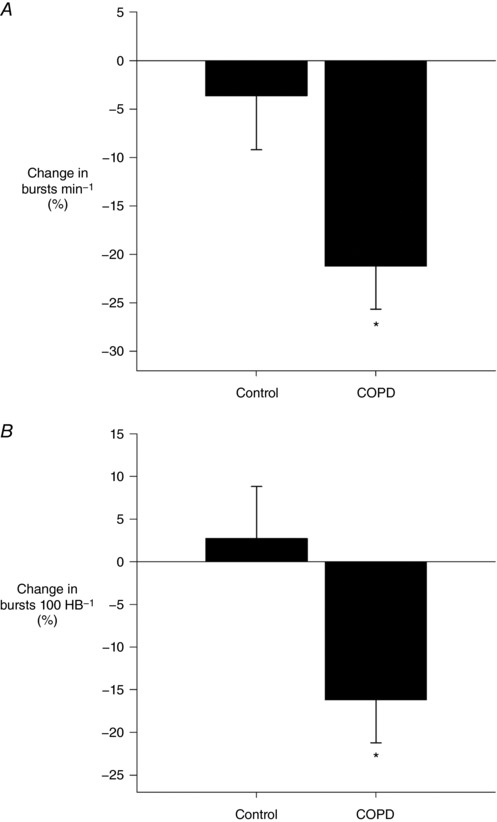
MSNA was reduced with hyperoxia in COPD. * P < 0.05 between groups.
Discussion
Carotid chemoreceptor inhibition with low‐dose dopamine reduced central and peripheral arterial stiffness, and increased vascular conductance, in COPD patients, while no change was observed in risk‐matched controls. Similarly, carotid chemoreceptor inhibition with inspired hyperoxia reduced both peripheral arterial stiffness and MSNA in COPD, while no effect was observed in controls. These data demonstrate the importance of the CC in contributing to the elevated arterial stiffness in COPD. There are multiple studies documenting elevated central PWV in COPD patients; however, the potential mechanism(s) for the increased PWV in COPD remain unclear (McAllister et al. 2007; Sabit et al. 2007; Maclay et al. 2009). This is the first study to demonstrate that central PWV can be reduced acutely in COPD. This work, combined with previous work examining peripheral PWV responses to CC inhibition, demonstrates that the CC has an important influence on PWV and CV risk in mild–moderate COPD patients (Stickland et al. 2016).
The CC has been shown to be a significant sensor for both the control of breathing and CV function (Sun et al. 1999; Stickland et al. 2007). Importantly, it is unlikely that central chemoreception contributed to the observed reduction in PWV with dopamine, as dopamine does not cross the blood–brain barrier and would not stimulate the rostral ventrolateral medulla (Zlokovic, 2008). It is often assumed that low‐dose dopamine causes vasodilatation through stimulation of dopamine‐1 vascular receptors. However, low‐dose dopamine does not cause vasodilatation in healthy controls (Stickland et al. 2011; Edgell et al. 2015), but vasodilatation is seen typically in conditions of high CC activity/sensitivity such as CHF (Stickland et al. 2007; Edgell et al. 2015) and at present COPD. It is acknowledged that the vasodilatation observed with dopamine in COPD could be due to the direct peripheral vasodilatory actions of dopamine (via direct stimulation of dopamine‐1 vascular receptors); however, it would be assumed that any direct peripheral vascular effects of dopamine would have also been observed in risk‐matched controls. Results from the current study show that CC activity/sensitivity was elevated in COPD, and dopamine reduced ventilation and caused vasodilatation in these patients, while controls showed no response to dopamine. These results strongly suggest that the cardiovascular effects of low‐dose dopamine in COPD are secondary to CC inhibition and not a central chemoreceptor or peripheral vascular effect.
Cardiovascular responses to carotid chemoreceptor inhibition
Arterial stiffness/PWV is determined by arterial wall material properties (e.g. elastin/collagen) and vessel diameter (O'Rourke et al. 2002). Neither short‐term hyperoxia nor dopamine would acutely affect arterial wall properties. Rather, our data demonstrate vasodilatation (increased brachial artery diameter and vascular conductance) with CC inhibition, indicating that the reduction in PWV with CC inhibition is likely secondary to increased vessel diameter. Changes in vessel diameter are highly influenced by MSNA (Laurent et al. 2006; Świerblewska et al. 2010) and increased MSNA has been linked with increased PWV, independent of age, body composition or blood pressure (Świerblewska et al. 2010). It is well documented that afferent carotid body discharge contributes to elevated SNA in health and CHF (Downing et al. 1962; Hardy et al. 1994; van de Borne et al. 1998), and inhibition of the CC reduces SNA and improves CV function in CHF (Sun et al. 1999; Stickland et al. 2007; Edgell et al. 2015). MSNA is chronically elevated in COPD compared to healthy individuals (Raupach et al. 2008; Andreas et al. 2014), and in the current study, CC inhibition significantly reduced both peripheral and central arterial stiffness, and MSNA, in COPD patients. Based on well‐established work linking the CC to MSNA, and MSNA to PWV, our data would indicate that the observed reduction in PWV with CC inhibition is likely the result of reduced MSNA.
Carotid–radial PWV reflects stiffness of the peripheral arteries, which are generally considered resistive vessels and thus under more smooth muscle sympathetic control compared to carotid–femoral PWV (i.e. central arterial stiffness). It is therefore not surprising that carotid–radial PWV had a larger reduction with dopamine than carotid–femoral PWV (0.9 ± 0.3 vs. 0.6 ± 0.3 m s−1, respectively) in COPD patients. A meta‐analysis concluded that a 1 m s−1 increase in carotid–femoral PWV elevated CV mortality risk by 15% (Vlachopoulos et al. 2010). Based on these data, the 0.6 m s−1 reduction in carotid–femoral PWV observed with CC inhibition in COPD patients in the current study would suggest an 8–9% reduction in risk of mortality with CC inhibition in COPD.
Chemoreflex
The reflex actions of both the central and peripheral (CC) chemoreceptors have been well characterized in other clinical conditions; however, there are limited studies in patients with COPD. Most of the previous work in COPD has focused on severe COPD patients who have complex physiological responses due to additional co‐morbidities (e.g. heart failure), severe airway obstruction and chronic hypoxaemia (Bradley et al. 1979; Heindl et al. 2001; Ponikowski et al. 2001). Importantly, the patients in our study had mild–moderate COPD, were non‐hypoxaemic and were compared to risk‐matched controls without COPD. While the ventilatory response to hyperoxia (CC activity) and hypoxia (CC sensitivity) were significantly greater in COPD patients, there was no difference in the hyperoxic hypercapnic ventilatory response compared to risk‐matched controls. These data suggest that while the peripheral (CC) chemoreceptors appear sensitized/activated in COPD, central chemoreception appears normal.
Limitations
Minute ventilation and peripheral arterial stiffness were reduced while breathing 100% O2 in COPD, and these findings are similar to previous work in COPD (Stickland et al. 2016). Despite this evidence of CC inhibition, inhaled hyperoxia did not have a similar effect to dopamine on central arterial stiffness and vascular conductance in COPD patients. It is important to note that arterial stiffness and blood flow measurements were taken in the second minute of the 2 min hyperoxic bout. It is possible that, consistent with previous work in healthy participants (Stickland et al. 2011), there was an acute vasodilatory response (<60 s) and a subsequent return to baseline, and that collecting data in the second minute resulted in our missing the transient vasodilatation. The lack of vasodilatation with hyperoxia may also be explained by autoregulation as the increased oxygen content with hyperoxia would reduce blood flow to maintain O2 delivery (Stickland et al. 2011). While the current results indicate that chronic suppression of CC activity may be an important target to reduce PWV and cardiovascular risk, care should be taken regarding the use of long‐term hyperoxia, as this may have important deleterious secondary consequences (Bartels et al. 2004).
At moderate‐to‐high doses (i.e. 5–20 μg kg−1 min−1), dopamine can stimulate α‐ and β‐receptors (Hoffman & Lefkowitz, 1990; Ciarka et al. 2007). β‐receptor stimulation would be expected to increase heart rate; however, heart rate was unaffected in both groups with 2 μg kg−1 min−1 dopamine infusion, suggesting an absence of β‐adrenergic stimulation. α‐adrenergic stimulation would result in vasoconstriction, reducing conductance and increasing blood pressure; however, the opposite was observed in COPD patients. Therefore, the cardiovascular responses observed with dopamine in COPD patients are unlikely to be explained by α‐ or β‐adrenergic stimulation.
Our groups were not matched for smoking history, and therefore we cannot separate the independent effect of smoking history vs. chronic airflow obstruction on chemoreception and vascular regulation.
While previous work has shown elevated resting MSNA in COPD (Raupach et al. 2008; Andreas et al. 2014), there was no between‐group difference in baseline MSNA in our small subset of participants with MSNA data (n = 7 per group). Unfortunately, we were not able to obtain MSNA data in a larger sample of COPD patients because of technical difficulties in obtaining and maintaining the MSNA signal. Based on the previous research demonstrating neurohumoral activation in COPD (Hofford et al. 1990; Andreas et al. 2005, 2014; Raupach et al. 2008) and the variability in baseline MSNA observed in our controls, we were likely underpowered to detect a difference in baseline MSNA. Although no between‐group difference in baseline MSNA was observed, our results combined with previous research examining carotid chemoreception and neurohumoral activation support the conclusion that the reduction in arterial stiffness was likely secondary to a reduction in MSNA.
Conclusions
CC inhibition reduced arterial stiffness and MSNA, and improved conductance in COPD. These data suggest that CC activity is increased at rest and contributes to elevated arterial stiffness in COPD. Our findings provide a potential mechanism for the increased PWV typically observed in COPD and demonstrate that the CC plays an important role in resting CV regulation in COPD. Interventions aimed at chronically reducing CC activity may be an effective strategy to reduce CV risk in COPD.
Additional information
Disclaimer
The views expressed in the submitted article are our own and not an official position of the institution.
Competing interests
None declared.
Author contributions
Conception or design of the work: D.B.P., V.T., M.B., M.K.S; acquisition, or analysis or interpretation of data for the work: D.B.P., C.D.S., S.É.C., D.P.F., T.L.B., E.Y.L.W., M.K.S.; drafting the work or revising it critically for important intellectual content: D.B.P., C.D.S., S.É.C., D.P.F., T.L.B., E.Y.L.W., V.T., M.B., M.K.S. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. This research was conducted through the Clinical Physiology Laboratory, Alberta Cardiovascular and Stroke Research Centre (ABACUS), Mazankowski Alberta Heart Institute.
Funding
This work was supported by Canadian Institutes of Health Research (106444 to M.K.S.).
Biographies
Devin Phillips is a Doctoral Student at the University of Alberta. His research investigates how autonomic dysfunction contributes to elevated cardiovascular risk in chronic lung disease. Devin completed his Master of Science at the University of Alberta and his Bachelor of Science at the University of Nevada, Las Vegas.

Michael Stickland received his Doctorate in 2004 from the University of Alberta, and completed postdoctoral training at the University of Wisconsin–Madison in 2006. He is currently a Professor in the Pulmonary Division within the Faculty of Medicine and Dentistry at the University of Alberta. Clinically, Dr Stickland is the Director of the G.F. MacDonald Centre for Lung Health, which delivers the primary pulmonary rehabilitation programme in Edmonton. His research interests in physiology include examining the cardiovascular consequences of chronic lung disease, the determinants of pulmonary gas exchange, and autonomic control of cardiovascular function in health and disease.
Edited by: Harold Schultz & John Osborn
Linked articles This article is highlighted by a Journal Club article by Bock. To read this Journal Club, visit https://doi.org/10.1113/JP276597.
References
- Andreas S, Anker SD, Scanlon PD & Somers VK (2005). Neurohumoral activation as a link to systemic manifestations of chronic lung disease. Chest 128, 3618–3624. [DOI] [PubMed] [Google Scholar]
- Andreas S, Haarmann H, Klarner S, Hasenfuss G & Raupach T (2014). Increased sympathetic nerve activity in COPD is associated with morbidity and mortality. Lung 192, 235–241. [DOI] [PubMed] [Google Scholar]
- Bartels MN, Jelic S, Basner RC, Ngai P, Gonzalez JM & De Meersman RE (2004). Supplemental oxygen increases arterial stiffness in chronic obstructive pulmonary disease. Respir Med 98, 84–89. [DOI] [PubMed] [Google Scholar]
- Bradley CA, Fleetham JA & Anthonisen NR (1979). Ventilatory control in patients with hypoxemia due to obstructive lung disease. Am Rev Respir Dis 120, 21–30. [DOI] [PubMed] [Google Scholar]
- Charkoudian N, Usselman CW, Skow RJ, Staab JS, Julian CG, Stickland MK, Chari RS, Khurana R, Davidge ST, Davenport MH & Steinback CD (2017). Muscle sympathetic nerve activity and volume‐regulating factors in healthy pregnant and nonpregnant women. Am J Physiol Heart Circ Physiol 313, H782–H787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarka A, Vincent JL & van de Borne P (2007). The effects of dopamine on the respiratory system: friend or foe? Pulm Pharmacol Ther 20, 607–615. [DOI] [PubMed] [Google Scholar]
- Dean JB, Mulkey DK, Henderson RA 3rd, Potter SJ & Putnam RW (2004). Hyperoxia, reactive oxygen species, and hyperventilation: oxygen sensitivity of brain stem neurons. J Appl Physiol (1985) 96, 784–791. [DOI] [PubMed] [Google Scholar]
- Dejours P (1962). Chemoreflexes in breathing. Physiol Rev 42, 335–358. [DOI] [PubMed] [Google Scholar]
- Ding Y, Li YL & Schultz HD (2011). Role of blood flow in carotid body chemoreflex function in heart failure. J Physiol 589, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing SE, Remensnyder JP & Mitchel JH (1962). Cardiovascular responses to hypoxic stimulation of the carotid bodies. Circ Res 10, 676–687. [DOI] [PubMed] [Google Scholar]
- Edgell H, McMurtry MS, Haykowsky MJ, Paterson I, Ezekowitz JA, Dyck JR & Stickland MK (2015). Peripheral chemoreceptor control of cardiovascular function at rest and during exercise in heart failure patients. J Appl Physiol (1985) 118, 839–848. [DOI] [PubMed] [Google Scholar]
- Edmunds AT, Godfrey S & Tooley M (1982). Cardiac output measured by transthoracic impedance cardiography at rest, during exercise and at various lung volumes. Clin Sci 63, 107–113. [DOI] [PubMed] [Google Scholar]
- Guyenet PG (2000). Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol 121, 147–162. [DOI] [PubMed] [Google Scholar]
- Hardy JC, Gray K, Whistler S & Leuenberger U (1994). Sympathetic and blood pressure responses to voluntary apnea are augmented by hypoxemia. J Appl Physiol (1985) 77, 2360–2365. [DOI] [PubMed] [Google Scholar]
- Heindl S, Lehnert M, Criee CP, Hasenfuss G & Andreas S (2001). Marked sympathetic activation in patients with chronic respiratory failure. Am J Respir Crit Care Med 164, 597–601. [DOI] [PubMed] [Google Scholar]
- Hoffman B & Lefkowitz R (1990). Catecholamines and sympathomimetic drugs In Goodman and Gilman's The Pharmacological Basis of Therapeutics, 8th edn, ed. Gilman A, Rall T, Nies A, Taylor P, pp. 187–220. Pergamon Press. [Google Scholar]
- Hofford JM, Milakofsky L, Wolfgang HV, Sacher RS, Savage JS & Pell S (1990). The nutritional status in advanced emphysema associated with chronic bronchitis: a study of amino acid and catecholamine levels. Am J Respir Crit Care Med 141, 902–908. [DOI] [PubMed] [Google Scholar]
- Jones NL, Robertson DG & Kane JW (1979). Difference between end‐tidal and arterial PCO2 in exercise. J Appl Physiol Respir Environ Exerc Physiol 47, 954–960. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Nishino T, Mokashi A & Mulligan E (1980). Interaction of dopamine and haloperidol with O2 and CO2 chemoreception in carotid body. J Appl Physiol Respir Environ Exerc Physiol 49, 45–51. [DOI] [PubMed] [Google Scholar]
- Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I & Struijker‐Boudier H; European Network for Non‐invasive Investigation of Large Arteries (2006). Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J 27, 2588–2605. [DOI] [PubMed] [Google Scholar]
- McAllister DA, Maclay JD, Mills NL, Mair G, Miller J, Anderson D, Newby DE, Murchison JT & MacNee W (2007). Arterial stiffness is independently associated with emphysema severity in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176, 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclay J, McAllister DA, Mills NL, Paterson F, Ludlam CA, Drost EM, Newby DE & MacNee W (2009). Vascular dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 180, 513–520. [DOI] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH & Schultz HD (2014). Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol 592, 391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye PC, Hanson MA & Torrance RW (1981). The effect on breathing of abruptly stopping carotid body discharge. Respir Physiol 46, 309–326. [DOI] [PubMed] [Google Scholar]
- O'Rourke MF, Staessen JA, Vlachopoulos C, Duprez D & Plante GE (2002). Clinical applications of arterial stiffness; definitions and reference values. Am J Hypertens 15, 426–444. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner W, Banasiak W, Poole‐Wilson PA, Piepoli MF & Coats AJS (2001). Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation 104, 544–549. [DOI] [PubMed] [Google Scholar]
- Porzionato A, Macchi V, De Caro R & Di Giulio C (2013). Inflammatory and immunomodulatory mechanisms in the carotid body. Respir Physiol Neurobiol 187, 31–40. [DOI] [PubMed] [Google Scholar]
- Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, Enright PL, Hankinson JL, Ip MS, Zheng J & Stocks J; ERS Global Lung Function Initiative (2012). Multi‐ethnic reference values for spirometry for the 3‐95‐yr age range: the global lung function 2012 equations. Eur Respir J 40, 1324–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach T, Bahr F, Herrmann P, Luethje L, Heusser K, Hasenfuss G, Bernardi L & Andreas S (2008). Slow breathing reduces sympathoexcitation in COPD. Eur Respir J 32, 387–392. [DOI] [PubMed] [Google Scholar]
- Read D (1967). A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann Med 16, 20–32. [DOI] [PubMed] [Google Scholar]
- Sabit R, Bolton CE, Edwards PH, Pettit RJ, Evans WD, McEniery CM, Wilkinson IB, Cockcroft JR & Shale DJ (2007). Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 175, 1259–1265. [DOI] [PubMed] [Google Scholar]
- Stickland MK, Fuhr DP, Edgell H, Byers BW, Bhutani M, Wong EY & Steinback CD (2016). Chemosensitivity, cardiovascular risk, and the ventilatory response to exercise in COPD. PLoS One 11, e0158341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickland MK, Fuhr DP, Haykowsky MJ, Jones KE, Paterson DI, Ezekowitz JA & McMurtry MS (2011). Carotid chemoreceptor modulation of blood flow during exercise in healthy humans. J Physiol 589, 6219–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickland MK, Miller JD, Smith CA & Dempsey JA (2007). Carotid chemoreceptor modulation of regional blood flow distribution during exercise in health and chronic heart failure. Circ Res 100, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH & Schultz HD (1999). Enhanced peripheral chemoreflex function in conscious rabbits with pacing‐induced heart failure. J Appl Physiol (1985) 86, 1264–1272. [DOI] [PubMed] [Google Scholar]
- Świerblewska E, Hering D, Kara T, Kunicka K, Kruszewski P, Bieniaszewski L, Boutouyrie P, Somers VK & Narkiewicz K (2010). An independent relationship between muscle sympathetic nerve activity and pulse wave velocity in normal humans. J Hypertens 28, 979–984. [DOI] [PubMed] [Google Scholar]
- van de Borne P, Oren R & Somers VK (1998). Dopamine depresses minute ventilation in patients with heart failure. Circulation 98, 126–131. [DOI] [PubMed] [Google Scholar]
- Vlachopoulos C, Aznaouridis K & Stefanadis C (2010). Prediction of cardiovascular events and all‐cause mortality with arterial stiffness: a systematic review and meta‐analysis. J Am Coll Cardiol 55, 1318–1327. [DOI] [PubMed] [Google Scholar]
- Wouters EFM (2002). Chronic obstructive pulmonary disease. 5: Systemic effects of COPD. Thorax 57, 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV (2008). The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. [DOI] [PubMed] [Google Scholar]