

Abstract
Key points
In anaesthetized rats, acute intermittent hypoxia increases sympathetic nerve activity, sympathetic peripheral chemoreflex sensitivity and central sympathetic–respiratory coupling.
Renin–angiotensin system inhibition prevents the sympathetic effects of intermittent hypoxia, with intermittent injections of angiotensin II into the systemic circulation replicating these effects.
Bilateral carotid body denervation reduces the sympathetic effects of acute intermittent hypoxia and eliminates the increases in chemoreflex sensitivity and sympathetic–respiratory coupling.
Pharmacological inhibition of the subfornical organ also reduces the sympathetic effects of acute intermittent hypoxia, although it has no effect on the increases in chemoreflex sensitivity and central sympathetic–respiratory coupling.
Combining both interventions eliminates the sympathetic effects of both intermittent hypoxia and angiotensin II.
Abstract
Circulating angiotensin II (Ang II) is vital for arterial pressure elevation following intermittent hypoxia in rats, although its importance in the induction of sympathetic changes is unclear. We tested the contribution of the renin–angiotensin system to the effects of acute intermittent hypoxia (AIH) in anaesthetized and ventilated rats. There was a 33.7 ± 2.9% increase in sympathetic nerve activity (SNA), while sympathetic chemoreflex sensitivity and central sympathetic–respiratory coupling increased by one‐fold following AIH. The sympathetic effects of AIH were prevented by blocking angiotensin type 1 receptors with systemic losartan. Intermittent systemic injections of Ang II (Int.Ang II) elicited similar sympathetic responses to AIH. To identify the neural pathways responsible for the effects of AIH and Int.Ang II, we performed bilateral carotid body denervation, which reduced the increase in SNA by 56% and 45%, respectively. Conversely, pharmacological inhibition of the subfornical organ (SFO), an established target of circulating Ang II, reduced the increase in SNA following AIH and Int.Ang II by 65% and 59%, respectively, although it did not prevent the sensitization of the sympathetic peripheral chemoreflex, nor the increase in central sympathetic–respiratory coupling. Combined carotid body denervation and inhibition of the SFO eliminated the enhancement of SNA following AIH and Int.Ang II. Repeated systemic injections of phenylephrine caused an elevation in SNA similar to AIH, and this effect was prevented by a renin inhibitor, aliskiren. Our findings show that the sympathetic effects of AIH are the result of RAS‐mediated activations of the carotid bodies and the SFO.
Keywords: sympathetic nervous system, hypoxia, angiotensin II, carotid body, subfornical organ, renin‐angiotensin system, sleep apnea
Key points
In anaesthetized rats, acute intermittent hypoxia increases sympathetic nerve activity, sympathetic peripheral chemoreflex sensitivity and central sympathetic–respiratory coupling.
Renin–angiotensin system inhibition prevents the sympathetic effects of intermittent hypoxia, with intermittent injections of angiotensin II into the systemic circulation replicating these effects.
Bilateral carotid body denervation reduces the sympathetic effects of acute intermittent hypoxia and eliminates the increases in chemoreflex sensitivity and sympathetic–respiratory coupling.
Pharmacological inhibition of the subfornical organ also reduces the sympathetic effects of acute intermittent hypoxia, although it has no effect on the increases in chemoreflex sensitivity and central sympathetic–respiratory coupling.
Combining both interventions eliminates the sympathetic effects of both intermittent hypoxia and angiotensin II.
Introduction
Chronic elevations in the activity of the sympathetic nerves controlling the cardiovascular system are a pathophysiological characteristic of neurogenic hypertension (Esler, 2010). In patients with obstructive sleep apnoea, there is evidence suggesting that recurrent hypoxemia during sleep contributes to the elevation in daytime sympathetic nerve activity (SNA) and hypertension associated with this condition (Peppard et al. 2000; Taylor et al. 2016). Similarly, adult rodents exposed to chronic intermittent hypoxia (CIH) develop hypertension (Fletcher et al. 1992a, b, 1999; Zoccal et al. 2007; Del Rio et al. 2016) and increased sensitivity of sympathoexcitatory reflexes (Huang et al. 2009; Silva & Schreihofer, 2011). The processes responsible for the effects of intermittent hypoxia on the SNA are not fully understood (Prabhakar et al. 2015; Kim et al. 2016a).
Experimental data suggest that peripheral renin–angiotensin system (RAS) activation and circulating angiotensin II (Ang II) contribute to hypertension in CIH‐treated rodents (Shell et al. 2016). For example, plasma renin is elevated in CIH‐treated rats (Fletcher et al. 1999) and prophylactic treatment with losartan, an angiotensin II type 1 receptor antagonist (AT1R), prevents hypertension caused by CIH (Fletcher et al. 1999; Marcus et al. 2010, 2012). Interestingly, AT1R antagonism does not normalize arterial pressure (AP) once hypertension is established (Zoccal et al. 2007), which suggests that AT1R binding does not support elevated AP in CIH‐treated rats.
Currently, the neural mechanisms by which RAS activation in CIH causes elevated SNA and hypertension remain unclear. Normally, the neural effects of circulating Ang II are mediated largely by neural structures that lie outside the blood–brain barrier (Smith & Ferguson, 2010), notably the carotid bodies (CB) and the subfornical organ (SFO) in the hypothalamus. The CB, which is critical for the cardiorespiratory response to hypoxia (Prabhakar et al. 2015) and contributes to the development of hypertension in CIH‐treated rats (Del Rio et al. 2016), expresses AT1Rs and is activated by Ang II in vitro (Allen, 1998; Peng et al. 2011). The SFO is arguably the most important neural structure for the autonomic and behavioural effects of circulating Ang II, and small interfering RNA knockdown of AT1R in the SFO reduces hypertension in CIH‐treated rats (Saxena et al. 2015).
Studies aiming to identify the mechanisms of the sympathetic changes caused by intermittent hypoxia have used acute intermittent hypoxia (AIH) in anaesthetized adult rats (Dick et al. 2007; Xing & Pilowsky, 2010; Yamamoto et al. 2015; Kim et al. 2016b). In the AIH model, there is a highly reproducible increase in mean SNA in the absence of changes in AP and inconsistent changes in central respiratory drive (Dick et al. 2007; Xing & Pilowsky, 2010). Similar to the CIH model, the underlying processes that lead to elevated SNA in AIH remain unclear. Considering the evidence implicating RAS signalling in cardiovascular effects of CIH, we examined the contribution of RAS signalling to the sympathetic effects of AIH, and subsequently, determined the contribution of the CB and the SFO to the sympathetic effects of AIH.
Methods
Ethical approval
All experimental procedures were executed in strict accordance with the guidelines set in the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, endorsed by the National Health and Medical Research Council of Australia. The present study was approved by the Sydney Local Health District Animal Welfare Committee (2015‐013). All experimental procedures conform to the principles and regulations as described in Grundy (2015).
Subjects and preparation
Male Sprague–Dawley rats (n = 114; 250–400 g) were acquired from the Animal Resource Centre (Canning Vale, WA, Australia) and were housed with ad libitum water and standard rodent chow at the Heart Research Institute Biological Facilities prior to the experiments. On the day of the experiment, rats were anaesthetized with urethane (1.3 g kg−1 i.p., with additional doses of 30 mg in 10% solution as required). Absences in the hind‐paw withdrawal reflex and the corneal reflex were used to confirm adequate anaesthesia beginning the surgery. Core temperature was maintained throughout the surgery and experimental procedures using a servo‐controlled heating pad set at 37.0 ± 0.5°C (CWE, Inc., Ardmore, PA, USA). Following completion of experiments, rats were given an additional 60 mg of urethane i.v. followed by 1 mL of 3 m potassium chloride to stop the heart.
Surgical procedures
The right carotid artery and jugular vein were cannulated using polyethylene tubing (PE50; Microtube Extrusions, North Rocks, NSW, Australia) for recording arterial blood pressure and infusing drugs and fluids, respectively. An ECG was recorded from leads implanted in the forepaws to determine heart rate. A tracheostomy was performed and a 14‐gauge cannula was sutured into the trachea to allow connection to a ventilator (Ugo Basile, Varese, Italy). Rats were artificially ventilated with medical oxygen (ventilation parameters: 70 cycles min–1, 1 mL 100 g–1). Expired CO2 was monitored (CapStar 100 CO2 analyser; CWE Inc.) and arterial blood gases and electrolytes (VetStat; IDEXX Laboratories, Westbrook, ME, USA) were measured intermittently (Tables 1 and 2). Ventilation was adjusted to maintain a constant expired CO2 and a P aCO2 of 40.0 ± 5 mmHg and a pH between 7.35 and 7.45.
Table 1.
Values for sympathetic nerve activity, mean arterial pressure and heart rate at before (i.e. baseline) and after the termination of the protocol (i.e. 60 min after) recording
| SNA (μV) | MAP (mmHg) | HR (beats min–1) | ||||
|---|---|---|---|---|---|---|
| Treatment groups | n | Baseline | Baseline | Post | Baseline | Post |
| Time control | 5 | 3.55 ± 0.36 | 99 ± 6 | 97 ± 6 | 457 ± 18 | 449 ± 17 |
| Losartan control | 4 | 3.44 ± 0.57 | 89 ± 6 | 88 ± 9 | 477 ± 12 | 467 ± 12 |
| AIH + Saline | 5 | 3.34 ± 0.25 | 101 ± 9 | 107 ± 8 | 446 ± 10 | 463 ± 11 |
| AIH + Losartan | 5 | 3.43 ± 0.24 | 88 ± 11 | 90 ± 6 | 439 ± 18 | 437 ± 9 |
| AIH + Aliskiren | 4 | 3.98 ± 0.50 | 98 ± 4 | 95 ± 4 | 431 ± 10 | 427 ± 8 |
| Int.Saline | 4 | 3.21 ± 0.11 | 110 ± 8 | 109 ± 4 | 436 ± 3 | 436 ± 7 |
| Int.Ang II | 5 | 3.18 ± 0.15 | 100 ± 5 | 110 ± 4 | 433 ± 5 | 448 ± 7 |
| Int.Ang II + Losartan | 4 | 3.25 ± 0.46 | 90 ± 5 | 91 ± 7 | 441 ± 16 | 432 ± 11 |
| Single Ang II | 4 | 3.17 ± 0.19 | 102 ± 5 | 105 ± 6 | 414 ± 19 | 414 ± 20 |
| Continuous Ang II | 4 | 3.17 ± 0.18 | 99 ± 8 | 101 ± 5 | 429 ± 7 | 432 ± 11 |
| AIH + Sham | 4 | 3.34 ± 0.40 | 101 ± 8 | 105 ± 6 | 463 ± 8 | 468 ± 11 |
| AIH + CBD | 6 | 3.21 ± 0.34 | 96 ± 4 | 99 ± 5 | 486 ± 7 | 476 ± 10 |
| AIH + SFOvehicle | 4 | 3.49 ± 0.33 | 92 ± 8 | 85 ± 9 | 439 ± 8 | 444 ± 10 |
| AIH + SFOmiss | 4 | 3.19 ± 0.13 | 102 ± 1 | 96 ± 3 | 471 ± 7 | 476 ± 10 |
| AIH + SFOiso | 6 | 3.28 ± 0.25 | 112 ± 5 | 105 ± 7 | 472 ± 5 | 471 ± 8 |
| AIH + CBD∙SFOiso | 6 | 3.71 ± 0.48 | 96 ± 3 | 91 ± 4 | 478 ± 7 | 460 ± 10 |
| Int.Ang II + Sham | 6 | 3.00 ± 0.27 | 104 ± 6 | 106 ± 5 | 450 ± 20 | 449 ± 18 |
| Int.Ang II + CBD | 5 | 3.50 ± 0.27 | 110 ± 3 | 114 ± 4 | 458 ± 10 | 461 ± 11 |
| Int.Ang II + SFOiso | 6 | 4.10 ± 0.35 | 102 ± 4 | 100 ± 5 | 484 ± 10 | 479 ± 11 |
| Int.Ang II + CBD∙SFOiso | 4 | 3.80 ± 0.54 | 91 ± 6 | 92 ± 5 | 464 ± 9 | 468 ± 13 |
| Int.Phenylephrine | 5 | 3.32 ± 0.41 | 101 ± 8 | 101 ± 13 | 426 ± 8 | 427 ± 3 |
| Int.Phenylephrine + Aliskiren | 4 | 3.10 ± 0.17 | 108 ± 6 | 112 ± 6 | 431 ± 6 | 428 ± 4 |
| Continuous phenylephrine | 4 | 3.21 ± 0.13 | 103 ± 7 | 102 ± 8 | 410 ± 3 | 407 ± 6 |
| Int.Sodium nitroprusside | 6 | 3.31 ± 0.26 | 115 ± 6 | 115 ± 7 | 433 ± 5 | 432 ± 8 |
Values are in mean ± SEM. One‐way ANOVA with Holm–Šidák correction was used for multiple comparisons between groups for baseline SNA, MAP and HR values. Two‐way ANOVA with Holm–Šidák correction was used for multiple comparisons between baseline and post‐protocol values for MAP and HR values. There were no differences between all comparisons.
Table 2.
Values for pulmonary carbon dioxide (), sodium (Na+), potassium (K+) and chloride (Cl−) levels prior to the start of the protocol (i.e. baseline conditions) and after the termination of the protocol (i.e. 60 min after recording)
| (mmHg) | Na+ (mmol L−1) | K+ (mmol L−1) | Cl− (mmol L−1) | |||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Post | Baseline | Post | Baseline | Post | Baseline | Post | |
| Hypoxia groups | ||||||||
| Time control | 43.0 ± 1.0 | 42.4 ± 0.5 | 145.4 ± 1.5 | 143.6 ± 1.5 | 4.2 ± 0.2 | 4.2 ± 0.2 | 112.4 ± 1.2 | 111.8 ± 1.0 |
| Losartan control | 42.3 ± 1.2 | 42.8 ± 0.9 | 142.0 ± 1.1 | 142.8 ± 1.3 | 4.1 ± 0.2 | 4.1 ± 0.1 | 110.5 ± 0.9 | 111.3 ± 1.1 |
| AIH + Saline | 41.2 ± 0.7 | 42.8 ± 0.7 | 142.2 ± 0.7 | 140.6 ± 0.7 | 3.9 ± 0.4 | 3.9 ± 0.3 | 109.4 ± 0.5 | 110.2 ± 0.4 |
| AIH + Losartan | 40.0 ± 0.7 | 41.0 ± 1.0 | 140.8 ± 0.9 | 141.2 ± 1.0 | 4.3 ± 0.2 | 4.2 ± 0.1 | 111.4 ± 1.2 | 112.8 ± 0.8 |
| AIH + Aliskiren | 41.5 ± 1.7 | 40.8 ± 1.6 | 142.0 ± 1.5 | 141.3 ± 0.8 | 3.8 ± 0.2 | 3.7 ± 0.1 | 109.5 ± 0.6 | 110.5 ± 0.9 |
| AIH + Sham | 41.3 ± 1.5 | 41.8 ± 1.4 | 141.3 ± 0.9 | 141.4 ± 0.7 | 3.7 ± 0.2 | 3.7 ± 0.2 | 110.0 ± 1.2 | 111.7 ± 1.2 |
| AIH + CBD | 40.3 ± 0.9 | 41.3 ± 1.3 | 139.8 ± 1.0 | 140.1 ± 0.7 | 4.2 ± 0.1 | 4.2 ± 0.1 | 112.0 ± 1.3 | 111.3 ± 0.6 |
| AIH + SFOvehicle | 41.0 ± 1.6 | 42.0 ± 0.9 | 139.3 ± 1.5 | 139.3 ± 0.5 | 4.1 ± 0.2 | 4.1 ± 0.2 | 109.0 ± 0.9 | 109.5 ± 0.9 |
| AIH + SFOmiss | 41.4 ± 1.4 | 41.0 ± 1.5 | 140.8 ± 1.1 | 141.2 ± 0.7 | 4.0 ± 0.1 | 4.0 ± 0.1 | 112.2 ± 1.0 | 111.4 ± 0.4 |
| AIH + SFOiso | 39.0 ± 1.5 | 40.5 ± 0.8 | 139.4 ± 1.5 | 140.3 ± 0.9 | 4.1 ± 0.2 | 4.2 ± 0.1 | 111.4 ± 1.4 | 111.0 ± 0.7 |
| AIH + CBD∙SFOiso | 41.3 ± 1.5 | 42.3 ± 0.7 | 141.2 ± 1.0 | 143.2 ± 0.9 | 4.1 ± 0.2 | 4.1 ± 0.1 | 110.2 ± 1.0 | 109.5 ± 1.0 |
| Drug treatment groups | ||||||||
| Int.Saline | 41.5 ± 1.7 | 40.8 ± 1.5 | 140.8 ± 0.8 | 140.3 ± 0.3 | 4.1 ± 0.2 | 4.1 ± 0.5 | 111.3 ± 1.7 | 110.3 ± 0.5 |
| Int.Ang II | 41.4 ± 1.1 | 40.4 ± 0.5 | 143.8 ± 2.4 | 141.6 ± 1.4 | 3.7 ± 0.5 | 3.7 ± 0.3 | 114.4 ± 2.8 | 111.6 ± 1.6 |
| Int.Ang II + Losartan | 42.3 ± 1.3 | 41.0 ± 0.7 | 145.3 ± 2.4 | 144.0 ± 1.5 | 3.6 ± 0.6 | 3.6 ± 0.3 | 111.5 ± 2.1 | 111.8 ± 1.5 |
| Int.Ang II + Sham | 40.6 ± 0.9 | 39.8 ± 0.6 | 141.8 ± 0.8 | 141.4 ± 0.6 | 4.0 ± 0.3 | 4.1 ± 0.2 | 108.6 ± 1.5 | 108.6 ± 1.4 |
| Int.Ang II + CBD | 40.8 ± 1.6 | 40.8 ± 0.7 | 141.2 ± 1.2 | 141.6 ± 0.7 | 4.0 ± 0.3 | 4.1 ± 0.2 | 111.8 ± 2.0 | 111.6 ± 1.3 |
| Int.Ang II + SFOiso | 39.3 ± 1.1 | 39.8 ± 0.9 | 142.0 ± 0.7 | 141.5 ± 0.6 | 4.4 ± 0.0 | 4.4 ± 0.0 | 110.3 ± 1.1 | 109.8 ± 0.9 |
| Int.Ang II + CBD∙SFOiso | 40.5 ± 0.9 | 40.5 ± 0.8 | 142.0 ± 1.4 | 141.3 ± 1.1 | 4.0 ± 0.2 | 4.0 ± 0.1 | 110.8 ± 1.3 | 111.3 ± 0.9 |
| Int.Phenylephrine | 40.7 ± 1.8 | 39.7 ± 0.9 | 139.3 ± 2.2 | 140.7 ± 0.3 | 4.4 ± 0.2 | 4.3 ± 0.0 | 109.0 ± 3.8 | 110.3 ± 2.9 |
| Int.Phenylephrine + Aliskiren | 42.7 ± 0.9 | 42.7 ± 0.6 | 139.0 ± 2.5 | 139.8 ± 2.4 | 4.5 ± 0.1 | 4.5 ± 0.1 | 111.3 ± 0.3 | 111.7 ± 0.5 |
| Int.Sodium nitroprusside | 40.0 ± 2.5 | 40.0 ± 1.3 | 142.3 ± 0.3 | 142.2 ± 0.5 | 4.4 ± 0.2 | 4.4 ± 0.2 | 110.7 ± 1.8 | 109.6 ± 1.9 |
Values are in mean ± SEM. One‐way ANOVA with Holm–Šidák correction was used for multiple comparisons between groups for baseline pulmonary carbon dioxide (), sodium (Na+), potassium (K+) and chloride (Cl−) levels. Two‐way ANOVA with Holm–Šidák correction was used for multiple comparisons between baseline and post‐protocol values for values. There were no differences between all comparisons.
Following ventilation, rats were bilaterally vagotomized and paralysed using pancuronium bromide (0.4 mg administered as a 0.2 ml bolus i.v. injection, followed by an infusion of 10% pancuronium bromide diluted in 0.9% saline, infused at a rate of 2 mL h−1; AstraZeneca, London, UK). Following paralysis, the depth of anaesthesia was determined by observing MAP changes in response to a strong tail pinch stimulus. Additional doses of urethane (30 mg in 10% solution) were delivered i.v. in cases where mean arterial pressure (MAP) increased or decreased by more than 10 mmHg. A retroperitoneal approach was used to access the left greater splanchnic nerve at a site proximal to the coeliac ganglion and a dorsal approach was used to access the left phrenic nerve. Nerve activities were recorded using bipolar silver electrodes.
Bilateral CB denervation (CBD)
To access the CB, the sternohyoid and sternocleidomastoid muscles were carefully retracted. The carotid bifurcations were exposed on both sides, the CB identified and the carotid sinus nerve was resected. Sham‐operated rats underwent the same surgical procedures to expose the CB, although the carotid sinus nerves were left intact.
SFO microinjection
A midline incision was made through the skin of the rat skull and a craniotomy was performed to provide access to the SFO (co‐ordinates: 1.35 mm caudal of bregma, midline, 4.2–4.3 mm ventral from the surface of the brain). To preserve the superior sagittal venous sinus, the initial penetration of the micropipette was performed 0.5 mm lateral of midline and advanced 3.0 mm ventrally before being shifted back to the midline and then advanced to the final target of the SFO. To inhibit the SFO, we injected the GABAa receptor agonist, isoguvacine (10 mm in PBS) (Sigma‐Aldrich, Castle Hill, NSW, Australia), 10 min prior to the start of AIH or intermittent Ang II and then again 1 h later after the completion of the protocol. Two injections were performed to ensure that the SFO was inhibited for the duration of the experiment. The injection volume was 100 nL per injection and the site of injection was marked by adding 1% Chicago Blue dye (Sigma‐Aldrich) to the injectate. PBS injections were performed in control groups for the effects of microinjection. Following these experiments, the brain was extracted and fixed in 10% neutral‐buffered formalin (Sigma‐Aldrich) for at least 72 h. The brain was coronally sectioned (80 μm), mounted on slides, and cover slipped before being examined to confirm the correct placement of injections in the SFO.
Gas delivery
Prior to the commencement of the AIH protocol and in between periods of hypoxia, rats were ventilated with 100% O2. The protocol for AIH was adopted from previous studies (Xing & Pilowsky, 2010; Kim et al. 2016b). Rats were subjected to 10 cycles of hypoxia lasting 45 s with 5 min of recovery between hypoxia cycles breathing 100% oxygen. The hypoxia gas consisted of 10% oxygen balanced with nitrogen (Coregas, Yennora, NSW, Australia). All gases were administered through the ventilator's passive air intake.
Drug administration
Intermittent (Int.) i.v. injections of Ang II (35 pmol per injection; Abcam, Melbourne, VIC, Australia), phenylephrine‐hydrochloride (2.5 μg per injection; Sigma‐Aldrich) and sodium nitroprusside‐dihydrate (2.5 μg per injection; Sigma‐Aldrich) were administered in 10 boluses of 0.1 mL, delivered with 5 min recovery intervals. Both Ang II and phenylephrine (350 pmol and 25 μg in 0.1 ml saline, respectively) were delivered as a single bolus or as an infusion over 10 min as dose and injection control groups, respectively. In experiments involving pre‐treatment with losartan‐potassium (2 mg kg−1; Sapphire Bioscience, Redfern, NSW, Australia) or Aliskiren‐Hemifumarate (1 mg kg−1; Selleckchem, Scoresby, VIC, Australia), drugs were delivered as an i.v. bolus 10 min before the initiation of AIH or Int.Ang II. To control for the potential changes in basal SNA as a result of changes in the depth of anaesthesia or pre‐treatment with Losartan or Aliskiren, we performed ‘time control’, ‘vehicle control’, and ‘pre‐treatment drug control’ experiments where rats were subjected to no intervention, vehicle injections (saline for i.v.; PBS for brain) or drug pre‐treatment alone, respectively.
Data acquisition and analysis
Data were obtained using an ADC system (CED 1401; Cambridge Electronic Design, Cambridge, UK) and acquisition and analysis software (Spike 2, version 8.07; Cambridge Electronic Design). The neurograms were amplified 10‐fold by a very low‐noise preamplifier (CWE, Inc.), band‐pass filtered (0.1–3 kHz) and then amplified a further 1000 times for phrenic nerve activity (PNA) and 2000 times for splanchnic nerve activity by a scaling amplifier (BMA‐400 AC/DC Bioamplifier; CWE Inc.). The analogue signal was then digitized (A/D converter 1401; Cambridge Electronic Design, Cambridge) and sampled at 5 kHz.
Splanchnic SNA
For analysis, splanchnic SNA was rectified and smoothed (τ = 1 s) and the level of SNA following death was subtracted from all values. In all experiments, SNA was evaluated 2 h from the protocol onset. Values of SNA in text refer to the mean activity over a 1 min interval taken immediately before AIH or Int.Ang II (baseline) and then again 2 h later (i.e. 60 min after the completion of AIH or Int.Ang II). The final value in the text refers to the change in SNA normalized to the baseline period calculated as SNA in μV (A) baseline before AIH or Int.Ang II and (B) 60 min after AIH or Int.Ang II:
Sympathetic chemoreflex response
The sympathetic chemoreflex response was calculated as the area under the curve (AUC) of the sympathetic response to hypoxia (45 s of 10% O2 balanced in N2) above the resting level of SNA prior to any intervention and again at the end of a 2 h recording period. As with basal SNA, we normalized the sympathetic chemoreflex to the response observed prior to any treatment. The equation used is:
PNA
PNA was rectified and smoothed (τ = 0.05 s). Phrenic nerve peak amplitude on the waveform across 10 respiratory bursts and frequency across this period were all measured at baseline before AIH or Int.Ang II protocol and 60 min after the termination of the protocol. Values for MAP and heart rate (HR) (mean value across a 1 min interval) were recorded at baseline and 60 min after the protocol to observe for any changes. If the antagonist was used, the baseline was taken following the injection of treatment.
Respiratory modulation of SNA
Respiratory modulation of SNA was assessed using cycle‐triggered averages of SNA triggered from the onset of integrated phrenic nerve bursts (minimum of 10 cycles). Under hyperoxic normocapnic conditions in this preparation, the most consistent pattern of respiratory–sympathetic coupling that we observed was a post‐inspiratory peak followed by a trough during the expiratory phase. We measured the difference between the post‐inspiratory peak and following trough under hyperoxic conditions as an index of respiratory modulation. We also measured PNA amplitude to assess central respiratory drive. These values were acquired prior to and 1 h after the AIH protocol and expressed as a percentage change from baseline.
Statistical analysis
Statistical analyses were performed using Prism version 7 (GraphPad Software, San Diego, CA, USA). Following tests for normality (Shapiro–Wilk test), statistical significance was determined by either an unpaired Student's t test where necessary and one‐way analysis of variance using a Holm–Šidák post hoc test with correction for multiple comparisons. Two‐way analysis of variance using a Holm‐Šidák post hoc test with correction for multiple comparisons was used where stated. P < 0.05 was considered statistically significant for differences. Grouped data are presented as the mean ± SEM unless otherwise noted.
Results
Blood gases, electrolytes and AP remain unchanged for all experimental conditions
Variables for AP and HR, and blood , pH and electrolytes were unchanged by AIH or any other procedures used in the present study (Tables 1 and 2). Oxygen saturation was measured during a 45 s hypoxia challenge (n = 12 challenges). Rats were hyperoxic at rest and reached an oxygen saturation nadir of 87.3 ± 0.7% during hypoxia trials (Table 3).
Table 3.
Values for , plasma haemoglobin and levels at baseline, during acute hypoxia and following re‐oxygenation
| Baseline | Hypoxia | Re‐oxygenation | |
|---|---|---|---|
| (mmHg) | 482.2 ± 11.8 | 67.6 ± 2.3**** | 475.6 ± 8.1†††† |
| Plasma haemoglobin (g dL–1) | 21.0 ± 0.4 | 24.2 ± 0.3**** | 20.7 ± 0.2†††† |
| (%) | 100.0 ± 0.0 | 87.3 ± 0.7**** | 100.0 ± 0.0†††† |
Values are the mean ± SEM. One‐way ANOVA with Holm–Šidák correction was used for multiple comparisons.
**** P < 0.0001 vs. baseline
†††† P < 0.0001 vs. hypoxia (n = 12)
Increased SNA and peripheral chemoreflex sensitivity following AIH are dependent on AT1Rs and renin activity
To evaluate the involvement of AT1Rs in the elevation of SNA following AIH, rats were administered either saline or losartan 10 min before the AIH protocol (Fig. 1 A and B). Rats pre‐treated with saline exhibited a significant increase in mean SNA 60 min after AIH (AIH + Saline vs. Time control, 33.7 ± 2.9% vs. 2.6 ± 2.2%; P < 0.0001) (Fig. 1 C), which was completely prevented by pre‐treating rats with losartan (−0.9 ± 4.1%; P < 0.0001 vs. AIH + Saline) (Fig. 1 A–C). Furthermore, the sympathetic response to hypoxia was approximately doubled following AIH in saline‐treated rats (AIH + Saline vs. Time control, 114.0 ± 19.7% vs. 11.5 ± 5.4%; P = 0.0003) (Fig. 1 D and E) and this was also completely prevented by pre‐treating rats with losartan (11.4 ± 10.4%; P = 0.0003 vs. AIH + Saline) (Fig. 1 D and E). For a more direct confirmation of the sympathetic effects of AIH being mediated by the RAS, we used aliskiren (1 mg kg−1), a drug that binds to renin preventing the conversion of angiotensinogen to angiotensin I, to inhibit the RAS. Administration of aliskiren fully abolished the elevation in SNA (−0.8 ± 4.6%; P = 0.0003 vs. AIH + Saline) (Fig. 1 F and G) and sympathetic chemoreflex sensitivity (6.0 ± 9.3%; P = 0.0027 vs. AIH + Saline) (Fig. 1 H) following AIH.
Figure 1. Enhancement of SNA following AIH is mediated by the RAS.
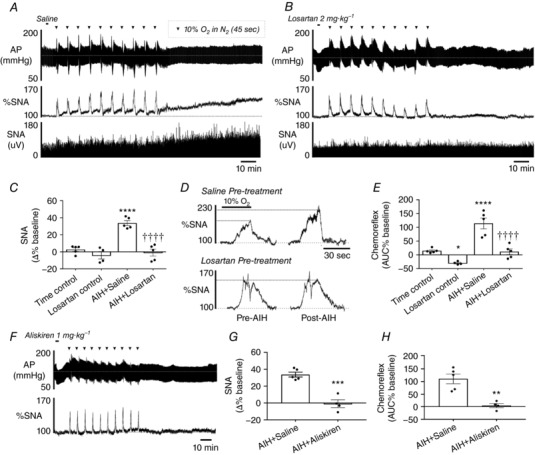
Effect of AIH on AP and SNA in (A) saline‐ and (B) losartan‐treated rats. C, group data for the change in SNA 60 min post‐AIH (**** P < 0.0001 vs. Time control; †††† P < 0.0001 vs. AIH + Saline). D, effect of losartan on the changes in the acute sympathetic chemoreflex caused by AIH. E, group data for the change in acute sympathetic chemoreflex (**** P < 0.0001 vs. Time control; †††† P < 0.0001 vs. AIH + Saline). F, effects of AIH on AP and SNA in a rat pre‐treated with aliskiren. G, group data for the change in SNA 60 min post‐AIH in aliskiren treated rats (*** P < 0.001 vs. AIH + Saline). H, grouped data of the effect of aliskiren on the changes in the acute sympathetic chemoreflex caused by AIH (** P < 0.01 vs. AIH + Saline).
Intermittent systemic injections of Ang II increases SNA and peripheral chemoreflex sensitivity
Next, we examined the effect of intermittent i.v. injections of Ang II delivered in a pattern replicating AIH (Int.Ang II; 35 pmol in 0.1 ml repeated 10 times at 5 min intervals). Int.Ang II produced an increase in mean SNA that was not observed when saline was administered in the same fashion (Int.Ang II vs. Int.Saline, 31.4 ± 5.7% vs 1.2 ± 2.7%; P = 0.0003) (Fig. 2 A and B). The overall kinetic and magnitude of the elevation in SNA after Int.Ang II was comparable to AIH (compare Figs 1 A and C and 2 A and B), whereas AP was unchanged from baseline (Table 1). Similar to AIH, Int.Ang II potentiated the sympathetic response to hypoxia (93.1 ± 13.1%; P < 0.0001 vs. Int.Saline) (Fig. 2 C and D). Similar to AIH, pre‐treating rats with losartan prior to Int.Ang II prevented the elevation in mean SNA (0.8 ± 3.7%; P = 0.0003 vs. Int.Ang II) (Fig. 2 A and B) and the sensitization of the sympathetic response to hypoxia (6.7 ± 10.9%; P < 0.0001 vs. Int.Ang II) (Fig. 2 C and D). Finally, an equivalent dose of Ang II (i.e. 350 pmol) given as a single injection or infused over a 10 min period had no effect on mean SNA (0.9 ± 2.6% and −0.9 ± 3.0%, respectively, n = 4 for both; P < 0.001 vs. Int.Ang II for both) (Fig. 2 B).
Figure 2. Intermittent i.v. injections of Ang II causes AT1R‐dependent SNA facilitation.
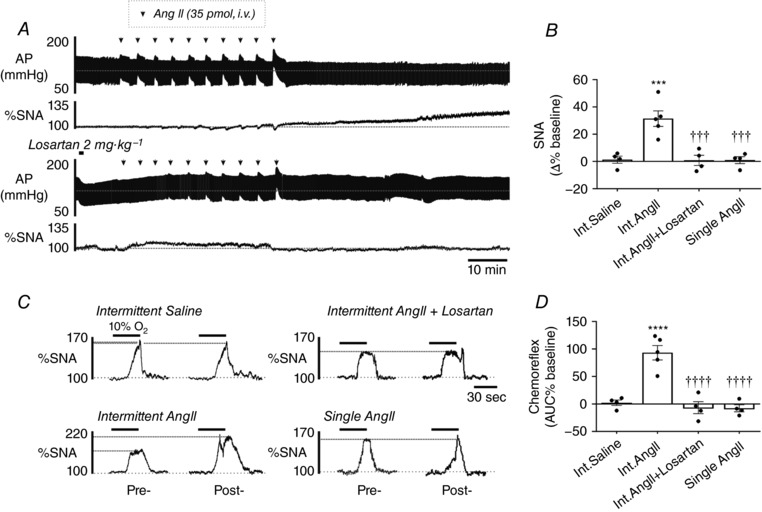
A, effects of intermittent i.v. injections of Ang II (Int.Ang II) on AP and SNA in rats untreated (upper trace) or pre‐treated (lower trace) with losartan. B, group data of the change in SNA 60 min after Int.Ang II (*** P < 0.001 vs. intermittent saline (Int.Saline), ††† P < 0.001 vs. Int.Ang II). C, changes in the acute sympathetic chemoreflex caused by Ang II and saline injection paradigms. D, group data for the change in acute sympathetic chemoreflex (**** P < 0.0001 vs. Int.Saline; †††† P < 0.0001 vs. Int.Ang II).
Confirmation of successful CBD and SFO microinjections
CBD was confirmed by the absence of PNA (49.4 ± 7.1% vs. 1.5 ± 1.7%; P = 0.0006, Sham vs. CBD), phrenic frequency (58.3 ± 9.9% vs. 1.4 ± 2.1%; P = 0.0013, Sham vs. CBD) and SNA (65.0 ± 5.4 vs. 12.2 ± 1.3%; P < 0.0001, Sham vs. CBD, n = 10 for Sham and n = 11 for CBD) responses to hypoxia (Fig. 3 Aa–Ad). Baroreflex function was confirmed by the presence of SNA fluctuations associated with pulsatile blood pressure (0.50 ± 0.1 μV vs. 0.59 ± 0.2 μV; P = 0.6881, Sham vs. CBD, n = 10 for Sham and n = 11 for CBD) (Fig. 3 Ba and Bb). Microinjections of the GABA receptor agonist isoguvacine or saline mixed 1% Chicago Blue dye (n = 22 with isoguvacine and n = 10 with saline) resulted in injection sites that covered the rostrocaudal extent of the SFO and included the ventral hippocampal commissure and, in some cases, the corpus callosum (example of the injection site shown in Fig. 3 C). We did not observe dye labelling in the median preoptic nucleus or dorsal thalamic nuclei below the fourth ventricle. In the course of establishing accurate stereotaxic co‐ordinates for the SFO, we performed injections in the fourth ventricle as indicated by the absence of a discrete injection spot and diffuse light staining of ependymal cells lining the third and fourth ventricle and the choroid plexus. We grouped these cases separately (SFOmiss) from microinjections that successfully targeted the SFO.
Figure 3. Confirmation of CBD and microinjection site in the SFO.
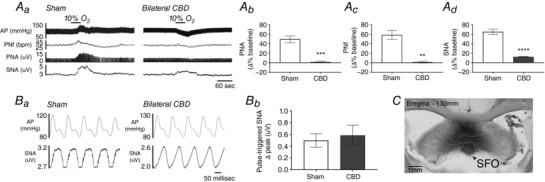
Aa, a 45 s hypoxia challenge on AP, PNf, PNA and SNA in a Sham‐operated (left) and CB denervated (right) rat. Grouped data quantifying the percentage change from baseline for (Ab) PNA, (Ac) PNf and (Ad) SNA in Sham‐operated vs. CB denervated rat (**** P < 0.0001; *** P < 0.001, ** P < 0.01 vs. Sham). Ba, baroreflex modulation of SNA in Sham and CBD rats. Bb, grouped data showing SNA fluctuation in Sham and CBD rats. C, an example of a successful injection in the SFO marked with Chicago blue dye in a coronal section of brain at Bregma level −1.33 mm. Scale bar = 1 mm.
Elevation of SNA following AIH requires functional CBs and SFO
Following CBD, the elevation in mean SNA following AIH was around half the magnitude of Sham‐operated rats (14.0 ± 1.2% vs. 32.2 ± 3.2%, CBD vs. Sham; P < 0.001) (Fig. 4 A and B). Conversely, inhibition of the SFO with the GABAA agonist, isoguvacine (SFOiso), resulted in a significantly smaller increase in mean SNA following AIH compared to rats injected with vehicle and then subject to AIH (SFOvehicle vs SFOiso; 17.0 ± 1.8% vs. 48.1 ± 3.3%; P < 0.0001) (Fig. 4 A and C). Inhibition of the SFO did not abrogate the enhanced sympathetic response to hypoxia observed in AIH‐treated rats (SFOvehicle vs SFOiso; 106.8 ± 15.7% vs. 108.0 ± 13.2%) (Fig. 4 D). When injections of isoguvacine were placed in the fourth ventricle (SFOmiss), we observed elevations in mean SNA following AIH (42.4 ± 5.8%) equivalent to rats injected with vehicle prior to AIH (P = 0.3101) (Fig. 4 C). Notably, mean SNA following AIH in rats with CBD or SFO inhibition was still significantly elevated compared to rats not subjected to AIH (AIH + CBD vs. Time control; P = 0.0209; AIH + SFOiso vs. Time control; P = 0.0059) (Fig. 4 C). However, combining CBD and SFO inhibition eliminated the elevation in mean SNA following AIH (5.9 ± 3.0%, P = 0.4493 vs. Time control) (Fig. 4 A and C).
Figure 4. SNA elevation caused by AIH requires the CBs and SFO.
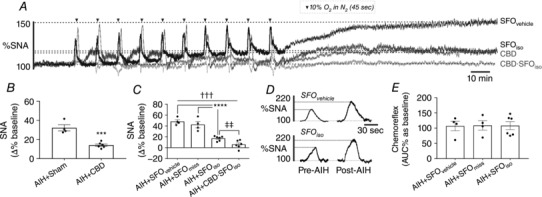
A, overlaid SNA from animals subjected to AIH with SFO microinjections of vehicle (SFOvehicle), isoguvacine (SFOiso), CBD, or combined CBD and SFO inhibition (CBD∙SFOiso). B, group data for the change in SNA 60 min post‐AIH in Sham‐operated and CBD rats (*** P < 0.001 vs. AIH + Sham). C, group data for the change in SNA 60 min post‐AIH in rats with vehicle and isoguvacine injected in the SFO (including cases in which the SFO was not successfully targeted, AIH + SFOmiss) and rats with CBD and injections of isoguvacine (**** P < 0.0001; ††† P < 0.001; ‡‡ P < 0.01). D, example of acute sympathetic chemoreflex before and after AIH in SFOvehicle (upper) and SFOiso (lower) rat. E, group data for the change in acute sympathetic chemoreflex.
Intermittent Ang II increases SNA via the CBs and the SFO pathways
Because the sympathetic effects of AIH require the CBs and the SFO, we speculated that the sympathetic effects of Int.Ang II may utilize similar pathways. We found that CBD reduced the elevation in SNA following Int.Ang II by 45% (CBD vs. Sham; 16.3 ± 1.9% vs. 28.8 ± 2.1%; P = 0.0001) (Fig. 5) and inhibition of the SFO in CB intact rats reduced the elevation in SNA following Int.Ang II by 59% (12.1 ± 1.0%, P < 0.0001 vs. Sham, also microinjected with PBS) (Fig. 5). Combined CBD and SFO inhibition in a separate series of rats fully eliminated the elevation in mean SNA caused by Int.Ang II (1.8 ± 0.5%; P < 0.0001 vs. CBD; P = 0.001 vs. SFOiso) (Fig. 5).
Figure 5. SNA elevation caused by Int.Ang II requires CBs and the SFO.
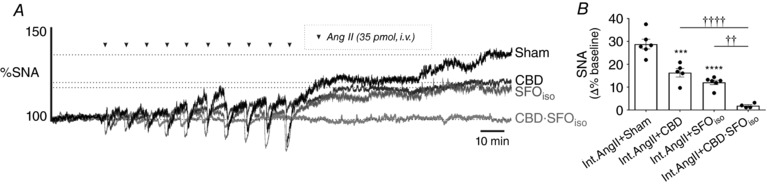
A, overlaid SNA from animals subjected to Int.Ang II that underwent Sham operation (Sham), CBD, SFO inhibition with isoguvacine (SFOiso), or combined CBD and SFO inhibition (CBD∙SFOiso). B, group data for the change in SNA 60 min post‐Int.Ang II (**** P < 0.0001 vs. Sham; *** P < 0.001 vs. Sham; †††† P < 0.0001; †† P < 0.01 vs. CBD∙SFOiso).
Enhanced central sympathetic–respiratory coupling following AIH requires AT1R and functional CBs but not the SFO
Previous studies have demonstrated that AIH increases the coupling between central respiratory drive and SNA (Dick et al. 2007; Xing & Pilowsky, 2010). Consistent with these studies, we found that AIH approximately doubles central sympathetic–respiratory coupling in rats pre‐treated with saline (98.6 ± 24.0% increase in modulation) (Fig. 6 A and B). By contrast, AIH did not increase central sympathetic–respiratory coupling in rats pre‐treated with losartan (7.0 ± 15.2%; P = 0.0017 vs. AIH + Saline) or after CBD (–3.3 ± 7.7%; P = 0.0027 vs. Sham). Inhibition of the SFO did not prevent the increased central sympathetic–respiratory coupling following AIH (83.5 ± 12.6%; P = 0.6826 vs. AIH + S) (Fig. 6 B). Phrenic amplitude was also compared between groups; AIH did not produce a noticeable change in phrenic amplitude (13.2 ± 2.3% vs. 19.3 ± 3.9%, Time control vs. AIH + Saline; P > 0.9999), whereas antagonizing AT1Rs caused a substantial reduction in phrenic nerve amplitude (19.3 ± 3.9% vs. −38.4 ± 10.5%, AIH + Saline vs. AIH + Losartan; P = 0.0009), with all other interventions having no discernible effects (Fig. 6 C).
Figure 6. Enhanced central sympathetic–respiratory coupling following AIH is dependent on AT1Rs and the CBs.
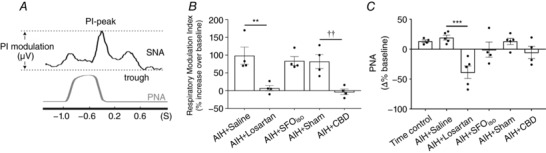
A, method for determining the respiratory modulation index. B, grouped data for the change in the respiratory modulation index 60 min after AIH (** P < 0.01 vs. AIH + Saline; †† P < 0.01 vs. Sham). C, change in PNA 60 min after AIH (*** P < 0.001 vs. AIH + Saline).
Intermittent injections of phenylephrine systemically cause SNA to enhance, which is renin‐dependent
One of the effects of acute hypoxia and systemic Ang II in CB intact rats is peripheral vasoconstriction. A transient increase in AP during acute hypoxia may cause restricted blood flow into the kidney, triggering the release of renin. To mimic this effect, we administered i.v. phenylephrine in a pattern matching AIH in CB intact rats (Int.Phenylephrine, 2.5 μg in 0.1 ml repeated 10 times at 5 min intervals). Phenylephrine caused a transient elevation in AP that was similar in magnitude to acute hypoxia and Ang II (Hypoxia vs. Ang II vs. Phenylephrine; 30 ± 5 mmHg vs. 24 ± 2 mmHg vs. 25 ± 6 mmHg respectively; P > 0.5 for all comparisons) (Fig. 7 Aa and Ab). As hypothesized, Int.Phenylephrine produced a progressive increase in mean SNA that had a similar kinetic and magnitude to AIH and Int.Ang II (35.9 ± 4.2%) (Fig. 7 B and C) and could be prevented by pre‐treating rats with aliskiren (1 mg kg−1 i.v.; −1.1 ± 2.2%; P < 0.0001 vs. Int.Phenylephrine) (Fig. 7 B and C). Finally, a continuous infusion of phenylephrine for 10 min did not increase mean SNA (−5.1 ± 6.1%, n = 4; P = 0.9655 vs. Int.Saline) (data not shown).
Figure 7. Intermittent i.v. injections of phenylephrine, but not sodium nitroprusside, causes a renin activity‐dependent elevation in SNA.
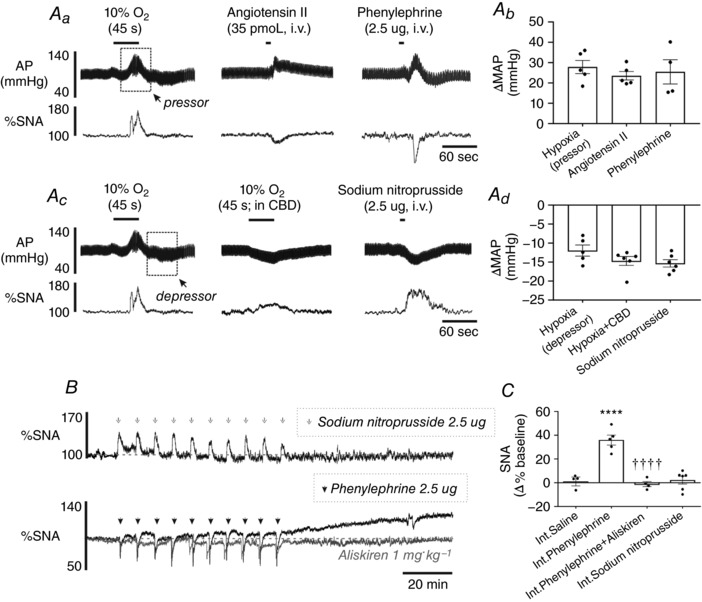
Aa, AP and SNA changes induced by hypoxia, Ang II (35 pmol, i.v.) and phenylephrine (2.5 μg, i.v.). Ab, grouped data comparing the peak increase in AP from baseline following hypoxia, Ang II and phenylephrine. Ac, AP and SNA changes induced by hypoxia, hypoxia in CBD and sodium nitroprusside (2.5 μg, i.v.). Ad, grouped data comparing the peak decrease in AP from baseline following hypoxia, hypoxia (CBD) and sodium nitroprusside. B, effects of intermittent i.v. injections of sodium nitroprusside (upper), phenylephrine (lower, black tace) and phenylephrine with aliskiren pre‐treatment (lower, grey trace). C, group data for the change in SNA 60 min post‐AIH (**** P < 0.0001 vs. Int.Saline, †††† P < 0.0001 vs. Int.Phenylephrine).
Elevation of SNA following AIH in CBD rats is not caused by hypotension
In the absence of the CBs, hypoxia causes hypotension and a modest increase in SNA presumably as a result of the unloading of arterial baroreflex (Fig. 7 Ac). We considered the possibility that intermittent unloading of the baroreceptors may be responsible for the increase in mean SNA after AIH in CB denervated rats. To test this possibility, we administered i.v. sodium nitroprusside (Int.Sodium nitroprusside) in a pattern that matches AIH in CB intact rats (2.5 μg in 0.1 ml repeated 10 times at 5 min intervals). Injections of sodium nitroprusside in CB intact rats produced similar falls in AP as hypoxia in CBD rats (Hypoxia + CBD vs. SNP, −12 ± 2 mmHg vs. ‐15 ± 1 mmHg; P = 0.1905) (Fig. 7 Ac and Ad) but failed to produce a sustained elevation in mean SNA 60 min after this protocol (2.2 ± 3.2% vs. 1.2 ± 2.7%, Int.Sodium nitroprusside vs. Int.Saline; P = 0.9966) (Fig. 7 B and C).
Discussion
We demonstrate that AIH in anaesthetized adult rats causes an elevation in SNA, sensitization of the sympathetic peripheral chemoreflex and an increase in central sympathetic–respiratory coupling, which are all prevented by pre‐treatment with an AT1R antagonist or inhibition of renin activity. We also show that the sympathetic effects of AIH can be recapitulated by injecting exogenous Ang II in a pattern matching AIH. The elevation in SNA following AIH and intermittent Ang II is the result of two distinct neural mechanisms involving a CB‐mediated increase in central sympathetic–respiratory coupling and enhanced chemoreflex sensitivity, as well as a SFO‐mediated increase in tonic SNA. Finally, we show that intermittent sodium nitroprusside does not enhance sympathetic discharge, whereas experimentally‐induced vasoconstriction with phenylephrine produces an elevation in SNA similarly to AIH and intermittent Ang II, with this effect also being dependent on RAS activity.
The sympathetic effects of AIH are mediated by the RAS
We demonstrate that the sympathetic effects of AIH are dependent on activation of the RAS. First, we show that AT1Rs and renin activity are necessary for the increase in SNA following AIH. Second, we show that systemic Ang II delivered in an AIH‐like pattern is sufficient to replicate the sympathetic effects of AIH and that this also requires AT1Rs. As reported previously (Peng et al. 2011), the intermittent pattern of Ang II administration is important for the effects that we observed because an equivalent dose of Ang II delivered by infusion, or as a single injection, did not produce any lasting effects on SNA. Together, these experiments suggest that the sympathetic effects of AIH are caused by transient surges in Ang II, and these surges engage mechanisms that cause a protracted increase in SNA via actions in the CNS. A similar pattern of RAS activation may be present in CIH because prophylactic losartan reduces or prevents hypertension in CIH‐exposed rats (Fletcher et al. 1999; Marcus et al. 2010, 2012), as well as in healthy humans exposed to 6 h of recurring hypoxia (Foster et al. 2010).
Whether or not AIH causes a chronic elevation in RAS activity that supports elevated SNA was not evaluated in the present study, although it is a possibility. However, studies have shown that administering AT1R antagonists after AIH in anaesthetized rats does not normalize mean SNA (Yamamoto et al. 2015) and losartan does not correct hypertension in CIH‐exposed rats (Zoccal et al. 2007). Furthermore, losartan application in superfused CBs effectively prevents the sensory facilitation caused by intermittent hypercapnic hypoxia but does not reverse this effect when applied afterwards (Roy et al. 2017). This evidence suggests that Ang II surges and AT1R activation during intermittent hypoxia activate a cascade of signalling that is subsequently insensitive to acute AT1R blockade. Intermittent hypoxia and Ang II cause transcriptional changes in the CB (Prabhakar et al. 2015) and the SFO (Zimmerman et al. 2002; Lob et al. 2013) that are associated with the production of ROS, which may explain the persistence of changes in SNA following AIH and Int.Ang II. Furthermore, several neuropeptides may contribute to the sympathetic effects of AIH, such as orexin (Kim et al. 2016b), serotonin (Dick et al. 2007; Bautista et al. 2012), vasopressin (Kc et al. 2010) and PACAP (Kakall et al. 2017); the release of these peptides during AIH would produce long‐lasting changes in CNS excitability that are unresponsive to acute AT1R blockade.
Chemosensory CB and SFO mediate distinct components of the sympathetic effects of AIH
We show that the CB and the SFO are required for the elevation of SNA following AIH and intermittent Ang II. Our supposition that the CB and the SFO are the sites of action for Ang II in AIH is supported by evidence indicating that AT1Rs are expressed by glomus cells of the CB (Allen, 1998; Lam et al. 2014) and neurons in the SFO (Mendelsohn et al. 1984; Castrén & Saavedra, 1989). Nevertheless, the site of action of Ang II in our model may be in the periphery, such as the blood vessel endothelial cells or the kidney (Marcus et al. 2012; Emans et al. 2016), and the activation of the CBs and SFO in AIH may be mediated by secondary messengers released from these tissues in response to AT1R binding, such as endothelin‐1 (Kanagy et al. 2001; Rey et al. 2006). Another outstanding issue not addressed in the present study is whether Ang II is derived from peripheral RAS activation, or from endogenous mechanisms for Ang II production in the SFO (Grobe et al. 2008) and the CBs (Leung et al. 2000; Lam & Leung, 2002, 2003; Lam et al. 2014). Notably, previous studies indicate that Ang II production in the CB is renin‐independent (Lam & Leung, 2002). We show that the sympathetic effects of AIH are prevented by inhibition of renin activity with aliskiren, which supports a role for peripheral RAS in AIH.
An interesting finding in the present study is that CBD eliminated the increase in sympathetic chemoreflex sensitivity and central sympathetic–respiratory coupling following AIH, with inhibition of the SFO having no effect on these parameters. It is important to note that, unlike models used to induce phrenic long‐term facilitation (Mitchell & Johnson, 2003), we do not regularly observe increased phrenic nerve activity following AIH, probably as a result of differences in AIH protocol. As such, increased central sympathetic–respiratory coupling following AIH is not caused by enhanced central respiratory drive, but instead relates to changes in coupling of respiratory and sympathetic neurons; our data show that the CB drives this effect. Conversely, the SFO probably elevates SNA through hypothalamic pathways that engage sympathetic neurons in the brainstem or spinal cord largely independent of the central respiratory network, such as descending inputs from neurons in the paraventricular nucleus of the hypothalamus (Holbein & Toney, 2015).
There is evidence that elevated SNA precedes the onset of hypertension in rodent models (Cabassi et al. 1998; Simms et al. 2009) and, as such, we speculate that the increase in SNA following AIH reflects the early stages of the sympathetic changes resulting in hypertension in CIH; our finding that the elevation in SNA after AIH is mediated by the CBs and SFO is consistent with this notion because hypertension in CIH is also dependent on the CBs and SFO. Del Rio et al. (2016) showed that CBD in CIH‐exposed rats with established hypertension normalizes AP and autonomic function, confirming that CIH has modest effects in the absence of the CBs. An earlier study demonstrated that the central administration of losartan or knockdown of AT1Rs in the SFO reduces the elevation of AP in CIH‐exposed rats (Saxena et al. 2015). Collectively, these studies support the notion that the effect of CIH on AP is mediated by recurrent activation of the CBs, leading to peripheral RAS activation and subsequent activation of the SFO, and possibly other neurons expressing AT1Rs (Faulk et al. 2016; Shell et al. 2016). This model largely accommodates our data, although we provide evidence that hypoxia causes RAS‐dependent sympathetic effects in the absence of the CBs.
Whether CB‐independent RAS activation during hypoxia is relevant in CIH models is not clear, although Del Rio et al. (2016) suggests that this is unlikely. There are considerable technical differences between the AIH and the CIH models that may influence the underlying processes that cause SNA to increase, most notably our use of anaesthetics, mechanical ventilation, and supplemental oxygen. Reporting on oxygen saturation during CIH is limited; the results of one study indicate that oxyhaemoglobin falls to 60–80% after less than 6 s of exposure to FiO2 of 3–5% (Fletcher et al. 1992b). Del Rio et al. (2016) exposed rats to FiO2 of 5% for 20 s, suggesting that these rats were probably subject to oxygen desaturations less than 60%. Oxygen desaturation in our AIH model is comparably mild (nadir of 87%), although our rats were ventilated at all other times with oxygen resulting in a stark elevation in baseline (≈470 mmHg). Thus, differences between the CIH and the AIH model could be related to more severe oxygen desaturations during CIH or a hyperoxia baseline in AIH. Hyperoxia has broad ranging effects on the CNS, including inducing oxidative stress and cerebrovascular acidosis (Dean et al. 2004). Most significantly, hyperoxia silences the CB, which eliminates the CB as a source of drive for the sustained increase in SNA following AIH. Thus, performing experiments over a background of hyperoxia may under‐emphasize the involvement of the CB in the sympathetic effects of intermittent hypoxia. Nevertheless, it is clear that the sympathetic effects of AIH and CIH share a dependency on RAS activation.
Systemic injections of phenylephrine produced a similar sympathoexcitatory effect to AIH
We provide evidence that peripheral vasoconstriction induced by activating α1‐adrenergic receptors with phenylephrine causes a protracted increase in SNA similar to AIH. The elevation in SNA following repetitive administrations of phenylephrine was prevented by blocking renin activity and thus is probably mediated by the same pathways as AIH. It is noteworthy that injections of phenylephrine cause inhibitions of sympathetic neurons in the rostral ventrolateral medulla and the spinal cord, which is the opposite to the excitatory effects elicited by hypoxia. Conversely, intermittent sodium nitroprusside had no lasting effects on mean SNA even though it strongly activates the central sympathetic network controlling the cardiovascular system. Collectively, the evidence shows that renin activity induced by peripheral vasoconstriction alone is sufficient to reproduce the sympathetic effects of AIH, whereas repeated sympathetic activation in the presence of exogenous nitric oxide (sodium nitroprusside) has no discernible effect on SNA.
Conclusions
The present study demonstrates the importance of the RAS for the sympathetic effects of AIH and suggests that transient repeated surges in Ang II cause immediate increases in SNA. The relationship between recurrent hypoxia and transient RAS activation may contribute to the association between apnoea/hypopnea index and daytime SNA in humans with obstructive sleep aponea (Peppard et al. 2000; Taylor et al. 2016). We also show that the sympathetic effects of AIH require both the CB and SFO and also that AIH has sympathetic effects in CB denervated rats. Considering that CBD is increasingly considered as a viable treatment for severe and resistant hypertension in humans (Paton et al. 2013) and the modulation of CB activity is proposed as a treatment for autonomic dysfunction in patients with obstructive sleep aponea (Del Rio et al. 2016), it will be important to fully understand the underlying mechanism responsible for sympathetic activation by hypoxia in the absence of the CBs.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SJK, AYF, PMP and SBGA conceived and designed the study. SJK and AYF performed the experiments. SJK and SBGA analysed the data, interpreted the results, prepared figures and drafted the manuscript. SJK, PMP and SBGA edited and revised the manuscript. All authors approved the final version of the manuscript submitted for publication.
Funding
Work in the authors’ laboratory is supported by the National Health and Medical Research Council of Australia Fellowship (PMP; 1024489) (NHMRC Project Grants: 1065485, 1082215) and the Heart Research Institute. SBGA is supported by the National Health and Medical Research Council of Australia (Grant GNT1052674) and the University of Sydney. SJK is supported by an Australian Postgraduate Award (APA SC0042) from the Australian Government and awarded by the University of Sydney, as well as a Heart Research Institute scholarship.
Acknowledgements
We thank M.J.M.Farnham for her ideas and contributions to some of these experiments.
Biography
Seung Jae Kim is a PhD candidate at the University of Sydney (Sydney Medical School & Heart Research Institute). He is currently co‐supervised by Professor Paul M. Pilowsky (University of Sydney) and Dr Stephen B.G. Abbott (University of Virginia). His work focuses on studying the neural mechanisms that cause sympathetic nerve activity to elevate, which later progresses into hypertension. Here, he demonstrates that the enhancement of sympathetic outflow following intermittent hypoxia is mediated by circulating angiotensin II acting at the carotid bodies and the subfornical organ. The two areas contribute equally to the overall sympathoexcitatory effects.

Edited by: Kim Barrett & Harold Schultz
References
- Allen AM (1998). Angiotensin AT1 receptor‐mediated excitation of rat carotid body chemoreceptor afferent activity. J Physiol 510, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista TG, Xing T, Fong AY & Pilowsky PM (2012). Recurrent laryngeal nerve activity exhibits a 5‐HT‐mediated long‐term facilitation and enhanced response to hypoxia following acute intermittent hypoxia in rat. J Appl Physiol (1985) 112, 1144–1156. [DOI] [PubMed] [Google Scholar]
- Cabassi A, Vinci S, Calzolari M, Bruschi G & Borghetti A (1998). Regional sympathetic activity in pre‐hypertensive phase of spontaneously hypertensive rats. Life Sci 62, 1111–1118. [DOI] [PubMed] [Google Scholar]
- Castrén E & Saavedra JM (1989). Angiotensin II receptors in paraventricular nucleus, subfornical organ, and pituitary gland of hypophysectomized, adrenalectomized, and vasopressin‐deficient rats. Proc Natl Acad Sci USA 86, 725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JB, Mulkey DK, Henderson RA, Potter SJ & Putnam RW (2004). Hyperoxia, reactive oxygen species, and hyperventilation: oxygen sensitivity of brain stem neurons. J Appl Physiol 96, 784–791. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Andrade DC, Lucero C, Arias P & Iturriaga R (2016). Carotid body ablation abrogates hypertension and autonomic alterations induced by intermittent hypoxia in rats. Hypertension 68, 436–445. [DOI] [PubMed] [Google Scholar]
- Dick TE, Hsieh YH, Wang N & Prabhakar N (2007). Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92, 87–97. [DOI] [PubMed] [Google Scholar]
- Emans TW, Janssen BJ, Pinkham MI, Ow CPC, Evans RG, Joles JA, Malpas SC, Krediet CTP & Koeners MP (2016). Exogenous and endogenous angiotensin‐II decrease renal cortical oxygen tension in conscious rats by limiting renal blood flow. J Physiol 594, 6287–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler M (2010). The 2009 Carl Ludwig Lecture: pathophysiology of the human sympathetic nervous system in cardiovascular diseases: the transition from mechanisms to medical management. J Appl Physiol 108, 227. [DOI] [PubMed] [Google Scholar]
- Faulk K, Shell B, Nedungadi TP & Cunningham JT (2016). Role of angiotensin‐converting enzyme 1 within the median preoptic nucleus following chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol 312, R245–R252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC, Bao G & Li R (1999). Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension 34, 309–314. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, Stauss H & Unger T (1992a). Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol 72, 1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Qian W, Miller Iii CC & Unger T (1992b). Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension 19, 555–561. [DOI] [PubMed] [Google Scholar]
- Foster GE, Hanly PJ, Ahmed SB, Beaudin AE, Pialoux V & Poulin MJ (2010). Intermittent hypoxia increases arterial blood pressure in humans through a renin‐angiotensin system‐dependent mechanism. Hypertension 56, 369. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Xu D & Sigmund CD (2008). An intracellular renin‐angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology 23, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbein WW & Toney GM (2015). Activation of the hypothalamic paraventricular nucleus by forebrain hypertonicity selectively increases tonic vasomotor sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 308, R351–R359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Lusina S, Xie T, Ji E, Xiang S, Liu Y & Weiss JW (2009). Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol 166, 102–106. [DOI] [PubMed] [Google Scholar]
- Kakall ZM, Pilowsky PM & Farnham MMJ (2017). PACAP(6‐38) or kynurenate microinjections into the RVLM prevent development of sympathetic long‐term facilitation following acute intermittent hypoxia. Am J Physiol Heart Circ Physiol 314, H563–H572. [DOI] [PubMed] [Google Scholar]
- Kanagy NL, Walker BR & Nelin LD (2001). Role of endothelin in intermittent hypoxia‐induced hypertension. Hypertension 37, 511–515. [DOI] [PubMed] [Google Scholar]
- Kc P, Balan KV, Tjoe SS, Martin RJ, LaManna JC, Haxhiu MA & Dick TE (2010). Increased vasopressin transmission from the paraventricular nucleus to the rostral medulla augments cardiorespiratory outflow in chronic intermittent hypoxia‐conditioned rats. J Physiol 588, 725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YJ, Kakall Z, Farnham MM & Pilowsky PM (2016a). Intermittent hypoxia‐induced cardiorespiratory long‐term facilitation: a new role for microglia. Respir Physiol Neurobiol 226, 30–38. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Pilowsky PM & Farnham MM (2016b). Intrathecal intermittent orexin‐A causes sympathetic long‐term facilitation and sensitizes the peripheral chemoreceptor response to hypoxia in rats. J Pharmacol Exp Ther 358, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SY & Leung PS (2002). A locally generated angiotensin system in rat carotid body. Regul Pept 107, 97–103. [DOI] [PubMed] [Google Scholar]
- Lam SY & Leung PS (2003). Chronic hypoxia activates a local angiotensin‐generating system in rat carotid body. Mol Cell Endocrinol 203, 147–153. [DOI] [PubMed] [Google Scholar]
- Lam SY, Liu Y, Ng KM, Liong EC, Tipoe GL, Leung PS & Fung ML (2014). Upregulation of a local renin‐angiotensin system in the rat carotid body during chronic intermittent hypoxia. Exp Physiol 99, 220–231. [DOI] [PubMed] [Google Scholar]
- Leung PS, Lam SY & Fung ML (2000). Chronic hypoxia upregulates the expression and function of AT(1) receptor in rat carotid body. J Endocrinol 167, 517–524. [DOI] [PubMed] [Google Scholar]
- Lob HE, Schultz D, Marvar PJ, Davisson RL & Harrison DG (2013). Role of the NADPH oxidases in the subfornical organ in angiotensin II‐induced hypertension. Hypertension 61, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Li Y‐L, Bird CE, Schultz HD & Morgan BJ (2010). Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol 171, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Philippi NR, Bird CE, Li Y‐L, Schultz HD & Morgan BJ (2012). Effect of AT1 receptor blockade on intermittent hypoxia‐induced endothelial dysfunction. Respir Physiol Neurobiol 183, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn FA, Quirion R, Saavedra JM, Aguilera G & Catt KJ (1984). Autoradiographic localization of angiotensin II receptors in rat brain. Proc Natl Acad Sci USA 81, 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GS & Johnson SM (2003). Neuroplasticity in respiratory motor control. J Appl Physiol 94, 358–374. [DOI] [PubMed] [Google Scholar]
- Paton JF, Sobotka PA, Fudim M, Engelman ZJ, Hart EC, McBryde FD, Abdala AP, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L & Nightingale A (2013). The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61, 5–13. [DOI] [PubMed] [Google Scholar]
- Peng Y‐J, Raghuraman G, Khan SA, Kumar GK & Prabhakar NR (2011). Angiotensin II evokes sensory long‐term facilitation of the carotid body via NADPH oxidase. J Appl Physiol 111, 964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Palta M & Skatrud J (2000). Prospective study of the association between sleep‐disordered breathing and hypertension. N Engl J Med 342, 1378–1384. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng Y‐J, Kumar GK & Nanduri J (2015). Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol 5, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R & Iturriaga R (2006). Contribution of endothelin‐1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res 1086, 152–159. [DOI] [PubMed] [Google Scholar]
- Roy A, Farnham MMJ, Derakhshan F, Pilowsky PM & Wilson RJA (2017). Acute intermittent hypoxia with concurrent hypercapnia evokes P2X and TRPV1 receptor dependent sensory long‐term facilitation in naïve carotid bodies. J Physiol 10.1113/JP275001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Little JT, Nedungadi TP & Cunningham JT (2015). Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia‐associated sustained increase in mean arterial pressure. Am J Physiol Heart Circ Physiol 308, H435–H446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell B, Faulk K & Cunningham JT (2016). Neural control of blood pressure in chronic intermittent hypoxia. Curr Hypertens Rep 18, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AQ & Schreihofer AM (2011). Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol 589, 1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms AE, Paton JFR, Pickering AE & Allen AM (2009). Amplified respiratory‐sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol 587, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM & Ferguson AV (2010). Circulating signals as critical regulators of autonomic state – central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol 299, R405–R415. [DOI] [PubMed] [Google Scholar]
- Taylor KS, Murai H, Millar PJ, Haruki N, Kimmerly DS, Morris BL, Tomlinson G, Bradley TD & Floras JS (2016). Arousal from sleep and sympathetic excitation during wakefulness. Hypertension 68, 1467–1474. [DOI] [PubMed] [Google Scholar]
- Xing T & Pilowsky PM (2010). Acute intermittent hypoxia in rat in vivo elicits a robust increase in tonic sympathetic nerve activity that is independent of respiratory drive. J Physiol 588, 3075–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Lalley P & Mifflin S (2015). Acute intermittent optogenetic stimulation of nucleus tractus solitarius neurons induces sympathetic long‐term facilitation. Am J Physiol Regul Integr Comp Physiol 308, R266–R275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR & Davisson RL (2002). Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91, 1038. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Oliveira FR, Antunes‐Rodrigues J & Machado BH (2007). Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol 92, 79–85. [DOI] [PubMed] [Google Scholar]