

Abstract
Key points
Activity‐dependent plasticity can be induced in carotid body (CB) chemosensory afferents without chronic intermittent hypoxia (CIH) preconditioning by acute intermittent hypoxia coincident with bouts of hypercapnia (AIH‐Hc).
Several properties of this acute plasticity are shared with CIH‐dependent sensory long‐term facilitation (LTF) in that induction is dependent on 5‐HT, angiotensin II, protein kinase C and reactive oxygen species.
Several properties differ from CIH‐dependent sensory LTF; H2O2 appears to play no part in induction, whereas maintenance requires purinergic P2X2/3 receptor activation and is dependent on transient receptor potential vanilloid type 1 (TRPV1) receptor sensitization.
Because P2X2/3 and TRPV1 receptors are located in carotid sinus nerve (CSN) terminals but not presynaptic glomus cells, a primary site of the acute AIH‐Hc induced sensory LTF appears to be postsynaptic.
Our results obtained in vivo suggest a role for TRPV1‐dependent CB activity in acute sympathetic LTF. We propose that P2X‐TRPV1‐receptor‐dependent sensory LTF may constitute an important early mechanism linking sleep apnoea with hypertension and/or cardiovascular disease.
Abstract
Apnoeas constitute an acute existential threat to neonates and adults. In large part, this threat is detected by the carotid bodies, which are the primary peripheral chemoreceptors, and is combatted by arousal and acute cardiorespiratory responses, including increased sympathetic output. Similar responses occur with repeated apnoeas but they continue beyond the last apnoea and can persist for hours [i.e. ventilatory and sympathetic long‐term facilitation (LTF)]. These long‐term effects may be adaptive during acute episodic apnoea, although they may prolong hypertension causing chronic cardiovascular impairment. We report a novel mechanism of acute carotid body (CB) plasticity (sensory LTF) induced by repeated apnoea‐like stimuli [i.e. acute intermittent hypoxia coincident with bouts of hypercapnia (AIH‐Hc)]. This plasticity did not require chronic intermittent hypoxia preconditioning, was dependent on P2X receptors and protein kinase C, and involved heat‐sensitive transient receptor potential vanilloid type 1 (TRPV1) receptors. Reactive oxygen species (O2·¯) were involved in initiating plasticity only; no evidence was found for H2O2 involvement. Angiotensin II and 5‐HT receptor antagonists, losartan and ketanserin, severely reduced CB responses to individual hypoxic‐hypercapnic challenges and prevented the induction of sensory LTF but, if applied after AIH‐Hc, failed to reduce plasticity‐associated activity. Conversely, TRPV1 receptor antagonism had no effect on responses to individual hypoxic‐hypercapnic challenges but reduced plasticity‐associated activity by ∼50%. Further, TRPV1 receptor antagonism in vivo reduced sympathetic LTF caused by AIH‐Hc, although only if the CBs were functional. These data demonstrate a new mechanism of CB plasticity and suggest P2X‐TRPV1‐dependent sensory LTF as a novel target for pharmacological intervention in some forms of neurogenic hypertension associated with recurrent apnoeas.
Keywords: carotid body, sensory long term facilitation, hypoxia‐hypercapnia
Key points
Activity‐dependent plasticity can be induced in carotid body (CB) chemosensory afferents without chronic intermittent hypoxia (CIH) preconditioning by acute intermittent hypoxia coincident with bouts of hypercapnia (AIH‐Hc).
Several properties of this acute plasticity are shared with CIH‐dependent sensory long‐term facilitation (LTF) in that induction is dependent on 5‐HT, angiotensin II, protein kinase C and reactive oxygen species.
Several properties differ from CIH‐dependent sensory LTF; H2O2 appears to play no part in induction, whereas maintenance requires purinergic P2X2/3 receptor activation and is dependent on transient receptor potential vanilloid type 1 (TRPV1) receptor sensitization.
Because P2X2/3 and TRPV1 receptors are located in carotid sinus nerve (CSN) terminals but not presynaptic glomus cells, a primary site of the acute AIH‐Hc induced sensory LTF appears to be postsynaptic.
Our results obtained in vivo suggest a role for TRPV1‐dependent CB activity in acute sympathetic LTF. We propose that P2X‐TRPV1‐receptor‐dependent sensory LTF may constitute an important early mechanism linking sleep apnoea with hypertension and/or cardiovascular disease.
Introduction
The carotid bodies (CBs) are the primary detectors of apnoea, responding vigorously to bouts of hypoxic‐hypercapnia (as occurs during prolonged apnoea) or bouts of hypoxia against a hypercapnic background (as occurs in most sleep apnoea patients). Vigorous activity of the CB caused by hypoxia with concurrent hypercapnia, in turn, initiates life‐saving cardiorespiratory and arousal reflexes (Mohan & Duffin, 1997; Duffin, 2010). However, compelling human data suggest that upregulation of CB activity and/or sensitivity is important to the aetiology of cardiorespiratory diseases, including hypertension and heart failure associated with sleep apnoea (Mansukhani et al. 2015). For example, sympathetic and ventilatory responses to hypoxia and hypercapnia are enhanced in sleep apnoea and/or borderline hypertensive patients compared to controls (Trzebski et al. 1982; Somers et al. 1988; Spicuzza et al. 2006; Imadojemu et al. 2007), whereas glomectomy or inactivation of the CB with excess oxygen (hyperoxia) mitigate sleep apnoea‐associated increases in sympathetic outflow and blood pressure (Somers & Abboud, 1993; Narkiewicz et al. 1998). Moreover, 20–30 min of intermittent hypoxia in healthy humans enhances sympathetic activity, including sympathetic sensitivity to hypoxia (Cutler et al. 2004; Leuenberger et al. 2007).
In accordance with the findings in humans, CB denervation prevents increases in sympathetic nerve activity (SNA) and/or increased blood pressure in animal models of sleep apnoea (chronic intermittent hypoxia) (Fletcher, 2000; Prabhakar et al. 2005; McBryde et al. 2013). The CBs from these disease models demonstrate functional changes. Peng et al. (2003) demonstrated that intact and isolated superfused CB preparations from rats exposed to 10 days of chronic intermittent hypoxia (CIH) undergo sensory long‐term facilitation (LTF), as characterized by a persistent increase in baseline afferent activity and augmented sensitivity to hypoxia, in response to 10 bouts of acute intermittent hypoxia (AIH). (Peng et al. 2003). Other studies report that rats exposed to AIH (e.g. ten 45 s periods of 10% O2 interspersed with 100% O2) have an enhanced chemoreflex response to hypoxia and/or demonstrate respiratory and/or sympathetic LTF without CIH preconditioning (Xie et al. 2000; Dick et al. 2007; Mahamad & Mitchell, 2007; Xing & Pilowsky, 2010), and we found qualitatively similar, although weaker, acute stimulus‐dependent responses in isolated perfused rat CBs (Cummings & Wilson 2005). Taken together, these studies suggest that the sensitivity of the CB might be enhanced during recurrent apnoeas, resulting in increased sympathetic activity and hypertension. Consequently, the CB has gained renewed attention as a possible target organ for the treatment of sleep apnoea and associated hypertension (DelRio et al. 2015).
The mechanism(s) responsible for the induction and maintenance phases of CB sensory LTF are not fully resolved. When sensory LTF is induced by a combination of CIH preconditioning and AIH, maintenance is probably contingent on a presynaptic (glomus cell) reactive oxygen species (ROS)‐dependent mechanism involving 5‐HT, NADPH oxidase/ROS signalling (Peng et al. 2009) and a T‐type voltage‐gated calcium channel, type 3.2 (CaV3.2) (Makarenko et al. 2016). However, sensory LTF induced by acute stimuli in the absence of CIH preconditioning may be mediated by a different mechanism.
Previously, we demonstrated that transient receptor potential vanilloid type 1 (TRPV1) receptors in CB chemosensory afferents are largely responsible for the anandamide and exquisite heat sensitivity of the CB (Roy et al. 2012). Because TRPV1 plays an important role in neuronal plasticity in the hippocampus and several sensory systems, and because, in some of these systems, ATP activation of P2X receptor leads to TRPV1 phosphorylation, we hypothesized that TRPV1 receptors contribute to CB sensory LTF (Bhave et al. 2003; Marsch et al. 2007; Li et al. 2008; Hasegawa et al. 2009; Shen et al. 2012; Lin et al. 2013; Saloman et al. 2013; Ruan et al. 2014; Yang et al. 2016).
In the present study, we report robust sensory LTF (increased baseline and augmented hypoxic responses) induced by acute recurrent apnoea‐like stimuli (10 bouts of acute hypoxia with a hypercapnic background, AIH‐SHc; or 10 bouts of acute intermittent hypoxia coincident with bouts of hypercapnia; AIH‐Hc) in perfused CBs isolated from naïve rats. We demonstrate that ATP, the primary neurotransmitter released from glomus cells and acting via P2X2/3 receptors, is necessary for the induction and maintenance of sensory LTF following AIH‐Hc; however, we also demonstrate that 50% of sensory LTF is mediated by TRPV1 receptors located on afferent fibres (postsynaptic to glomus cells). Unlike P2X2/3, TRPV1 plays no role in the responses to individual hypoxic‐hypercapnic challenges prior to sensory LTF induction, suggesting that TRPV1 is a primary effector of CB chemosensory plasticity.
Methods
Ethical approval
CB experiments were performed in accordance with The Canadian Council of Animal Care guidelines and were approved by the Animal Care Committee of the Cumming School of Medicine, University of Calgary, Canada. Sympathetic experiments were performed in accordance with Macquarie University Animal Care Committees and were approved by The Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. All animal procedures conform with the principles and regulations as described by Grundy (2015).
Experimental animals
CB ex vivo experiments were conducted on 126 male adult Sprague–Dawley Albino rats (150–250 g) purchased from Charles River (Quebec, Canada) and in vivo sympathetic experiments were conducted on 29 adult male Sprague–Dawley rats (350–500 g; Animal Resource Centre, Perth, Australia). Animals were housed conventionally in temperature and humidity controlled facilities with food and water available ad libitum.
Solutions and chemicals
Physiological buffer for dissection and perfusion comprised (in mm): 115 NaCl, 4 KCl, 24 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 10 d‐glucose and 12 sucrose (Sigma‐Aldrich, Oakville, Ontario, Canada). The chemicals used were: ATP, ketanserin and polyethylene glycol (PEG)‐catalase (Sigma‐Aldrich); suramin, 2,4,6‐trinitrophenol (TNP)‐ATP, AMG9810 [(2E)‐N‐(2,3‐dihydro‐1,4‐benzodioxin‐6‐yl)‐3‐(4‐[1,1‐dimethylethyl]phenyl)‐2‐propenamide], GF109203X (bisindolylm‐aleimide) and AIP (autocamtide‐2‐related inhibitory peptide) (Tocris Bioscience, Bristol, UK); losartan (Cayman Chemical Company, Ann Arbor, MI, USA); and MnTMPyP [5,10,15,20‐Tetrakis(1‐methylpyridinium‐4‐yl)‐21H,23H‐porphyrin manganese(III) pentachloride] (Calbiochem, San Diego, CA, USA).
Ex vivo carotid body‐carotid sinus nerve (CB‐CSN) preparation
This preparation was used to characterize and determine the mechanism responsible for CB sensory LTF induced by AIH with concurrent hypercapnia. Rats were heavily anaesthetized with 5% isoflurane by inhalation in air and then decapitated (lower cervical level). Immediately after killing the animal, the carotid bifurcation, including the CB, CSN and superior cervical ganglion, was quickly removed en bloc for in vitro arterial perfusion as described previously (Roy et al. 2012). The carotid bifurcation was then transferred to physiological buffer equilibrated with 95% O2 and 5% CO2. After incubating for a minimum of 20 min in ice‐cold buffer, the preparation was transferred en bloc to a temperature‐regulated recording chamber, the common carotid artery was immediately cannulated for luminal perfusion with physiological buffer equilibrated with 100 Torr and 35 Torr (balanced N2) and the CSN was freed and de‐sheathed. A peristatic pump and heat exchanger were used to supply physiological saline via the cannula at a flow rate of ∼15 ml min−1 (the pressure at the tip of the cannula was ∼100 mmHg) and to maintain the temperature at 37 ± 0.5°C. We established that sensory LTF occurred whether or not the effluent was recirculated (see Results); therefore, in most experiments, the effluent was recirculated for convenience. Perfusate was equilibrated with computer‐controlled gas mixtures. Mixtures were monitored using precision CO2 and O2 gas analysers (models CA‐2A and PA1B, respectively; Sable Systems, Las Vegas, NV, USA) and balanced with N2. Unless otherwise stated, 100 Torr (normoxia) and 35 Torr (normocapnia; yielding pH ∼7.4) were used. Chemosensory afferent activity was recorded extracellularly from the CSN (multifibre) using a platinum‐hook electrode and a differential AC amplifier (Model 1700; AM Systems Inc., Sequim, WA, USA). The neural activity was amplified, filtered (300 Hz low cut‐off, 5 kHz high cut‐off) and displayed on an oscilloscope (model 2230; Tektronix, Beaverton, OR, USA); raw, rectified and integrated (200 ms time constant; MA‐821; CWE, Inc., Ardmore, PA, USA) data were stored on a computer using an A/D board (Digidata 1322A; Axon Instruments, Axon Instruments, Foster City, CA, USA) and data acquisition software (Axoscope, version 9.0; Molecular Devices, Sunnyvale, CA, USA) at 5 kHz.
Experimental protocols (ex vivo preparation)
Once stable recordings had been achieved, preparations were exposed to a brief hypoxic challenge (4 min, 60 Torr); only preparations showing a clear‐cut increase in activity were used. Preparations were perfused for an additional 60 min with normoxic‐normocapnic buffer before initiation of the experimental protocol.
Exposure to AIH‐Hc or AIH‐SHc
AIH‐Hc experiments were performed on naive rats, not having prior exposure to CIH. The AIH‐Hc paradigm consisted of ten 1 min bouts of hypoxia‐hypercapnia ( = 40 Torr; = 60 Torr) interspersed with 5 min of normoxia‐normocapnia ( = 100 Torr; = 35 Torr). The AIH‐SHc paradigm consisted of ten 1 min bouts of hypoxia ( = 60 Torr) interspersed with 5 min of normoxia ( = 100 Torr) with a hypercapnic background ( = 50 Torr). The preparation was perfused with normoxic‐normocapnic buffer immediately after the last stimulus. Sensory LTF was defined as the difference between CSN activity at baseline (pre‐stimulus) and 60 min after completion of the AIH‐Hc or AIH‐SHc protocols. Time control experiments were performed by challenging the CB with 10 episodes of intermittent normoxia‐normocapnia.
Exposures to AIH or AIHc
Separate experiments were performed in naive rats to determine whether sensory LTF can also be evoked by stimuli other than AIH‐Hc. AIH or AIHc paradigms consisted of ten 1 min bouts of hypoxia ( = 40 Torr) or hypercapnia ( = 60 Torr). Each bout was separated by 5 min of normoxia‐normocapnia.
Exposure to ACH‐Hc
Separate experiments were performed to examine the cumulative effects of hypoxia‐hypercapnia to determine the importance of stimulus pattern. The ACH‐Hc paradigm consisted of 10 min of continuous hypoxia‐hypercapnia ( = 40 Torr; = 60 Torr).
Data analysis (ex vivo preparation)
Data were analysed offline using custom software (written by RJAW). CSN activity was divided into 60 s time bins and the activity in each bin was rectified and summed (expressed as integrated neural discharge). The neural responses for different conditions in the protocol were normalized to the baseline (normoxic) condition. All data were expressed as the mean ± SEM. One‐way ANOVA with Holm–Šidák post hoc tests were used to analyse the data (SigmaStat, version 2.03; Systat Software Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
In vivo anaesthetized rat preparation
This preparation was used to determine the role of the CB and TRPV1 receptors in sympathetic LTF induced by AIH‐Hc. Experiments were conducted on adult male Sprague–Dawley rats (350–500 g; Animal Resource Centre). Rats (n = 29) were anaesthetized with urethane (1.3 g kg−1 in a 10% solution i.p.). Complete details of the surgical preparation and data acquisition methods are provided elsewhere (Farnham et al. 2008). Briefly, the core temperature was maintained at 37 ± 0.5°C. The right carotid artery and jugular vein were cannulated for measurement of mean arterial pressure (MAP) and the administration of drugs and fluids, respectively. Tracheostomy was performed to permit artificial ventilation. The left greater splanchnic sympathetic nerve and left phrenic nerve were isolated and activity was recorded using silver hook electrodes. Data were acquired using a CED 1401 ADC system and Spike 2 acquisition and analysis software, version 7.12 (Cambridge Electronic Design, Cambridge, UK). All animals were bilaterally vagotomized, ventilated with either room air (n = 14) or hyperoxia (n = 15) and paralysed (0.8 mg kg−1 pancuronium bromide i.v., followed by an infusion of 0.8 mg kg−1 h−1 of pancuronium in 0.9% saline at a rate of 2 ml h−1; Astra Zeneca, Australia). Supplemental anaesthetic (0.2–0.4 ml of 10% urethane) was administered (i.v.), if necessary, to maintain a surgical plane of anaesthesia, which was assessed by monitoring if there was a change in blood pressure >10 mmHg in response to a tail or paw pinch.
Experimental protocols (in vivo preparation)
Recordings were allowed to stabilize until 30 min of stable nerve activity was recorded. All rats had 0.2 ml of arterial blood withdrawn for respiratory blood gas (O2 and CO2) and pH analysis (electrolyte and blood gas analyser; IDEXX Laboratories, Lenexa, KS, USA) 10 min prior to AIH‐Hc. The AIH‐Hc protocol consisted of 10 bouts (1 min each) of ventilation with a gas mixture of 8% CO2 and 5.4% O2 in N2, interspersed at intervals of 5 min. This was performed in two groups of animals, against a backdrop of either normoxic or hyperoxic conditions (Table 1). Arterial blood gas was analysed at 30 min, 60 min and 90 min following the 10th hypoxia‐hypercapnia bout (Table 1). At 60 min post‐AIH‐Hc, a single hypoxia‐hypercapnia bout was administered with 5 min of recovery. At this point, the vehicle control solution [dimethylsulphoxide (DMSO)] or AMG9810 (100 mg kg−1) was administered i.p. and recordings were maintained for another 30 min. A final arterial blood gas analysis and hypoxia‐hypercapnia stimulus was performed before killing the rats with 0.5 ml of 3 m KCl.
Table 1.
, O2 saturation, , pH and MAP measurements pre, post‐AIH‐Hc and during drug treatment
| Background O2 | Treatment time | (Torr) | O2 saturation (%) | (Torr) | pH | MAP (mmHg) |
|---|---|---|---|---|---|---|
| Normoxia | Pre‐AIH‐Hc | 86.8 ± 7.1 | 94.3 ± 2.0 | 39.7 ± 2.1 | 7.48 ± 0.02 | 99 ± 3 |
| 30 min | 83.5 ± 4.1 | 93.0 ± 1.4 | 38.8 ± 1.6 | 7.49 ± 0.02 | 107 ± 6 | |
| 60 min | 81.7 ± 4.4 | 92.0 ± 1.3 | 39.3 ± 1.9 | 7.48 ± 0.01 | 102 ± 3 | |
| 90 min DMSO | 79.3 ± 4.4 | 92.0 ± 1.5 | 39.7 ± 1.7 | 7.46 ± 0.01 | 106 ± 3 | |
| Pre‐AIH‐Hc | 81.2 ± 5.1 | 92.4 ± 1.6 | 41.5 ± 1.8 | 7.48 ± 0.02 | 90 ± 3 | |
| 30 min | 79.0 ± 5.8 | 91.4 ± 2.4 | 41.8 ± 1.5 | 7.52 ± 0.04 | 93 ± 2 | |
| 60 min | 80.3 ± 3.8 | 92.8 ± 0.8 | 40.6 ± 1.0 | 7.48 ± 0.01 | 96 ± 3 | |
| 90 min AMG9810 | 79.3 ± 4.4 | 92.0 ± 1.5 | 39.7 ± 1.7 | 7.47 ± 0.01 | 94 ± 3 | |
| Hyperoxia | Pre‐AIH‐Hc | 496.8 ± 10.8 | 100 ± 0 | 42.3 ± 1.9 | 7.42 ± 0.01 | 113 ± 6 |
| 30 min | 455.8 ± 36.6 | 100 ± 0 | 41.0 ± 2.3 | 7.41 ± 0.01 | 108 ± 8 | |
| 60 min | 469.5 ± 21.3 | 100 ± 0 | 40.3 ± 1.2 | 7.41 ± 0.01 | 105 ± 6 | |
| 90 min DMSO | 469.8 ± 24.8 | 100 ± 0 | 43.7 ± 2.8 | 7.39 ± 0.02 | 112 ± 8 | |
| Pre‐AIH‐Hc | 491.0 ± 7.1 | 100 ± 0 | 40.7 ± 1.3 | 7.43 ± 0.01 | 110 ± 3 | |
| 30 min | 467.0 ± 13.9 | 100 ± 0 | 41.2 ± 1.2 | 7.43 ± 0.01 | 119 ± 4 | |
| 60 min | 483.0 ± 18.2 | 100 ± 0 | 41.0 ± 1.1 | 7.43 ± 0.01 | 117 ± 4 | |
| 90 min AMG9810 | 492.3 ± 10.6 | 100 ± 0 | 40.2 ± 1.3 | 7.42 ± 0.01 | 113 ± 3 |
Data analysis (in vivo preparation)
Mean arterial pressure, heart rate and SNA, were analysed from 1 min blocs taken 1 min prior to and also 30 min, 60 min and 90 min after AIH‐Hc. Statistical analysis was conducted using Prism, version 5.01 (GraphPad Software Inc., San Diego, CA, USA). The changes in responses from 60 min to 90 min after AIH‐Hc were compared between vehicle control and AMG9810 using a one‐way ANOVA with post hoc Holm–Šidák tests.
Results
AIH with concurrent hypercapnia induces CB sensory LTF
The effects of AIH‐Hc on chemosensory activity were examined in ex vivo perfused CBs from six naϊve rats (without prior conditioning to CIH exposures). CSN activity increased progressively with each successive hypoxia‐hypercapnia stimulus (Fig. 1 A). Terminating each AIH‐Hc bout with normoxia‐normocapnia promptly returned the CSN activity to baseline; the baseline incremented only slightly between bouts. After the last (10th) AIH‐Hc bout, baseline CSN activity gradually increased. This increase in baseline (i.e. sensory LTF) lasted at least 60 min (pre AIH‐Hc baseline vs. baseline 60th minute post AIH‐Hc: 1.00 ± 0.01 vs. 1.82 ± 0.13; P < 0.001; n = 6) (Fig. 2 A). There was no difference in the magnitude of sensory LTF regardless of whether the effluent was recirculated or not (n = 6) (1.82 ± 0.13 vs. 1.71 ± 0.09; P = 0.73) (Fig. 1 B and Fig. 2 F). No sensory LTF was evident in time controls (n = 5) (Fig. 2 A). During the course of our investigation into the mechanisms involved in the induction and maintenance of sensory LTF, 63 experiments were conducted using AIH‐Hc in which we could assess the induction of sensory LTF without drug intervention; of these, six failed to show sensory LTF (10.5%) (Fig. 1 D). All failures either had a progressive decline or initial progressive increase followed by a decline in peak CSN activity with each AIH‐Hc exposure (Fig. 1 C). Overall, however, there was no significant relationship between the change in CSN response from the first to last bout of AIH‐Hc and the magnitude of sensory LTF 60 min after the last bout (r 2 = 0.02; P = 0.19) (Fig. 1 E).
Figure 1. AIH‐Hc causes sensory LTF in ex vivo CB from naïve rat.
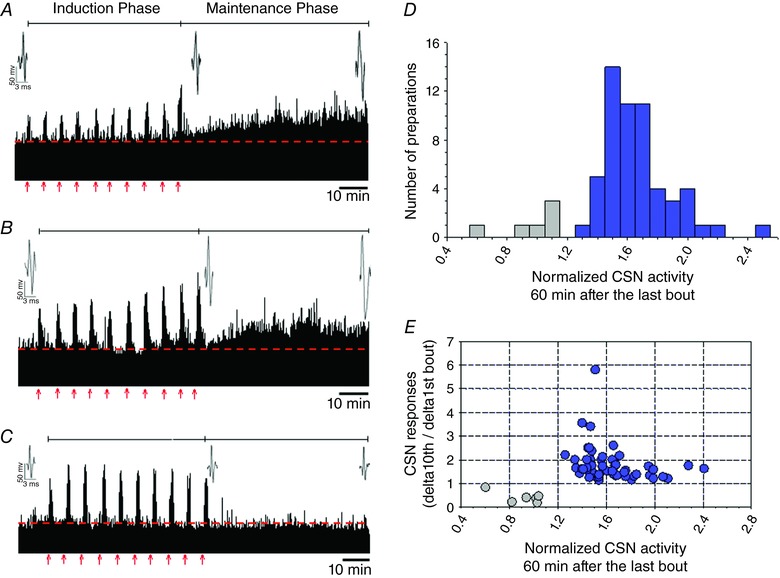
Examples of rectified CB sensory activity (A–B) from three preparations exposed to ten 1 min bouts of hypoxia‐hypercapnia (red arrows; each interspersed with 5 min of normoxia‐normocapnia). Insets represent action potentials from a single unit. During AIH‐Hc (i.e. the induction phase), most preparations had augmenting responses with each bout (A, recirculating effluent; B, non‐recirculating effluent). We observed a small number of preparations with decrementing responses, C, after AIH‐Hc (i.e. during the maintenance phase), CSN activity increased in (A) and (B) (indicative of sensory LTF) but not in (C) (no sensory LTF). D, magnitude of CSN activity 60 min after the last hypoxia‐hypercapnia bout (n = 63). In total, 57 preparations demonstrated sensory LTF (blue); in the remaining six, sensory LTF was absent (grey). E, scatter plot showing the relationship between the change in CSN activity between the first and last (10th) bout of hypoxia‐hypercapnia compared to the CSN activity at 60 min after the last bout. Only preparations in which responses increased with bouts demonstrated sensory LTF (blue). However, the degree of augmentation did not predict sensory LTF magnitude (P = 0.06; r 2 = 0.02). Note that CSN activity is normalized to the baseline preceding the first bout (dashed line). [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 2. Sensory LTF in ex vivo CBs from naïve rats depends on stimulus pattern and strength.
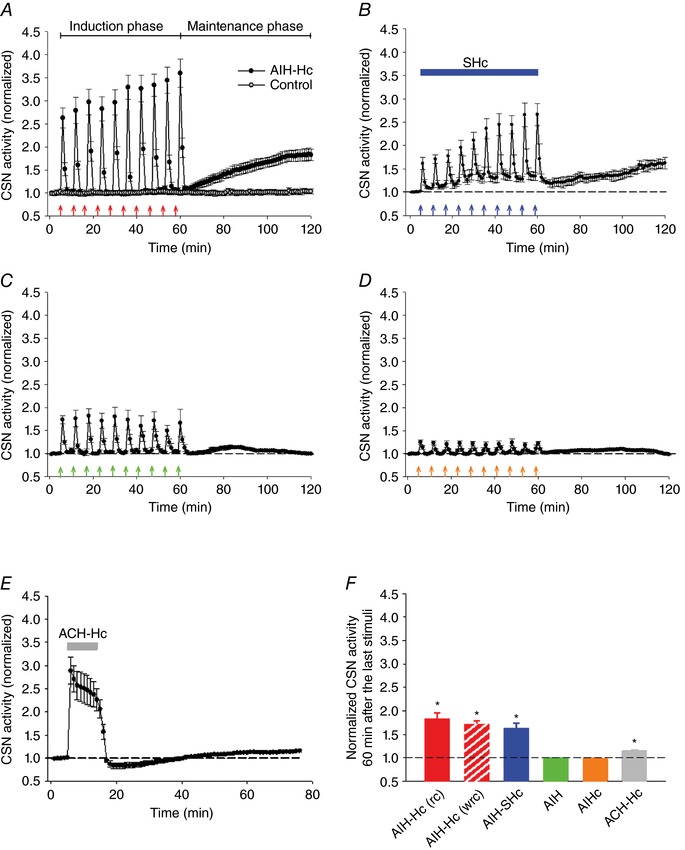
Average integrated CSN activity in response to: (A) no gaseous challenge (control; open circles) or AIH‐Hc (red arrows; closed circles); (B) 10 bouts of hypoxia ( = 60 Torr; blue arrows) with concurrent sustained Hc (SHc) ( = 50 Torr; blue horizontal bar); (C) 10 bouts of hypoxia ( = 40 Torr; green arrows); (D) 10 bouts of Hc ( = 60 Torr; orange arrows); and (E) 10 min of acute continuous hypoxia‐hypercapnia (ACH‐Hc) ( = 40 Torr & = 60 Torr; grey bar). F, summary of CSN activity 60 min after the last stimuli. Note that concurrent hypercapnia, which enhances the acute response of the CB to hypoxia, was necessary to induce sensory LTF. * P < 0.05 compared to baseline (dashed line) (one‐way ANOVA). All data are the mean ± SEM (n = 6 per group). rc, recirculated effluent; wrc, without recirculated effluent.
To determine whether sensory LTF can be induced by stimulating the CB with moderate AIH and sustained hypercapnia, i.e. AIH‐SHc, naive CBs were challenged with 10 episodes of moderate AIH ( = 60 Torr) with a hypercapnia background ( = 50 Torr). CSN activity increased progressively with each bout of AIH. During induction, baseline activity remained elevated after termination of each AIH challenge as a result of the background hypercapnia (Fig. 2 B). Activity levels 60 min after the last challenge with the preparation receiving normoxia‐normocapnia perfusate demonstrated that the combination of moderate AIH with sustained hypercapnia also resulted in sensory LTF (pre AIH baseline 1.00 ± 0.02 vs. baseline 60th minute post AIH 1.62 ± 0.12, P < 0.05, n = 6) (Fig. 2 B).
To determine whether sensory LTF can also be induced by stimulating the CBs with either AIH or AIHc alone; normal CBs were subjected to ten episodes of AIH ( = 40 Torr) or AIHc ( = 60 Torr). Each bout of hypoxia or hypercapnia augmented the CSN activity; however, the characteristic progressive increase in CSN activity seen following AIH‐Hc was absent (AIH: 1.01 ± 0.02 vs. 0.99 ± 0.01; P = 0.32; n = 6; AIHc: 0.99 ± 0.01 vs. 0.98 ± 0.01; P = 0.53; n = 6) (Fig. 2 C and D).
To determine whether the temporal pattern of the stimuli is critical for inducing sensory LTF, we tested the effects of an ACH‐Hc, equivalent to the cumulative dose during AIH‐Hc. ACH‐Hc elicited a robust increase in CSN activity followed by post stimuli depression on returning to normoxia‐normocapnia. At 60 min after ACH‐Hc, sensory LTF was minimal (baseline pre‐stimulus vs. 60th minute post stimulus: 1.01 ± 0.01 vs. 1.15 ± 0.02; n = 6; P < 0.05) (Fig. 2 E) and only a fraction of that induced by AIH‐Hc (1.15 ± 0.02 vs. 1.82 ± 0.13; n = 6; P < 0.05) or AIH with a background of hypercapnia (1.15 ± 0.02 vs. 1.62 ± 0.12; n = 6; P < 0.05). The above observations confirm and extend our previous findings (Cummings & Wilson, 2005) suggesting that induction of sensory LTF is possible without CIH pre‐conditioning and depends on a diverse stimulus patterning and strength (Fig. 2 F).
Sensory LTF increases CB sensitivity to hypoxia and temperature
To determine whether sensory LTF increases the sensitivity of the CB to hypoxia, ex vivo CBs were subjected to brief (3 min) hypoxic challenges ( = 60 Torr) pre AIH‐Hc and 60 min after the 10th AIH‐Hc bout. The data shown in Fig. 3(A and B) show a marked increase in CSN activity to acute hypoxia during the maintenance phase of sensory LTF (ΔHx pre‐sensory LTF vs. ΔHx during sensory LTF: 0.56 ± 0.06 vs. 0.85 ± 0.07; P < 0.001; n = 5). In another set of experiments, we assessed the temperature sensitivity of CBs subjected to AIH‐Hc treatment (Fig. 3 C and D). At 1 h after the 10th AIH‐Hc bout, the CB response to an increase in basal perfusate temperature from 37°C to 39°C for 1 min was enhanced (ΔT pre‐sensory LTF vs. ΔT during sensory LTF: 0.30 ± 0.04 vs. 0.52 ± 0.05; P < 0.001; n = 5).
Figure 3. AIH‐Hc induced sensory LTF sensitizes responses to hypoxia and heat of CBs from naïve rats.
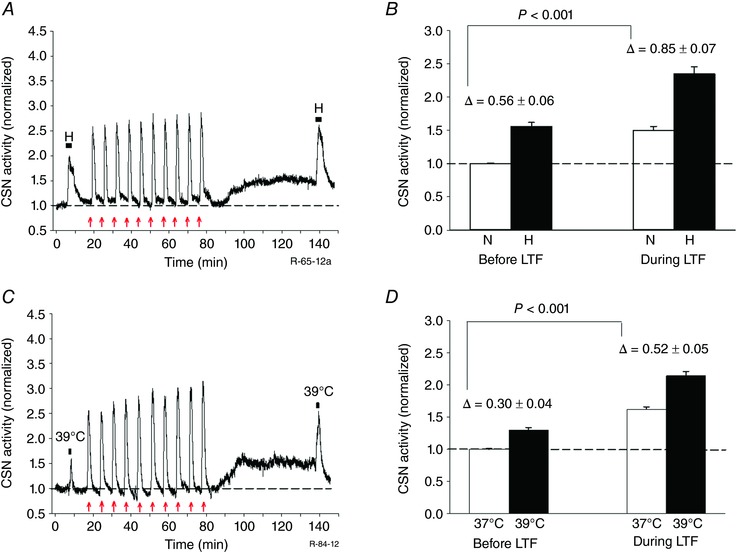
A and C, examples of integrated CSN responses to acute hypoxia ( = 60 Torr for 3 min; black horizontal bars) and temperature challenge (37°C to 39°C for 1 min; black horizontal bars), respectively, before and 60 min after 10 episodes of AIH‐Hc (red arrows). Note the increased hypoxic and temperature responses during the maintenance phase of sensory LTF compared to control (pre‐AIH‐Hc). B and D, summary of the effects of hypoxia and temperature challenges, respectively, before and during sensory LTF. Data are the mean ± SEM (n = 5 per group). Comparison between delta changes before and during sensory LTF. N, normoxia; H, hypoxia.
Maintenance of CB sensory LTF involves ionotropic P2X receptors
Because ATP and P2X2/3 purinergic receptors play important roles in hypoxic and hypercapnic responses at the level of the CB (Prasad et al. 2001; Zhang et al. 2000) and because non‐selective P2X receptor antagonist PPADS (pyridoxalphosphate‐6‐azophenyl‐2′,4′‐disulphonic acid) reduces phrenic LTF 60 min post AIH (Sibigtroth & Mitchell, 2011), we predicted the involvement of ATP and P2X receptors in sensory LTF. As shown in Fig. 4 A suramin (a broad spectrum P2X receptor blocker) (100 μm) applied 60 min post AIH‐Hc during the maintenance phase of sensory LTF, abolished sensory LTF within 10 min (sensory LTF pre suramin vs. sensory LTF post suramin: 1.60 ± 0.11 vs. 1.03 ± 0.02; P < 0.001; n = 5). To determine the subtypes of P2X receptors involved in the maintenance of sensory LTF, experiments were carried out using specific P2X receptor blocker TNP‐ATP, which selectively inhibits P2X2/3. TNP‐ATP (10 μm) abolished sensory LTF (sensory LTF pre TNP‐ATP vs. sensory LTF post TNP‐ATP: 1.55 ± 0.10 vs. 0.94 ± 0.03; n = 5; P < 0.001) (Fig. 4 B). Overall, these data suggest that ATP release and activation of P2X2/3 are critical for the maintenance of sensory LTF induced by AIH‐Hc.
Figure 4. AIH‐Hc induced sensory LTF involves P2X receptors and increases sensitivity to ATP.
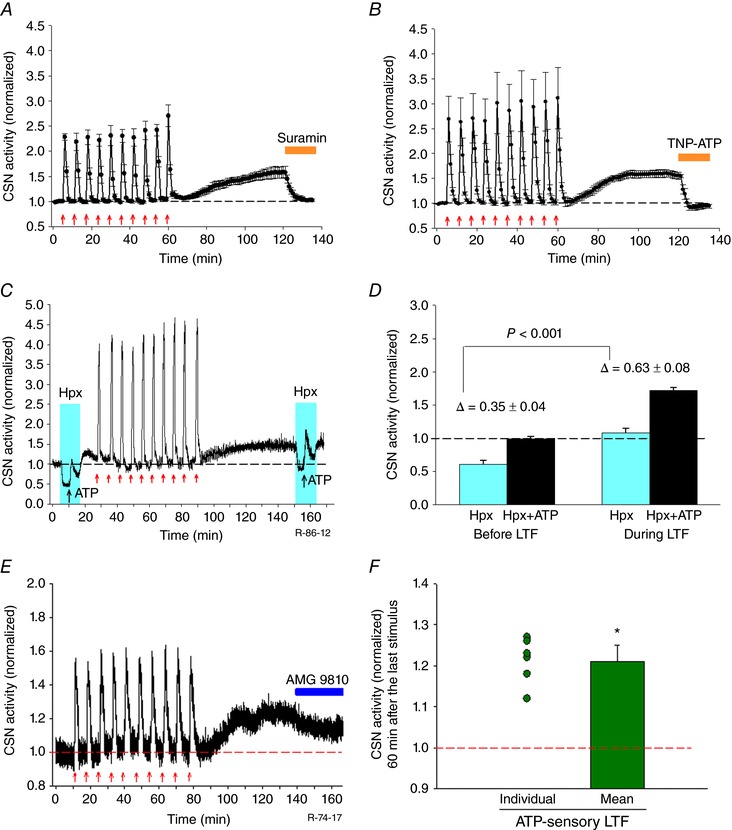
A and B, average integrated CSN activity showing sensory LTF induced by AIH‐Hc is eliminated by broad spectrum purinergic P2X receptor antagonist suramin and P2X 2/3 receptor‐specific antagonist TNP‐ATP, respectively (orange bars; mean ± SEM; n = 6). C, integrated CSN activity from one preparation showing the response to a bolus of ATP (1 ml of 100 μM ATP for 1 min; black arrows) in hyperoxia ( ∼ 500 Torr; blue areas) before and during sensory LTF. Prior to AIH‐Hc, hyperoxia, by silencing the CB, possibly eliminates presynaptic ATP release from glomus cells (their primary neurotransmitter) and all but abolishes CSN activity; this is transiently reversed by exogenous ATP, presumably acting on postsynaptic P2X receptors. After AIH‐Hc, hyperoxia reduces but does not abolish CSN activity and the ATP response is enhanced. D, summary of CSN activity in hyperoxia and with ATP challenges, before and during sensory LTF (mean ± SEM; n = 5); Comparison between delta changes with ATP, before and during sensory LTF. Hpx, hyperoxia. E, CB sensory responses to repetitive applications of 100 μm ATP (ten 1 min boluses at 5 min intervals; as indicated by the red arrows) produced mild sLTF; application of AMG 9810 (10 μm) partially suppressed sLTF (Fig. 5). F, individual scatter plot (n = 6) and average data of sensory LTF 60 min after the last ATP pulse (mean ± SEM; n = 6; * P < 0.05 compared to baeline). Hpx, hyperoxia.
To determine whether maintenance of sensory LTF is a result of presynaptic (glomus cells) or postsynaptic (CSN terminals) effects, we used hyperoxia ( = 500 Torr) to silence glomus cell activity and tested the postsynaptic responses to exogenous ATP challenges. As expected, hyperoxia before AIH‐Hc reduced CSN activity close to zero; however, after AIH‐Hc, hyperoxia reduced but failed to abolish CSN activity (before AIH‐Hc vs. after AIH‐Hc; 0.61 ± 0.05 vs. 1.08 ± 0.07; P = 0.003; n = 5). In both cases and as expected, adding a 1 ml bolus of ATP (100 μm at 1 ml min−1) during hyperoxia to activate postsynaptic purinergic receptors transiently stimulated CSN activity. Of note, however, the ATP challenge during the LTF maintenance phase was significantly more efficacious (ΔATP vs. ΔATP + sensory LTF: 0.35 ± 0.04 vs. 0.63 ± 0.08; P < 0.001; n = 5) (Fig. 4 c and d). These data suggest that a postsynaptic mechanism participates in the CB plasticity evoked by AIH‐Hc.
If postsynaptic P2X receptor activation is critical for AIH‐Hc induced sensory LTF, spaced application of exogenous ATP should evoke sensory LTF of the CB. Accordingly, 10 repeated applications of a 1 ml bolus of ATP (100 μm at 1 ml min−1 without recirculation of effluent) induced sensory LTF (0.99 ± 0.01 vs. 1.21 ± 0.04; P < 0.05; n = 6) (Fig. 4 E and F). However, the magnitude of sensory LTF was significantly less than the sensory LTF seen with AIH‐Hc (1.21 ± 0.04 vs. 1.82 ± 0.13; P < 0.001; n = 6).
Maintenance of CB sensory LTF involves TRPV1 receptors
To assess the role of TRPV1 receptors in AIH‐Hc evoked sensory LTF, we used the TRPV1 antagonist AMG9810 (10 μm) (Gavva et al. 2005; Roy et al. 2012) 60 min after the 10th AIH‐Hc bout during the maintenance phase of sensory LTF. Figure 5 A shows that 30 min of AMG9810 application suppressed CB sensory LTF by ∼50% and the remaining was suppressed by suramin. Thus, AMG 9810 reduces sensory LTF (sensory LTF vs. sensory LTF + AMG9810: 1.86 ± 0.20 vs. 1.48 ± 0.12; P = 0.017; n = 5; Fig. 5 B) but does not reduce it to baseline (1.48 ± 0.12 vs. 1.00 ± 0.01; P = 0.004; n = 5) (Fig. 5 B). This partial blockade was not related to the dose of AMG 9810 because, in preliminary studies, increasing the dose of the antagonist did not improve the suppressive effect. In another set of experiments, AMG 9810 (10 μm) application failed to produce any effect on the augmented CSN activity induced by 10 min of ACH‐Hc (ACH‐Hc vs. ACH‐Hc + AMG9810: 2.77 ± 0.5 vs. 2.77 ± 0.5; P = 0.97; n = 6) (Fig. 5 C and D), suggesting that sustained stimulus has no postsynaptic influence on TRPV1 receptors. Overall, the results indicate that both P2X and TRPV1 receptor activation is involved in sensory LTF.
Figure 5. AIH‐Hc induced sensory LTF involves heat‐sensitive TRPV1 receptors and increases CB heat sensitivity.
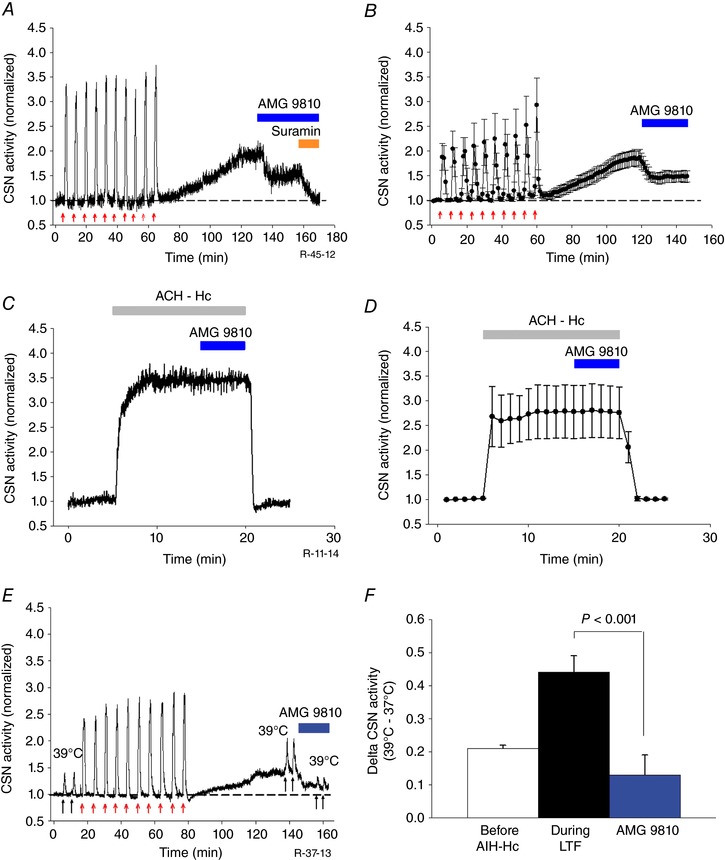
A, integrated CSN activity from one preparation showing ∼50% of sensory LTF induced by AIH‐Hc is inhibited by TRPV1 receptor antagonist AMG 9810 (10 μm; blue bar). The facilitated activity that remained was completely suppressed by suramin (100 μm; orange bar). B, average integrated CSN activity showing the effect of AMG9810 on sensory LTF (mean ± SEM; n = 5). C and D, integrated CSN activity from one preparation and average data (mean ± SEM; n = 6; P = 0.97) showing that AMG 9810 had no effect on the response to 10 min of acute continuous hypoxia‐hypercapnia (ACH‐Hc) (grey bar). E, integrated CSN activity from one preparation showing responses to temperature challenges (37 to 39°C for 1 min each; black arrows) before, 60 min after 10 episodes of AIH‐Hc (red arrows) and, subsequently, with partial inhibition of sensory LTF with TRPV1 antagonist AMG9810 (10 μm). F, summary of CSN responses following temperature challenge. Data are the mean ± SEM; n = 5.
To further establish the involvement of TRPV1 receptors in AIH‐Hc induced sensitization of the CB, we exploited the temperature sensitivity of the TRPV1 channel (Caterina et al. 1997; Roy et al. 2012) and tested whether the increase in temperature sensitivity (Fig. 3 C) during the maintenance phase of sensory LTF was mediated via TRPV1 receptor sensitization. AMG 9810 (10 μm) reduced sensory LTF (as above) and blocked almost half of the CSN response to a temperature challenge from 37o to 39°C (sensory LTF ΔT vs. sensory LTF ΔT+AMG9810: 0.44 ± 0.05 vs. 0.13 ± 0.06; P < 0.001; n = 5) (Fig. 5 E and F).
Maintenance of CB sensory LTF involves TRPV1 receptors in vivo
To assess the role of TRPV1 receptors in AIH‐Hc evoked sensory LTF in vivo, we used the TRPV1 antagonist AMG9810 (100 mg kg−1) (Gavva et al. 2005) 60 min after the 10th AIH‐Hc bout, during the maintenance phase of sympathetic LTF. Under both normoxic and hyperoxic conditions, and pH were maintained within physiologically normal levels (Table 1). AIH‐Hc caused an increase in splanchnic SNA under both hyperoxic (64.1 ± 12%) and normoxic (60.7 ± 9.4%) conditions at 60 min after the 10th AIH‐Hc (Fig. 6 C). Vehicle control (100% DMSO; 1 ml kg−1) or AMG9810 (100 mg kg−1) was administered i.p. and recordings were maintained for another 30 min. Animals that received the vehicle control showed a continuing increase in splanchnic SNA, whereas the animals treated with AMG9810 showed a reduction but only under normoxic conditions (Fig. 6 A, B and D). Compared to the activity present at 60 min following the 10th AIH‐Hc, the DMSO treatment group increased activity by a further ∼50% under both normoxic and hyperoxic conditions (Fig. 6 E). The SNA of the hyperoxic AMG9810 group also increased by a further 30.7 ± 18.3%, whereas the SNA of the AMG9810 treated normoxic group was significantly decreased by 32.0 ± 10.1% (P = 0.01; n = 6) (Fig. 6 E). Thus, under the in vivo condition, TRPV1 receptors contribute to sympathetic LTF only when the CB is active.
Figure 6. Effect of TRPV1 antagonism on AIH‐Hc‐induced sympathetic LTF in naïve rats in vivo .
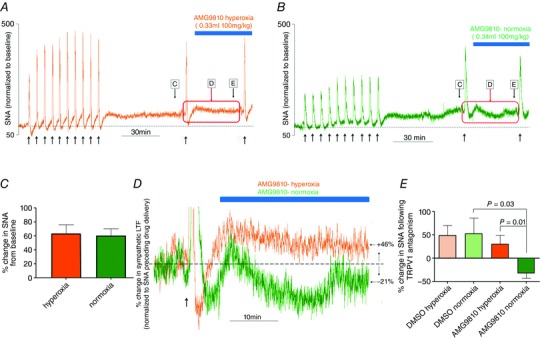
A and B, integrated sympathetic responses to AIH‐Hc from two preparations with hyperoxic (orange trace) and normoxic (green trace) backgrounds, respectively. Note the sustained increase in sympathetic activity (indicative of sympathetic LTF) following the last bout of hypoxia‐hypercapnia. C, the magnitude of sympathetic LTF 60 min post‐AIH‐Hc was not different between preparations given normoxia or hyperoxia backgrounds. D, TRPV1 antagonist AMG9810 reduced sympathetic LTF, in preparations with a normoxic background (green) when the CB was functional but not in preparations with a hyperoxic background (orange). E, summary data showing percentage change in sympathetic activity following DMSO or AMG9810 administration in preparations with normoxic and hyperoxic backgrounds. AMG9810 significantly decreased sympathetic LTF under normoxic conditions but had no effect under hyperoxic conditions. Data are the mean ± SEM (n = 6).
Effects of anti‐hypertensive drugs ketanserin and losartan on the AIH‐Hc evoked sensory LTF
Previously, 5‐HT2 and angiotensin (Ang) type 1 (AT1) receptors were shown to be required for sensory LTF induced by AIH in CIH preconditioned animals (Peng et al. 2006, 2011). Therefore, we examined whether 5‐HT2 and AT1 receptors also contribute to CB sensory LTF induced by AIH‐Hc in naïve animals. 5‐HT2 receptor antagonist ketanserin (1 μm) applied throughout the experiment (i.e. during the induction and maintenance phases of sensory LTF) caused a progressive decline in CSN activity with each AIH‐Hc bout and abolished manifestation of sensory LTF as indicated by a reduction in CSN activity (pre AIH‐Hc vs. 60 min post AIH‐Hc: 1.01 ± 0.01 vs. 0.88 ± 0.03; P = 0.002; n = 5) (Fig. 7 A). However, when applied 60 min after the 10th AIH‐Hc challenge, during the sensory LTF maintenance phase, LTF was unaffected (sensory LTF vs. ketanserin + sensory LTF: 1.56 ± 0.08 vs. 1.54 ± 0.10; P = 0.48; n = 5) (Fig. 7 B). Losartan (3 μm), an AT1 receptor antagonist, had similar effects (Fig. 7 C and D). Thus, it appears that both 5‐HT2 and AT1 receptor activation is necessary to induce but not to maintain sensory LTF.
Figure 7. Involvement of 5‐HT2 and AT1 receptors, and PKC activation in AIH‐Hc induced sensory LTF in CBs from naïve rats.
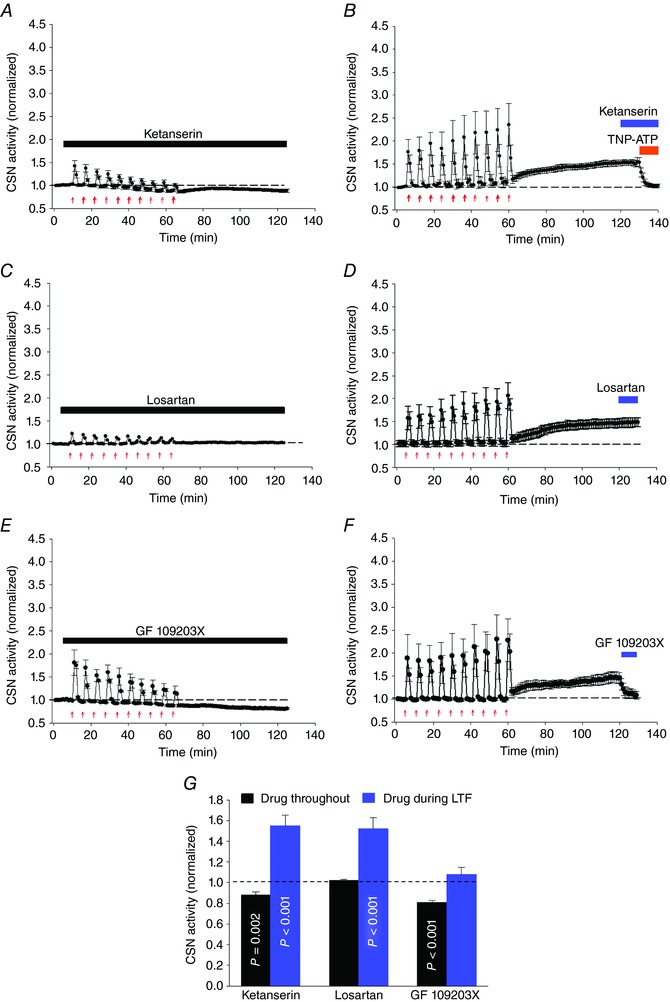
Average integrated CSN activity (mean ± SEM; n = 5) showing that the anti‐hypertensive drugs ketanserin (A, 1 μm; 5‐HT2 receptor antagonist) and losartan (C, 3 μm; AT1 receptor antagonist) applied throughout the experiment diminished responses to hypoxia‐hypercapnia bouts (red arrows) and prevented sensory LTF. B and D, ketanserin and losartan, respectively, had no effect during the maintenance phase (i.e. once sensory LTF was established), whereas TNP‐ATP (P2X 2/3 receptor‐specific antagonist) completely abolished sensory LTF (as shown in Fig. 4 B). E, PKC inhibitor (GF 109203X; 10 μm) applied throughout the experiment also diminished responses to hypoxia‐hypercapnia bouts and prevented sensory LTF. F, when applied during the maintenance phase, GF 109203X potently inhibited sensory LTF (mean ± SEM; n = 5). G, summary data; P values were determined by comparing CSN activities with drugs applied throughout/after sensory LTF and the basal responses before AIH‐Hc (dashed line). Data are the mean ± SEM; n = 5).
AIH‐Hc induced sensory LTF involves protein kinase C (PKC) activation
To determine whether sensory LTF induced by AIH‐Hc involves increased phosphorylation through PKC activation, the effects of GF109203X (10 μm), a potent and selective inhibitor of PKC (Toullec et al. 1991) during the induction and maintenance of sensory LTF were examined. As shown in Fig. 7 E, GF 109203X applied throughout the experiment completely prevented sensory LTF, with a prominent decrease in baseline activity (pre AIH‐Hc vs. 60 min post AIH‐Hc: 1.01 ± 0.02 vs. 0.83 ± 0.02; P < 0.001; n = 5). To establish whether or not increased phosphorylation was essential to sustain sensory LTF, GF 109203X was applied 60 min after AIH‐Hc during the maintenance phase of sensory LTF. As shown in Fig. 7 F, GF 109203X blocked sensory LTF, demonstrating that ongoing phosphorylation is required to maintain sensory LTF (sensory LTF vs. sensory LTF + GF109203X: 1.51 ± 0.12 vs. 1.08 ± 0.07; P < 0.001; n = 5). Because CaMKII has been implicated in phosphorylation of TRPV1 receptor (Jung et al. 2004) and is present in the CB (Pokorski et al. 2012), we tested the effects of the CaMKII inhibitor AIP (2 μm) in three preparations, although it had no apparent effect on the maintenance phase of sensory LTF (sensory LTF vs. sensory LTF + AIP: 1.72 ± 0.12 vs. 1.73 ± 0.13; P = 0.95; n = 3). Thus, phosphorylation mediated by PKC is essential to induce and maintain sensory LTF evoked by AIH‐Hc in naïve CB. Overall, the data indicate that the anti‐hypertensive drugs ketanserin and losartan cannot prevent sensory LTF once it is established, although PKC mediated phosphorylation is essential for the development and maintenance of sensory LTF (Fig. 7 G).
ROS scavenger does not block sensory LTF when applied post AIH‐Hc
To determine whether ROS (O2·¯) are involved in induction and maintenance of sensory LTF, we used MnTMPyP (25 μm), a membrane permeable O2¯ scavenger. When applied throughout the entire experiment, the CSN responses after the first bout of AIH‐Hc were not incremental but rather indicated a decline in activity with subsequent bouts. Moreover, manifestation of sensory LTF 60 min after the last bout was significantly reduced (baseline 1.00 ± 0.03 vs. sensory LTF + MnTMPyP 1.10 ± 0.05; P = 0.008; n = 5) (Fig. 8 A). ROS therefore probably did not play an important role in the acute response of the CB to hypoxia‐hypercapnia but are essential for sensory LTF induction. These findings are consistent with the role of ROS described for CIH‐dependent CB sensory LTF (Peng et al. 2009). However, when MnTMPyP was applied 60 min after the 10th AIH‐Hc episode, during the maintenance phase of sensory LTF, LTF was not diminished; indeed, CSN activity was stimulated (sensory LTF vs. sensory LTF+ MnTMPyP: 1.52 ±0.12 vs. 1.82 ± 0.20; P = 0.03; n = 5) (Fig. 8 B). These data suggest that ROS (O2·¯) are not essential for sensory LTF maintenance and may limit the sensory LTF magnitude (Fig. 8 E).
Figure 8. ROS (O2·¯), but not H2O2, is involved in sensory LTF induction evoked by AIH‐Hc in naïve CB.
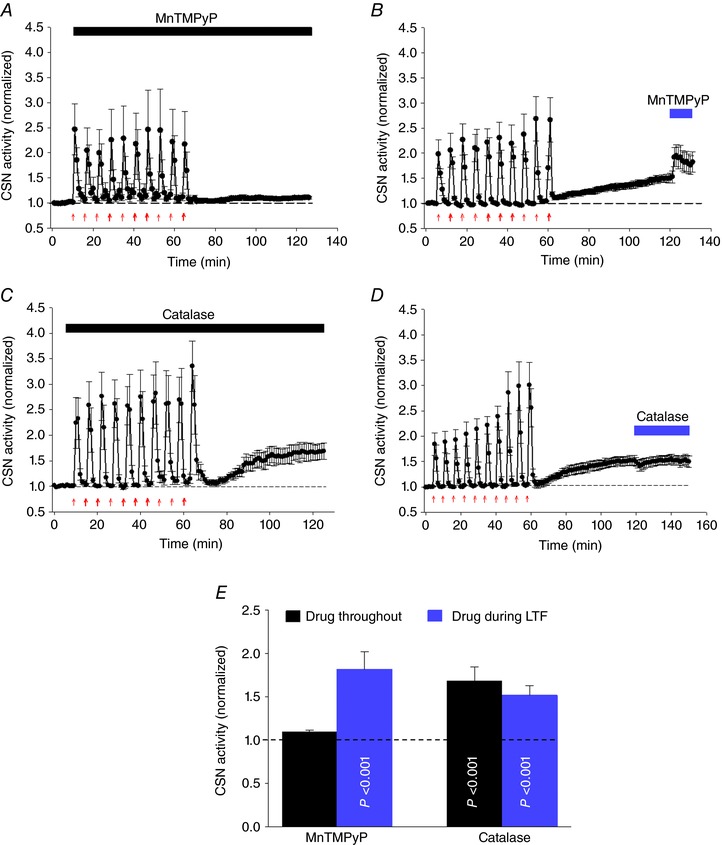
A and B, average integrated CSN activity (mean ± SEM; n = 5) showing that ROS scavenger MnTMPyP (25 μm) applied throughout (red arrows) prevents sensory LTF but failed to suppress sensory LTF 60 min after the last hypoxia‐hypercapnia bout. C and D, average integrated CSN activity (mean ± SEM; n = 5) demonstrating the H2O2 scavenger PEG‐catalase (200 U ml−1) had no effect when applied either the induction or maintenance phase of sensory LTF. E, summary data of MnTMPyP and catalase applied throughout and after sensory LTF induction. P values were determined by comparing CSN activities with basal responses (dashed line) before AIH‐Hc stimuli. Data are the mean ± SEM; n = 5.
H2O2 is not involved in sensory LTF induced by AIH‐Hc
PEG‐catalase, a membrane permeant enzyme that breaks down H2O2 releasing O2 (George, 1947) is reported to reverse sensory LTF induced by AIH in CIH preconditioned animals (Peng et al. 2009). However, when tested in naïve CBs exposed to AIH with concurrent hypercapnia, PEG‐catalase (200 U ml–1) had no effect whatsoever on sensory LTF if applied during the entire experiment (i.e. during both the induction and maintenance phases; baseline vs. sensory LTF + PEG‐catalase: 1.00 ± 0.01 vs. 1.69 ± 0.16; P < 0.05; n = 5) (Fig. 8 C) and caused only a transient decrease in activity when applied during the maintenance phase (sensory LTF vs. sensory LTF+ PEG‐catalase: 1.52 ± 0.10 vs. 1.50 ± 0.11; P = 0.51, n = 5) (Fig. 8 D). These data suggest that H2O2 is not involved in this CB plasticity mechanism (Fig. 8 E).
Discussion
The primary novel findings of the present study are that a robust and long‐lasting activity‐dependent plasticity in CB can be induced by AIH‐Hc without CIH‐preconditioning. This sensory LTF requires P2X 2/3 receptor activation and is dependent on TRPV1 receptors. As P2X 2/3 and TRPV1 receptors are located in petrosal sensory afferents (not O2‐sensitive glomus cells) (Prasad et al. 2001) and repeated pulses of ATP induced mild sensory LTF, one of the main sites of plasticity appears to be postsynaptic. Our data suggest that several features of this plasticity are shared by CIH‐dependent sensory LTF because the induction of plasticity is dependent on 5‐HT, Ang II, PKC and ROS signalling (Peng et al. 2003, 2006, 2009, 2011). However, H2O2 appears to play no part in sensory LTF induction and 5‐HT, Ang II, ROS and H2O2 are not required for sensory LTF maintenance (Fig. 9). Because our in vivo experiments highlight a role for TRPV1‐dependent CB activity in sympathetic LTF, we suggest that TRPV1‐dependent sensory LTF constitutes an important mechanistic link contributing to the well‐documented association between sleep apnoea, sympathoexcitation, hypertension and cardiovascular disease.
Figure 9. Schematic diagram comparing mechanisms of sensory LTF induction and maintenance by AIH requiring CIH pre‐conditioned and AIH‐Hc in CB from naïve rats.
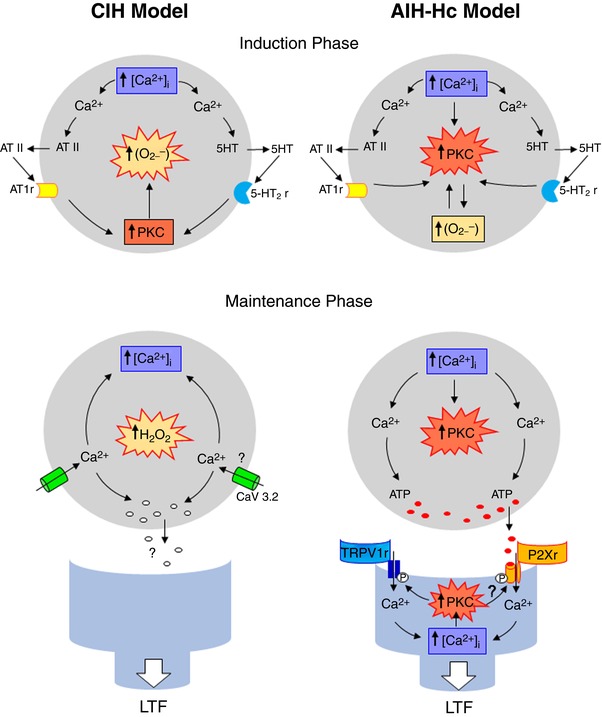
Although sensory LTF requiring CIH pre‐conditioning is largely dependent on O2.− and subsequent production of H2O2, sensory LTF resulting from AIH with concurrent hypercapnia is largely dependent on PKC. Moreover, our novel data suggest that sensory LTF resulting from AIH with concurrent hypercapnia is P2X‐dependent, involves TRPV1 and is predominantly postsynaptic.
Role of hypercapnia in sensory LTF in the naïve CB
Peng et al. (2003) reported that only CIH‐preconditioned CBs manifest sensory LTF in response to AIH in vitro (i.e. the superfused CB). This phenomenon was not evident in CBs extracted from naïve animals. Our demonstration of robust sensory LTF induced without preconditioning may be the consequence of preparation differences (we used a perfused CB preparation in which arterial and were precisely controlled and could be changed quickly) or our choice of acute stimuli. Notably, we could induce robust sensory LTF with either AIH against a background of hypercapnia or with AIH‐Hc against a background of normoxic normocapnia. However, we could not induce robust sensory LTF with AIH alone, AIHc alone or following ACH‐Hc. These results demonstrate the need to combine AIH with hypercapnia in the naïve CB and also show that the composition, pattern and duration of gaseous stimuli are all important for the induction of sensory LTF (Cummings & Wilson, 2005). These CB data parallel our in vivo data demonstrating the involvement of the CB in induction of sympathetic LTF with AIH‐Hc.
Nonetheless, the importance of hypoxia vs. hypercapnia in inducing sympathetic LTF warrants further study. In the clinical setting, chronic and severe hypercapnia is common in sleep apnoea, and the sizable decrease in that occurs with each apnoea is accompanied by increased (Lanphier & Rahn, 1963; Chin et al. 1997). In healthy humans, ventilatory and sympathetic LTF may be enhanced and/or only evident when AIH is presented with an elevated baseline level of CO2 (Morgan et al. 1995; Harris et al. 2006). However, in rats, AIH and CIH alone can induce sympathetic LTF. With CIH, where sympathetic LTF is accompanied by hypertension, Fletcher (2000) reports that adding CO2 to CIH does not further elevate sympathetic LTF or blood pressure (Bao et al. 1997; Lesske et al. 1997; Dick et al. 2007; Xing & Pilowsky 2010). These results are somewhat unexpected because hypercapnia might be expected to increase sympathetic LTF based on: (i) hypercapnia increasing the CB and ventilatory responses to hypoxia (Lahiri & Delaney, 1975; Wilson & Teppema, 2016) and (ii) Fletcher (2000) and coworkers (Bao et al. 1997) showing that the addition of inspired CO2 decreases the caused by inspired hypoxia and augments the effects of each hypoxic bout on sympathetic activity and blood pressure (Bao et al. 1997). Because acute intermittent electrical stimulation of the CSN nerve in naïve animals also causes respiratory (and presumably sympathetic) LTF (Hayashi et al. 1993) and hypercapnia is required for AIH‐induced sensory LTF in naïve animals (present study), we propose that sympathetic LTF results from central and/or CB plasticity (with CB plasticity requiring CIH precondition or concurrent hypercapnia), with sympathetic LTF having an upper cap that limits sympathetic LTF magnitude and gives rise to CB/central redundancy. The concept of competing sites of plasticity is reminiscent of that proposed for respiratory LTF (Hayashi et al. 1993) and is discussed further below in the context of our in vivo data.
In the present study, we show that CB sensory LTF comprises two distinct phases: induction and maintenance that require different cellular processes. This is reminiscent of the role of 5‐HT and adenosine receptors in the maintenance of phrenic LTF, for which 5‐HT and/or adenosine receptor activation is necessary to initiate but not maintain phrenic LTF following episodic hypoxia (Fuller et al. 2001; Fields & Mitchell, 2015). Also, activation of spinal orexin receptors is critical for enhancing chemoreflex phrenic responses to hypoxia after AIH (Kim et al. 2016b).
Mechanism of induction
Our data demonstrate that the induction of sensory LTF in naïve CBs bears some resemblance to that proposed for sensory LTF requiring CIH preconditioning (Peng et al. 2003). Peng et al. (2006, 2011) demonstrated that sensory LTF mirroring that requiring CIH could be induced by repetitive applications of 5‐HT and Ang II in the naïve CB. In our model, 5‐HT2 and Ang II receptor antagonists caused a profound decrease in the response magnitude to AIH‐Hc stimuli and completely abolished sensory LTF (Fig. 7 A and C). The importance of endogenous 5‐HT2 and Ang II receptor activation in sensory LTF suggests that the induction of sensory LTF is dependent on ongoing PKC‐mediated phosphorylation (Peng et al. 2006). Indeed, the PKC blocker GF109203X decreased the response magnitude to consecutive stimuli in our preparation and abolished the induction of sensory LTF in both naïve and CIH‐preconditioned CBs. Using the ROS scavenger (MnTMPyP), Peng et al. (2003, 2009) also demonstrated that ROS (O2·¯) are necessary for the induction of CIH‐preconditioned sensory LTF. Similarly, we show that MnTMPyP reduces consecutive responses to stimulus and prevents the induction of sensory LTF (Fig. 8 A). Thus, 5‐HT2, Ang II, PKC and O2·¯ mediated‐signalling influences the magnitude of response to individual stimuli and strongly influences the induction of both forms of sensory LTF. We therefore suggest that the mechanism of sensory LTF induction in CIH‐ preconditioned and naϊve rats is similar, sharing a dependence on PKC‐modulation of stimulus amplitude (Fig. 9, induction phase). However, PEG‐catalase had no effect on induction of sensory LTF in the present study and, as such, our data do not support a role for H2O2, proposed as the mechanistic lynch pin between O2·¯ and glomus cell Cav3.2 modification in CIH‐preconditioned sensory LTF (Makarenko et al. 2016).
Maintenance of sensory LTF
The release of ATP from glomus cells resulting in activation of postsynaptic P2X receptors on the terminals of chemosensory afferents plays an essential role in the acute hypoxic response of the CB (Prasad et al. 2001; Rong et al. 2003). We show that activation of P2X receptors is also important for sensory LTF maintenance because sensory LTF could be almost completely reversed with suramin. The effects of P2X receptor antagonism may reflect presynaptic plasticity resulting in an increased release of ATP and/or a postsynaptic mechanism causing an increased responsiveness to ATP. Although we cannot exclude a presynaptic mechanism, three lines of evidence suggest plasticity occurs postsynaptically. (i) The sensory response to exogenous test pulse of ATP, presumably caused by activation of P2X receptors on sensory afferents and not P2Y receptor on Type II cells (Murali and Nurse, 2016), was increased following sensory LTF induction (Fig. 4 C and D). This may be a consequence of modification of the expression or function of P2X receptors or it may reflect plasticity downstream. (ii) Sensory LTF could be induced by replacing bouts of gaseous stimuli with exogenous pulses of ATP (Fig. 4 E and F). We note that the magnitude of sLTF induced with pulses of ATP was less than that with AIH‐Hc, probably reflecting differences in the temporal pattern and magnitude of ATP within the synaptic cleft. Consistent with this explanation, Peng et al. (2011) were unable to demonstrate this phenomenon in their en bloc preparation, possibly because they used fewer pulses of ATP (5 bouts compared to 10 bouts) and there were also increased wash in and washout times associated with superfusion compared to perfusion. (iii) Perhaps the strongest argument for postsynaptic plasticity within the CB is the involvement of TRPV1 receptors that are located exclusively on postsynaptic sensory afferents (Roy et al. 2012). Indeed, blocking TRPV1 during the maintenance phase with AMG9810 reduced sensory LTF by half and, unlike P2X receptors, TRPV1 plays no significant role in acute hypoxic‐hypercapnic responses (Fig. 5 C and D). This suggests that AIH‐Hc leads to postsynaptic TRPV1 receptor modification and persistent activation. In other systems where P2X and TRPV1 receptors co‐localize, the activation of purinoceptors by ATP augments ionic currents induced by activation of TRPV1 receptors, presumably via TRPV1 phosphorylation (Bhave et al. 2003; Saloman et al. 2013). Consistent with the possibility that TRPV1 phosphorylation is a critical step in sensory LTF, we demonstrate that both the induction and maintenance phases of sensory LTF could be reversed by the general PKC inhibitor, GF 109203X (Fig. 7 E and F).
Interestingly, PKC activation can lead to generation of ROS (O2·¯ and H2O2) (Lee et al. 2004) and O2·¯ and H2O2 can activate P2X and TRPV1 receptors (Shen et al. 2012; Lin et al. 2013; Ruan et al. 2014). Moreover, the explanation proposed by Peng et al. (2009) for the changing role of O2·¯ in sensory LTF (MnTMPyP being important for the initiation but not maintenance of sensory LTF requiring CIH preconditioning) is that O2·¯ becomes ineffective when dismutated to H2O2, and H2O2 is responsible for maintaining sensory LTF. Thus, PEG‐catalase, a H2O2 scavenger, applied 15 min after the onset of sensory LTF, completely prevented sensory LTF requiring CIH preconditioning (Peng et al. 2009). Motivated by these observations, we tested the importance of ROS in TRPV1‐dependent sensory LTF. Although we also found that O2·¯contributed to the induction phase but not the maintenance phase, we found no evidence for a role for H2O2 during the induction phase. PEG‐catalase had no effect whatsoever during induction and, during the maintenance phase, it caused only a transient decrease in CSN (Fig. 8 C and D), consistent with the brief oxygen burst probably occurring with catalase application (George, 1947) and a concomitant brief inhibition of glomus cells. We therefore conclude that the role of ROS in sensory LTF induced by AIH with concurrent hypercapnia is limited to a role for O2·¯ in regulating the response magnitude during the induction phase, participating only indirectly in the plasticity mechanism (Fig. 9).
Sensory LTF increases the activity and sensitivity of the CB
During the maintenance phase of sensory LTF, in addition to increased tonic CSN activity, we observed an increased responsiveness to acute hypoxia (Fig. 3 A and B), heat (change from 370 to 39°C) (Fig. 3 C and D) and ATP (Fig. 4 C and D) challenges. This is reminiscent of the increased sensory responsiveness to hypoxia reported during sensory LTF in CIH‐preconditioned CBs, suggesting identical functional outcomes between the forms of sensory LTF induced by AIH with concurrent hypercapnia and CIH (Peng et al. 2003). With regard to breathing, increasing CB sensitivity is expected not only to increase the ability of an animal to respond to a single apnoea, but also to destabilize breathing, making apnoeas more probable; this may be partially compensated by the increased CB baseline activity that would be expected to increase the CB CO2 reserve (Smith et al. 2003). However, as our in vivo data suggest, TRPV1‐dependent CB sensory LTF can contribute to sympathetic LTF. In a clinical setting, a long‐lasting increase in sympathetic tone would probably lead to target organ damage (vessels, heart, brain and kidney in particular), chronic metabolic acidosis, obesity, insulin resistance and neurogenic hypertension.
Multiple sites of plasticity involved in sympathetic LTF
Our in vivo data demonstrate that sympathetic LTF is only sensitive to AMG9810 under conditions that support sensory LTF (i.e. with CBs in a background of normoxia); AMG9810 was without effect on sympathetic LTF when CB activity between and after bouts of AIH‐Hc was suppressed (i.e. with CBs in a background of hyperoxia) (Fig. 6). These data are entirely consistent with a direct role for CB TRPV1‐mediated sensory LTF in sympathetic LTF when the CBs are fully functional. Moreover, the persistence of sympathetic LTF when sensory LTF is suppressed corroborates the concept discussed above in that there are additional presumed central sites capable of plasticity, located downstream of the CB (Xing et al. 2014; Yamamoto et al. 2015; Kim et al. 2016a, b).
Because the magnitude of sympathetic LTF is similar with or without the direct contribution of CBs to plasticity (Fig. 6 C–E), we propose that sympathetic LTF, regardless of the loci of plasticity, is capped. Accordingly, with sensory LTF suppressed, this upper cap is reached entirely by central plasticity but, when CBs are fully functional, plasticity in the form of sensory LTF contributes substantially.
With regard to the sympathetic LTF capping mechanism, this may be mediated entirely centrally. However, we note that sympathetic LTF has no effect on blood pressure in the anaesthetized in vivo preparation (Dick et al. 2007; Xing & Pilowsky 2010; present study). This leads us to speculate that the capping mechanism is baroreflex‐related, imposing the maximum long‐term (i.e. tens of minutes) increase in sympathetic activity possible without a corresponding increase in blood pressure. Accordingly, should a sustained increase in sympathetic activity above the cap occur, the resulting sustained increase in blood pressure is expected to trigger feedback inhibition, thus returning long‐term sympathetic activity to or below the cap and long term blood pressure back to resting.
Conclusions
The present study demonstrates plasticity in CB chemo‐afferent activity induced by AIH with concurrent hypercapnia, without CIH pre‐conditioning. Several features of this plasticity are shared with CIH‐dependent sensory LTF; however, other features are unique. Sensory LTF induced by AIH with concurrent hypercapnia is dependent on activation/sensitization of P2X and TRPV1 receptors, which are located in CB chemo‐afferent terminals, suggesting that one of the main sites of plasticity is postsynaptic. Sensory LTF probably has adaptive and maladaptive effects on the ability to respond to repeated apnoeas: over the short term, sensory LTF may be beneficial for mounting responses to hypoxia, whereas, over the long term, it probably destabilizes breathing and paves the way for neurogenic hypertension. As such, the present study provides a possible mechanistic role of the CB in cardiovascular disease and indicates P2X‐TRPV1 receptors as being novel targets for non‐surgical intervention in humans with uncontrolled hypertension.
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Author contributions
AR and RJAW conceived and designed the experiments. AR performed the en bloc experiments and analysis. FD performed the analysis. RJAW wrote the software to analyse CSN activity. MJF performed the in vivo experiments and analysis. AR, MJF and RJAW prepared the figures. AR, MJF and RJAW wrote the first draft of the manuscript. PP provided important edits. All experiments were performed in the laboratories of RJAW and PP. The final version of the manuscript submitted for publication was approved by all of the authors. All authors agree to be accountable for all aspects of the work with respect to ensuring that the questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by CIHR (RJAW) and NHMRC (PMP, MMJF, RJAW; Grant # 1065485). RJAW is an AIHS Senior Scholar.
Edited by: Kim Barrett & Harold Schultz
This is an Editor's Choice article from the 1 August 2018 issue.
Linked articles This article is highlighted by a Journal Club article by Amorim & de Deus. To read this Journal Club, visit https://doi.org/10.1113/JP275889.
References
- Bao G, Randhawa PM & Fletcher EC (1997). Acute blood pressure elevation during repetitive hypocapnic and eucapnic hypoxia in rats. J Appl Physiol 82, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu H‐J, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS & Gereau RW (2003). Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc Natl Acad Sci USA 100, 12480–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD & Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389, 816–824. [DOI] [PubMed] [Google Scholar]
- Chin K, Hiras M, Kuriyama T, Fukui M, Kuno K, Sagawa Y & Ohi M (1997). Changes in the arterial PCO2 during a single night's sleep in patirnts with obstructive sleep apnea. Intern Med 36, 454–460. [DOI] [PubMed] [Google Scholar]
- Cummings KJ & Wilson RJA (2005). Time‐dependent modulation of carotid body afferent activity during and after intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol 288, R1571–R1580. [DOI] [PubMed] [Google Scholar]
- Cutler MJ, Swift NM, Keller DM, Wasmund WL, Burk JR & Smith ML (2004). Periods of intermittent hypoxic apnea can alter chemoreflex control of sympathetic nerve activity in humans. Am J Physiol Heart Circ Physiol 287, H2054–H2060. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Iturriaga R & Schultz HD (2015). Editorial: carotid body: a new target for rescuing neural control of cardiorespiratory balance in disease. Front Physiol 6, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Hsieh Y‐H, Wang N & Prabhakar N (2007). Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92, 87–97. [DOI] [PubMed] [Google Scholar]
- Duffin J (2010). The role of the central chemoreceptors: a modeling perspective. Respir Physiol Neurobiol 173, 230–243. [DOI] [PubMed] [Google Scholar]
- Farnham MMJ, Li Q, Goodchild AK & Pilowsky PM (2008). PACAP is expressed in sympathoexcitatory bulbospinal C1 neurons of the brain stem and increases sympathetic nerve activity in vivo. Am J Physiol Regul Integr Comp Physiol 294, R1304–R1311. [DOI] [PubMed] [Google Scholar]
- Fields DP & Mitchell GS (2015). Spinal metaplasticity in respiratory motor control. Front Neural Circuits 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC (2000). Effect of episodic hypoxia on sympathetic activity and blood pressure. Respir Physiol 119, 189–197. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL & Mitchell GS (2001). Phrenic long‐term facilitation requires 5‐HT receptor activation during but not following episodic hypoxia. J Appl Physiol 90, 2001–2006. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Kilonsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC & Treanor JJ (2005). AMG 9810 [(E)‐3‐(4‐t‐butylphenyl)‐N‐(2,3‐dihydrobenzo[b][1,4]dioxin‐6‐yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther 313, 474–484. [DOI] [PubMed] [Google Scholar]
- George P (1947). Reaction between catalase and hydrogen peroxide. Nature 159, 41–43. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DP, Balasubramaniam A, Badr MS & Mateika JH (2006). Long‐term facilitation of ventilation and genioglossus muscle activity is evident in the presence of elevated levels of carbon dioxide in awake humans. Am J Physiol Regul Integr Comp Physiol 291, R1111–R1119. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Kohro Y, Tsuda M & Inoue K (2009). Activation of cytosolic phospholipase A2 in dorsal root ganglion neurons by Ca2+/calmodulin‐dependent protein kinase II after peripheral nerve injury. Mol Pain 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Coles SK, Bach KB, Mitchell GS & McCrimmon DR (1993). Time‐dependent phrenic nerve responses to carotid afferent activation: intact vs. decerebellate rats. Am J Physiol 265, R811–R819. [DOI] [PubMed] [Google Scholar]
- Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS & Leuenberger UA (2007). Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest 131, 1406–1413. [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H & Oh U (2004). Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin‐dependent kinase II regulates its vanilloid binding. J Biol Chem 279, 7048–7054. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim YJ, Kakall Z, Farnham MMJ & Pilowsky PM (2016a). Intermittent hypoxia‐induced cardiorespiratory long‐term facilitation: a new role for microglia. Respir Physiol Neurobiol 226, 30–38. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Pilowsky PM & Farnham MMJ (2016b). Intrathecal intermittent orexin‐A causes sympathetic long‐term facilitation and sensitizes the peripheral chemoreceptor response to hypoxia in rats. J Pharmacol Exp Ther 358, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S & DeLaney RG (1975). Stimulus interaction in the responses of carotid body chemoreceptor single afferent fibers. Respir Physiol 24, 249–266. [DOI] [PubMed] [Google Scholar]
- Lanphier EH & Rahn H (1963). Alveolar gas exchange during breath holding with air. J Appl Physiol 478, 478–482. [DOI] [PubMed] [Google Scholar]
- Lee HB, Yu MR, Song JS & Ha H (2004). Reactive oxygen species amplify protein kinase C signaling in high glucose‐induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int 65, 1170–1179. [DOI] [PubMed] [Google Scholar]
- Lesske J, Fletcher EC, Bao G & Unger T (1997). Hypertension caused by chronic intermittent hypoxia‐influence of chemoreceptors and sympathetic nervous system. J Hypertens 15, 1593–1603. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Hogeman CS, Quraishi SA, Quraishi S, Linton‐Frazier L & Gray KS (2007). Short‐term intermittent hypoxia enhances sympathetic responses to continuous hypoxia in humans. J Appl Physiol 103, 835–842. [DOI] [PubMed] [Google Scholar]
- Li H‐B, Mao R‐R, Zhang J‐C, Yang Y, Cao J & Xu L (2008). Antistress effect of TRPV1 channel on synaptic plasticity and spatial memory. Biol Psychiatry 64, 286–292. [DOI] [PubMed] [Google Scholar]
- Lin Y‐J, Hsu H‐H, Ruan T & Kou YR (2013). Mediator mechanisms involved in TRPV1, TRPA1 and P2X receptor‐mediated sensory transduction of pulmonary ROS by vagal lung C‐fibers in rats. Respir Physiol Neurobiol 189, 1–9. [DOI] [PubMed] [Google Scholar]
- Mahamed S & Mitchell GS (2007). Is there a link between intermittent hypoxia‐induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol 92, 27–37. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Ahmmed GU, Peng Y‐J, Khan SA, Nanduri J, Kumar GK, Fox AP & Prabhakar NR (2016). CaV3.2 T‐type Ca2+ channels mediate the augmented calcium influx in carotid body glomus cells by chronic intermittent hypoxia. J Neurophysiol 115, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani MP, Wang S & Somers VK (2015). Chemoreflex physiology and implications for sleep apnoea: insights from studies in humans. Exp Physiol 100, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsch R, Foeller E, Rammes G, Bunck M, Kössl M, Holsboer F, Zieglgänsberger W, Landgraf R, Lutz B & Wotjak CT (2007). Reduced anxiety, conditioned fear, and hippocampal long‐term potentiation in transient receptor potential vanilloid type 1 receptor‐deficient mice. J Neurosci 27, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJA, Sobotka PA & Paton JFR (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 1–11. [DOI] [PubMed] [Google Scholar]
- Mohan R & Duffin J (1997). The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol 108, 101–115. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Crabtree DC, Palta M & Skatrud JB (1995). Combined hypoxia and hypercapnia evokes long‐lasting sympathetic activation in humans. J Appl Physiol 79, 205–213. [DOI] [PubMed] [Google Scholar]
- Murali S & Nurse CA (2016). Purinergic signalling mediates bidirectional crosstalk between chemoreceptor type I cells and glial‐like type II cells of the rat carotid body. J Physiol 594, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Montano N, Dyken ME, Phillips BG & Somers VK (1998). Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation 97, 943–945. [DOI] [PubMed] [Google Scholar]
- Peng Y‐J, Overholt JL, Kline D, Kumar GK & Prabhakar NR (2003). Induction of sensory long‐term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100, 10073–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y‐J, Yuan G, Jacono FJ, Kumar GK & Prabhakar NR (2006). 5‐HT evokes sensory long‐term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol (Lond) 576, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y‐J, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK & Prabhakar NR (2009). NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci 29, 4903–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y‐J, Raghuraman G, Khan SA, Kumar GK & Prabhakar NR (2011). Angiotensin II evokes sensory long‐term facilitation of the carotid body via NADPH oxidase. J Appl Physiol 111, 964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorski M, Sakagami H & Okada Y (2012). Calcium/calmodulin‐dependent protein kinases in the carotid body: an immunohistochemical study. Springerplus 1, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng Y‐J, Jacono FJ, Kumar GK & Dick TE (2005). Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32, 447–449. [DOI] [PubMed] [Google Scholar]
- Prasad M, Fearon IM, Zhang M, Laing M, Vollmer C & Nurse CA (2001). Expression of P2X2 and P2X3 receptor subunits in rat carotid body afferent neurones: role in chemosensory signalling. J Physiol (Lond) 537, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong W, Gourine AV, Cockayne DA, Xiang Z, Ford APDW, Spyer KM & Burnstock G (2003). Pivotal role of nucleotide P2X2 receptor subunit of the ATP‐gated ion channel mediating ventilatory responses to hypoxia. J Neurosci 23, 11315–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Mandadi S, Fiamma M‐N, Rodikova E, Ferguson EV, Whelan PJ & Wilson RJA (2012). Anandamide modulates carotid sinus nerve afferent activity via TRPV1 receptors increasing responses to heat. J Appl Physiol 112, 212–224. [DOI] [PubMed] [Google Scholar]
- Ruan T, Lin Y‐J, Hsu T‐H, Lu S‐H, Jow G‐M & Kou YR (2014). Sensitization by pulmonary reactive oxygen species of rat vagal lung C‐fibers: the roles of the TRPV1, TRPA1, and P2X receptors. PLoS ONE 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloman JL, Chung M‐K & Ro JY (2013). P2X₃ and TRPV1 functionally interact and mediate sensitization of trigeminal sensory neurons. Neuroscience 232, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M‐Y, Luo Y‐L, Yang C‐H, Ruan T & Lai CJ (2012). Hypersensitivity of lung vagal C fibers induced by acute intermittent hypoxia in rats: role of reactive oxygen species and TRPA1. Am J Physiol Regul Integr Comp Physiol 303, R1175–R1185. [DOI] [PubMed] [Google Scholar]
- Sibigtroth CM & Mitchell GS (2011). Carotid chemoafferent activity is not necessary for all phrenic long‐term facilitation following acute intermittent hypoxia. Respir Physiol Neurobiol 176, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Nakayama H & Dempsey JA (2003). The essential role of carotid body chemoreceptors in sleep apnea. Can J Physiol Pharmacol 81, 774–779. [DOI] [PubMed] [Google Scholar]
- Somers VK & Abboud FM (1993). Chemoreflexes–responses, interactions and implications for sleep apnea. Sleep 16, S30–S33. [PubMed] [Google Scholar]
- Somers VK, Mark AL & Abboud FM (1988). Potentiation of sympathetic nerve responses to hypoxia in borderline hypertensive subjects. Hypertension 11, 608–612. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Bernardi L, Balsamo R, Ciancio N, Polosa R & Di Maria G (2006). Effect of treatment with nasal continuous positive airway pressure on ventilatory response to hypoxia and hypercapnia in patients with sleep apnea syndrome. Chest 130, 774–779. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand‐Perret T, Ajakane M, Baudet V, Boissin P, Boursier E & Loriolle F (1991). The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266, 15771–15781. [PubMed] [Google Scholar]
- Trzebski A, Tafil M, Zoltowski M & Przybylski J (1982). Increased sensitivity of the arterial chemoreceptor drive in young men with mild hypertension. Cardiovasc Res 16, 163–172. [DOI] [PubMed] [Google Scholar]
- Wilson RJA & Teppema LJ (2016). Integration of central and peripheral respiratory chemoreflexes. Compr Physiol 6, 1005–1041. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Crabtree DC, Puleo DS, Goodman BM & Morgan BJ (2000). Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol 89, 1333–1339. [DOI] [PubMed] [Google Scholar]
- Xing T & Pilowsky PM (2010). Acute intermittent hypoxia in rat in vivo elicits a robust increase in tonic sympathetic nerve activity that is independent of respiratory drive. J Physiol (Lond) 588, 3075–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing T, Pilowsky PM & Fong AY (2014). Mechanism of sympathetic activation and blood pressure elevation in humans and animals following acute intermittent hypoxia. Prog Brain Res 209, 131–146. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Lalley P & Mifflin S (2015). Acute intermittent optogenetic stimulation of nucleus tractus solitarius neurons induces sympathetic long‐term facilitation. Am J Physiol Regul Integr Comp Physiol 308, R266–R275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C‐H, Zhuang W‐L, Shen Y‐J, Lai CJ & Kou YR (2016). NADPH oxidase‐derived ROS induced by chronic intermittent hypoxia mediates hypersensitivity of lung vagal C fibers in rats. Front Physiol 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhong H, Vollmer C & Nurse CA (2000). Co‐release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol (Lond) 525, 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]