

Abstract
The retrotrapezoid nucleus (RTN) regulates breathing in a CO2‐ and state‐dependent manner. RTN neurons are glutamatergic and innervate principally the respiratory pattern generator; they regulate multiple aspects of breathing, including active expiration, and maintain breathing automaticity during non‐REM sleep. RTN neurons encode arterial /pH via cell‐autonomous and paracrine mechanisms, and via input from other CO2‐responsive neurons. In short, RTN neurons are a pivotal structure for breathing automaticity and arterial homeostasis. The carotid bodies stimulate the respiratory pattern generator directly and indirectly by activating RTN via a neuronal projection originating within the solitary tract nucleus. The indirect pathway operates under normo‐ or hypercapnic conditions; under respiratory alkalosis (e.g. hypoxia) RTN neurons are silent and the excitatory input from the carotid bodies is suppressed. Also, silencing RTN neurons optogenetically quickly triggers a compensatory increase in carotid body activity. Thus, in conscious mammals, breathing is subject to a dual and interdependent feedback regulation by chemoreceptors. Depending on the circumstance, the activity of the carotid bodies and that of RTN vary in the same or the opposite directions, producing additive or countervailing effects on breathing. These interactions are mediated either via changes in blood gases or by brainstem neuronal connections, but their ultimate effect is invariably to minimize arterial fluctuations. We discuss the potential relevance of this dual chemoreceptor feedback to cardiorespiratory abnormalities present in diseases in which the carotid bodies are hyperactive at rest, e.g. essential hypertension, obstructive sleep apnoea and heart failure.
Keywords: central respiratory chemoreceptor, carotid body, optogenetics
Introduction
The retrotrapezoid nucleus (RTN) is a small group of medulla oblongata neurons that regulate breathing in a CO2‐dependent manner. Since 2004 a number of laboratories, including ours, have investigated the possibility that this nucleus might be the main hub of the central respiratory chemoreflex as well as the principal CNS site where CO2 is sensed for the purpose of breathing regulation. The experimental evidence is briefly updated here (for prior reviews see Guyenet & Bayliss, 2015 and Guyenet et al. 2016).
For consistency with the main theme of the 2017 ISAC meeting, a large portion of our report focuses on how the carotid bodies, the body's main oxygen sensors (Prabhakar & Semenza, 2015), and central chemoreceptors cooperate in regulating arterial . For instance, the notion that, in conscious mammals, poikilocapnic hypoxia silences central respiratory chemoreceptors via alkalosis has long been accepted in spite of scant cellular evidence. We highlight here recent experimental data showing that RTN is silenced by hypoxia, pointing out that this result is consistent both with the postulated central respiratory chemoreceptor function of this nucleus and with the theory that hypoxia leads to a silencing of central respiratory chemoreceptors.
Finally, we discuss how recent findings concerning the interactions between carotid bodies and RTN may help explain the cardiorespiratory anomalies associated with various diseases in which the carotid bodies are hyperactive at rest, e.g. hypertension, heart failure, obstructive sleep apnoea.
The retrotrapezoid nucleus (RTN) – anatomical definition
The definition of RTN that we currently advocate relies on congruent developmental, biochemical and physiological evidence that was complemented more recently by RNA‐Seq and ISH data (Shi et al., 2017). This cell group resides in the parafacial region, mainly below the facial motor nucleus and consists of around ∼700 glutamatergic neurons in mice (∼1900 in rats) that express homeobox transcription factor Phox2b and NK1 receptors, and lack mRNA for tyrosine‐hydroxylase, choline acetyltransferase, glutamic acid decarboxylases, glycine transporter‐2 and tryptophan hydroxylase (Guyenet & Bayliss, 2015; Guyenet et al. 2016). These neurons have a common developmental lineage (Egr‐2, Phox2b, Atoh‐1) (Ramanantsoa et al. 2011; Ruffault et al. 2015) and similar late embryonic (Thoby‐Brisson et al. 2009), postnatal (Onimaru et al. 2014) and adult biochemical characteristics (Guyenet et al. 2016). The terms embryonic parafacial oscillator (Thoby‐Brisson et al. 2009), Phox2b‐positive parafacial neurons (Onimaru et al. 2014) and RTN neurons (Ramanantsoa et al. 2011; Ruffault et al. 2015; Guyenet et al. 2016) designate what are probably three developmental stages of the same neurons. In addition to expressing Phox2b, RTN neurons can be distinguished from surrounding neurons by the presence of neuromedin B mRNA and, typically (80–90%) by the presence of both GPR4 and TASK‐2 (putative physiological proton detectors) (Fig. 1 A–C).
Figure 1. Anatomy of the retrotrapezoid nucleus.
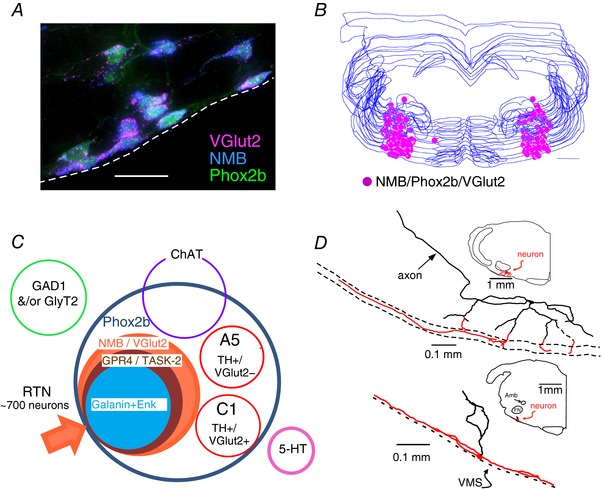
A, cluster of RTN neurons containing mRNA transcripts for VGlut2 and NMB. These neurons are immunoreactive for eGFP, the latter denoting the presence of Phox2b (transverse section, JX‐99 – Phox2b‐eGFP mouse; dashed line identifies the ventral medullary surface (VMS). Scale bar = 25 μm. Unpublished work of R. L. Stornetta. Every neuron is triple‐labelled. B, computer‐assisted mapping of RTN neurons (NMB+) in one mouse. Neurons identified in a one‐in‐three series of 30 μm‐thick transverse sections between 6.65 and 5.63 mm behind bregma. Scale bar = 500 μm (from Shi et al. 2017, with modifications). C, Venn diagram representing the various cell populations located in the parafacial region of the mouse. RTN neurons are defined as positive for Phox2b, VGlut2 and NMB. A least 90% of these neurons contain putative proton sensor TASK‐2 and ∼82% contain GPR4 (Shi et al. 2017). D, location and dendritic structure of two RTN neurons from rat recorded and labelled juxtacellularly in vivo. Note presence of extensive dendrites in the marginal layer (ML) regardless of the position of the cell body (adapted from Mulkey et al. 2004).
RTN neurons, despite their small number, are heterogeneous; for example, prepro‐galanin or prepro‐enkephalin transcripts are detectable in only 82 and 67% of these neurons, respectively, and some neurons express only one of the two proton sensors (Shi et al., 2017). We stress that our definition of RTN is based primarily on developmental and biochemical similarities (including pH sensitivity) between neurons. It does not imply that every RTN neuron has the same function, or that every RTN neuron responds equally to changes in /pHa. In fact, as illustrated by the accompanying Venn diagram (Fig. 1 C), 10–20% of the parafacial neurons that have the biochemical markers previously used to characterize RTN chemoreceptors do not express detectable levels of proton sensors in mice. These neurons do not express Fos after hypercapnia either (Shi et al., 2017). In brief, the vast majority of RTN neurons do have biochemical and physiological properties consistent with a central chemoreceptor function but around 10–20% do not (Fig. 1 C). Finally, a single marker, neuromedin B mRNA, identifies RTN chemoreceptors with 80–90% fidelity in mice (Shi et al., 2017).
RTN innervates selectively four respiratory‐related regions: the ventral respiratory column in its entirety, the Kölliker‐Fuse region, select lateral parabrachial nuclei and the ventrolateral portion of the nucleus of the solitary tract (Fig. 2 A) (Bochorishvili et al. 2012). In rodents, most RTN neurons have extensive dendrites just below the ventral surface of the brain (Fig. 1 D). This anatomical peculiarity suggests that these neurons could also respond to the chemical composition of the cerebrospinal fluid, CO2 included.
Figure 2. Retrotrapezoid nucleus: axonal projections and CO2 sensing mechanisms.
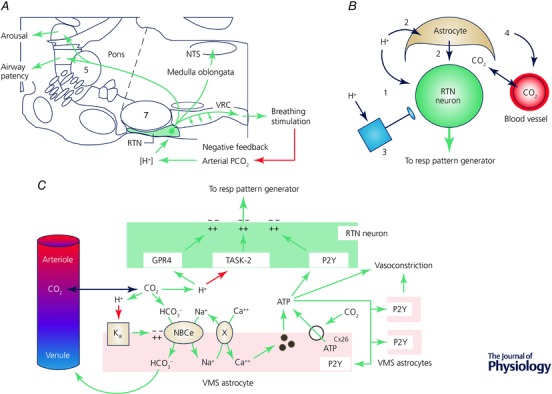
A, brain regions targeted by RTN neurons. Schematic summary of results from Bochorishvili et al. (2012). B, CO2 detection by RTN neurons. Four mechanisms are thought to contribute to the exquisite sensitivity of RTN neurons to hypercapnia in vivo: (1) direct effect of proton on RTN neurons, (2) paracrine effect of protons mediated via acid‐sensitive astrocytes, (3) excitatory inputs from other pH‐sensitive neurons, (4) CO2‐induced vasoconstriction. The relative importance of each of the 4 mechanisms is unsettled. C, cell autonomous and paracrine mechanisms underlying the CO2 sensitivity of RTN neurons. The cell‐autonomous neuronal response to protons is mediated by TASK‐2 (Kcnk5) and GPR4. RTN is surrounded by astrocytes that are depolarized by local acidification. The depolarization activates an electrogenic sodium–bicarbonate transporter (NBCe) which alkalizes the glial cytoplasm and simultaneously acidifies the extracellular space. This extracellular acidification likely potentiates the intrinsic response of RTN neurons to a given change in arterial pH/. Sodium entry via NBCe activation also stimulates sodium–calcium exchange. The rise in intracellular calcium causes exocytosis of gliotransmitters such as ATP. ATP may also be released through connexin26 hemichannels opened by carbamylation. ATP recruits neighbouring astrocytes and may depolarize RTN neurons. Acid‐induced ATP release (from astrocytes or other unidentified cells) causes vasoconstriction, reduced wash‐out of metabolically produced CO2 and further acidification. Key supporting references: Mulkey et al. (2004); Erlichman & Leiter, (2010); Gestreau et al. (2010); Gourine et al. (2010); Huckstepp et al. (2010); Wenker et al. (2010); Wang et al. (2013); Kumar et al. (2015); Turovsky et al. (2016); Hawkins et al. (2017).
Mechanisms responsible for the pH/CO2 sensitivity of RTN neurons
Four mechanisms are thought to contribute to the CO2‐sensitivity of RTN neurons (Fig. 2 B): (1) a cell‐autonomous effect of protons on these neurons, (2) a paracrine effect mediated via surrounding CO2‐responsive astrocytes, (3) mono‐ or polysynaptic inputs from other CO2‐responsive neurons and (4) a vasoconstrictor effect of CO2. Type‐3 mechanisms include a polysynaptic input from the carotid bodies to RTN and direct inputs from CO2 activated CNS serotonergic, noradrenergic or orexinergic neurons.
The intrinsic response of RTN neurons to [H+] is a depolarization mediated via proton sensors TASK‐2 and GPR4 (Kumar et al. 2015; for review, see Guyenet et al. 2016). Surrounding astrocytes are also depolarized by acidification but via a different mechanism (KIR4.1/5.1) (Gourine et al. 2010; Wenker et al. 2010). Astrocyte depolarization may further enhance the acidification of the RTN region relative to that of the plasma via the phenomenon called depolarization‐induced alkalization, thereby increasing the sensitivity of RTN neurons to changes in arterial pH. Astrocytes may also activate RTN neurons by releasing ATP, prostaglandin E2 or d‐alanine (Gourine et al. 2010; Beltran‐Castillo et al. 2017; Forsberg et al. 2017; see legend to Fig. 2 C for additional references and further details regarding these mechanisms). Finally, in the RTN region, hypercapnia may produce arteriolar constriction (Hawkins et al. 2017). This newly identified and unexpected vasoconstrictor effect of CO2 may be unique to the ventrolateral medulla. In vivo, vasoconstriction likely causes further CO2 retention, parenchymal acidification and ultimately activation of RTN neurons (Xie et al. 2006; Hawkins et al. 2017). The relative importance of the four mechanisms listed above is yet to be clearly determined and could vary depending on the characteristics of the hypercapnic stimulus (e.g. intensity, duration).
Evidence that RTN neurons mediate the central respiratory chemoreflex
The conclusion that RTN neurons mediate the central component of the ventilatory response to changes in arterial (Fig. 2 A) relies on four congruent pieces of evidence. In anaesthetized rats, RTN neurons increase their firing rate on exposure to hypercapnia (0.5 Hz per 0.01 change in arterial pH (pHa)), including when the carotid bodies or the nucleus of the solitary tract are disabled (Mulkey et al. 2004; Kumar et al. 2015; Wakai et al. 2015; Fig. 3 A). Optogenetic activation of RTN produces very large increases in breathing and RTN inhibition elicits major breathing reductions (Abbott et al. 2011; Basting et al. 2015; Holloway et al. 2015; Fig. 3 B). The breathing reduction caused by inhibiting RTN is linearly related to pHa below a threshold of ∼7.5; in quietly resting animals, these cells do not contribute to ventilation above this pH level (Fig. 3 C and D). Thus, RTN neurons encode (and pHa) almost linearly in vivo with a pHa recruitment threshold at around pH 7.5 and, most importantly, they appear to drive breathing in proportion to their discharge rate. A fourth and crucial piece of evidence is that RTN lesions performed by genetic means or via local injection of NK1R selective saporin‐based toxins produce profound reductions of the hypercapnic ventilatory reflex (Nattie & Li, 2002; Dubreuil et al. 2008; Ramanantsoa et al. 2011; Takakura et al. 2014).
Figure 3. RTN‐dependent and ‐independent activation of breathing by the carotid bodies.
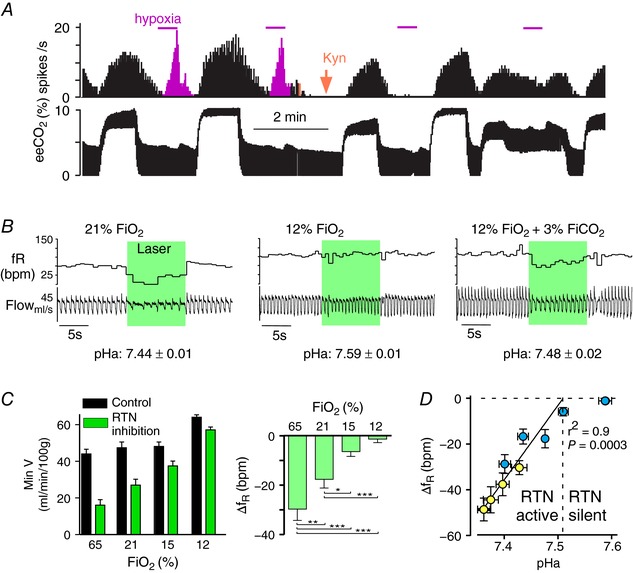
A, single‐unit recording evidence that carotid body stimulation can activate RTN. This RTN neuron was activated by hypercapnia (eeCO2: end‐expiratory CO2) and by brief hypoxia (magenta). Administration of kynurenate i.c.v. (orange arrow), a blocker of excitatory glutamatergic synapses, eliminated the effect of hypoxia selectively whereas the effect of hypercapnia, which is primarily mediated by cell‐autonomous and paracrine effects of CO2, persisted (redrawn from Mulkey et al. 2004). B, evidence that the carotid bodies activate breathing via a pathway that bypasses RTN. Bilateral opto‐inhibition of RTN neurons (conscious rats) reduces breathing under normoxia (21% ) but has no effect under hypoxia (12% ). Breathing rate and amplitude increased under hypoxia despite RTN being silent, demonstrating that the CBs activated breathing via a pathway that bypasses RTN. The addition of a small amount of CO2 restored the breathing reduction caused by RTN inhibition, suggesting that hypoxia‐induced hyperventilation had inhibited RTN via the resultant respiratory alkalosis. Average plasma pH identified in 6 rats is shown below the representative examples (modified from Basting et al. 2015). C, breathing reduction elicited by bilateral optoinhibition of RTN is reduced under hypoxia. Left panel, minute volume before (black bars) and during RTN inhibition (green bars); right panel change in breathing frequency elicited by RTN inhibition. At 12% , breathing is activated but RTN no longer contributes to breathing (right panel). At 15% breathing is unchanged from room air but, as indicated in the right panel, a much smaller portion of the respiratory drive originates from RTN (modified from Basting et al. 2015). D, relationship between arterial pH and effect of RTN inhibition on breathing frequency (f R) and plasma pH was manipulated with hypoxia. Respiratory alkalosis was compensated by adding CO2 (blue symbols) or by inducing metabolic acidosis (acetazolamide, yellow symbols). Above pHa 7.5, RTN inhibition has no effect on breathing consistent with single‐unit evidence that the neurons are silent (e.g. panel B). Below pH 7.5, the breathing reduction elicited by RTN inhibition (change in breathing frequency is depicted) is a linear function of arterial pH, consistent with single‐unit evidence (not shown) that RTN neurons are increasingly active (modified from Basting et al. 2015).
RTN neurons drive multiple aspects of breathing, including active expiration
Optogenetic stimulation of RTN neurons in quietly awake rodents increases breathing frequency and inspiratory amplitude; phasic activation of RTN also entrains the breathing cycle and elicits active expiration (Fig. 4 A; Abbott et al. 2011; Burke et al. 2015; Holloway et al. 2015). These effects require the release of glutamate by RTN neurons (Holloway et al. 2015). Optogenetic activation of RTN also reduces expiratory flow immediately following inspiration (Fig. 4 A). This phenomenon (expiratory brake) could be caused by enhanced diaphragmatic contraction and/or increased laryngeal resistance during the post‐inspiratory phase of the breathing cycle.
Figure 4. RTN activates multiple breathing parameters in a state‐dependent fashion.
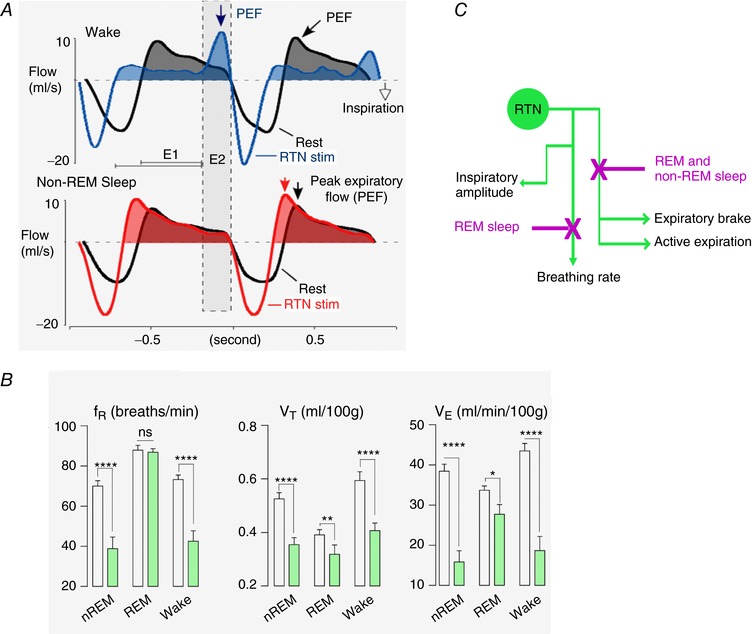
A, unilateral phasic optogenetic activation of RTN (channelrhodopsin‐2; trains of 3 light pulses; trains delivered at a rate slightly above resting breathing frequency) during quiet waking entrains the respiratory pattern generator, increases inspiratory amplitude, produces active expiration (note the distinctive peak‐expiratory flow, PEF, during late expiratory phase, E2) and activates the expiratory brake (note reduced expiratory flow during early expiration, E1). During non‐REM sleep (same rat), RTN activation still entrained the pattern generator and increased inspiratory amplitude but active expiration and the expiratory brake were no longer elicited (from Burke et al. 2015, with slight modifications). B, bilateral optogenetic inhibition of RTN (archaerhodopsin, 10 s light pulses; green bars) reduces respiratory frequency (f R) and tidal volume (V T) during quiet resting and non‐REM sleep. During REM sleep RTN inhibition has no effect on f R but still reduces V T, albeit to a lesser degree. This evidence indicates that RTN controls breathing frequency only when the pattern generator is auto‐rhythmic (slow wave sleep or quiet awake state) but not when it is externally driven, as in REM sleep. Adapted from Basting et al. 2015). C, schematic representation of the four known effects of RTN on breathing and their differential regulation by the state of vigilance.
In the arterially perfused rat preparation, non‐selective inhibition of the RTN region or selective pharmacogenetic inhibition of RTN neurons eliminates the expiratory abdominal muscle outflow elicited by hypercapnia (Abdala et al. 2009; Marina et al. 2010). Thus, both gain (Burke et al. 2015) and loss of function experiments (Marina et al. 2010) show that RTN neurons stimulate multiple aspects of breathing, including active expiration. Whether RTN neurons and the proposed parafacial ‘oscillator for active expiration’ are one and the same, overlapping or entirely separate neuronal populations remains to be determined (Janczewski & Feldman, 2006; Pagliardini et al. 2011; Huckstepp et al. 2015, 2016; de Britto & Moraes, 2017).
The effect of RTN stimulation on breathing is state dependent
Optogenetic activation of RTN neurons produces substantially different effects on breathing depending on the state of vigilance (Fig. 4). The forced expiration elicited during the E2 phase when rats are quietly awake disappears during non‐REM sleep and the peak expiratory flow migrates to the early part of the expiratory phase (E1) denoting a simultaneous reduction of the expiratory brake (Burke et al. 2015; Fig. 4 A). However, during this sleep phase, RTN stimulation still increases inspiratory rate and amplitude (Burke et al. 2015). In other words, the network responsible for active expiration and post‐inspiratory brake is less responsive to excitatory inputs from RTN during non‐REM sleep than during quiet resting. Reduced release of wake‐promoting transmitters (e.g. noradrenaline, serotonin, orexin) at strategic sites of the breathing network (abdominal motor and/or premotor neurons, expiratory ‘oscillator’) could underlie this reduced excitability.
The transition from non‐REM to REM sleep brings about a different set of changes (Burke et al. 2015). As the rat enters REM sleep, RTN stimulation immediately ceases to elicit any effect on breathing frequency whereas inspiratory amplitude is still elevated (Burke et al. 2015). The converse is also true; selective optogenetic inhibition of RTN neurons reduces breathing rate and amplitude profoundly during non‐REM sleep whereas, during REM sleep, inspiratory amplitude alone is decreased (Fig. 4 B) (Burke et al. 2015). Finally, during quiet waking or non‐REM sleep, the breathing cycle can be faithfully entrained by phasic stimulation of RTN at rates above the resting breathing frequency but no such entrainment can be produced during REM sleep (Burke et al. 2015). This change probably denotes a fundamental difference in the way inspiration is generated in different arousal states. During anaesthesia, quiet waking and non‐REM sleep, inspiration is probably generated autonomously by the relatively regular discharges of the preBötzinger complex and its surround (Kam et al. 2013; Rybak et al. 2014). During REM sleep, as during voluntary control, the expiratory phase is likely controlled by inputs originating outside this circuitry (Richter & Smith, 2014). The optogenetic evidence obtained in rats indicates that RTN neurons regulate breathing frequency only when the pontomedullary respiratory circuit is in auto‐rhythmic mode (anaesthesia, quiet rest and non‐REM as opposed to active behaviour or REM sleep) (Fig. 4 B; Abbott et al. 2011; Burke et al. 2015). Thus, besides driving inspiratory amplitude and active expiration, RTN is a key regulator of breathing auto‐rhythmicity. Frequency regulation, like inspiratory amplitude control is mediated primarily by glutamate and possibly by direct projections of RTN to the rhythmogenic neurons of the preBötzinger complex (Bochorishvili et al. 2012; Holloway et al. 2015). The simplest, though yet untested, mechanism could be that RTN increases the breathing rate by accelerating the inter‐burst depolarization rate of preBötzinger complex rhythmogenic neurons.
The mechanism by which RTN controls breathing frequency is speculative at this point. However, the loss of frequency control by RTN during REM sleep is consistent with breathing frequency being virtually insensitive to hypercapnia during this stage of sleep (reviewed by Coote, 1982; additional citations in Horner et al. 2002; Burke et al. 2015). The effect of hypoxia (HVR) and that of pulmonary afferents (Hering‐Breuer reflex) on breathing frequency is also greatly diminished during this sleep stage.
In short, in rodents, RTN neurons and CNS hypercarbia elicit similar effects on a broad range of respiratory motor outflows (Abbott et al. 2011). This is consistent with the notion that RTN controls lung ventilation by modulating inspiratory, expiratory and post‐inspiratory (glottis, diaphragm) muscle activity in an orderly manner according to the intensity of the chemoreceptor stimulus. Like those of CO2, the effects of RTN on breathing are profoundly state dependent, the most dramatic aspect being the loss of effect of RTN on breathing frequency during REM sleep (Fig. 4 C for summary).
The carotid bodies stimulate breathing via and independently of RTN
Except during REM sleep, carotid bodies and central chemoreceptors provide a substantial tonic drive to breathe, even at rest under normoxic and normocapnic conditions (Smith et al. 1995; Pan et al. 1998; Dahan et al. 2007; Burke et al. 2015). Hypoxia and hypercapnia have supra‐additive effects on breathing, which are at least partly caused by the robust potentiative effect of acid and low on the activity of carotid body afferents (Daristotle & Bisgard, 1989; Kumar & Prabhakar, 2012). Centrally, carotid body afferent activity and central chemoreceptors have additive or supra‐additive effects on breathing in intact and unanaesthetized mammals whereas hypo‐additive effects are observed in an arterially perfused rat preparation (Duffin & Mateika, 2013; Teppema & Smith, 2013; Wilson & Day, 2013; Smith et al. 2015).
An excitatory polysynaptic pathway between carotid body afferents and RTN neurons has been identified by single unit recordings in anaesthetized rats (Fig. 5; pathway 1); under normocapnia, RTN neurons are powerfully excited by brief hypoxia or by intravenous administration of cyanide (Fig. 3 A; Mulkey et al. 2004; Takakura et al. 2006). This activation requires the integrity of the carotid sinus nerves and the nucleus of the solitary tract (NTS); it may be mediated by a direct excitatory connection between NTS second‐order neurons and RTN (Fig. 5, pathway 1; Takakura et al. 2006). The existence of this connection suggests that the carotid bodies activate breathing in part by stimulating RTN.
Figure 5. RTN‐dependent (1) and RTN‐independent pathways (2) from the carotid bodies to the respiratory pattern generator (RPG).
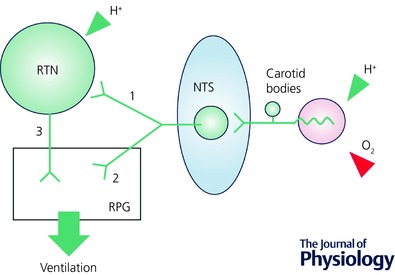
The carotid bodies also undoubtedly stimulate breathing via pathways that bypass RTN (pathway 2, Fig. 5). Experiments conducted in an arterially perfused preparation indicated that the carotid bodies can activate breathing when brain perfusate is reduced to levels that should be well below central chemoreceptor threshold (Fiamma et al. 2013). Evidence that the carotid bodies can stimulate breathing via pathways that bypass RTN was also obtained in unanaesthetized rats. RTN inhibition (optogenetic) reduces breathing rate and amplitude in rats breathing room air or CO2‐enriched air but produces no effect in rats exposed to 12% inspired O2 fraction () implying that the RTN had been silenced during hypoxia; the addition of a minimal amount of CO2 ( 3%, in 12% ) restored opto‐inhibition of breathing, indicating that RTN neurons were reactivated (Fig. 3 B and C; Basting et al. 2015). Accordingly, in an intact rat, the respiratory alkalosis elicited by hypoxic hyperventilation appears to reduce the excitability of RTN neurons to such a degree that they no longer respond to the excitatory input from the carotid bodies. Yet, despite RTN being silenced, breathing is maintained or even elevated if is low enough (∼12% or less in Sprague‐Dawley rats). This can only be explained by the existence of strong inputs from the carotid bodies to the respiratory pattern generator that bypass RTN.
In short, in unanaesthetized mammals, humans included, the interaction between carotid bodies and central chemoreceptors is at least additive and often potentiative. The carotid bodies activate the respiratory pattern generator via RTN and independently of this nucleus. The indirect pathway through RTN could perhaps explain why the interaction between carotid body afferents and central chemoreceptors can be supra‐additive.
Circumstances under which the activity of RTN and carotid bodies vary in the opposite direction
Under most physiological conditions, the activity of RTN and carotid bodies changes in parallel (e.g. during hypoventilation, when both increase). However, there are occasions when their activity is regulated in opposite directions. One such case, discussed in the previous paragraph in another context, is poikilocapnic hypoxia; increasing degrees of carotid body activation and the ensuing respiratory alkalosis lead to correspondingly larger inhibition of RTN neurons until these cells become silent, somewhere around 12% in conscious rats (Fig. 3 B and C).
A second example is when RTN is selectively inhibited using optogenetics in conscious rats breathing room air; breathing returns quickly towards control and a breathing overshoot is observed after the optical inhibition is terminated (Fig. 6 A). Breathing recovery and overshoot are primarily mediated by an increase in respiratory frequency (f R) and are absent when the carotid bodies are either silenced by hyperoxia (65% ) or surgically denervated (Fig. 6 A and B). Thus, within this specific time frame, both recovery and overshoot of breathing following RTN inhibition result from carotid body activation rather than from reactivation of RTN or other central chemoreceptors (interpretation in Fig. 6 C; Basting et al. 2016).
Figure 6. Carotid body activation rapidly restores breathing during RTN inhibition.
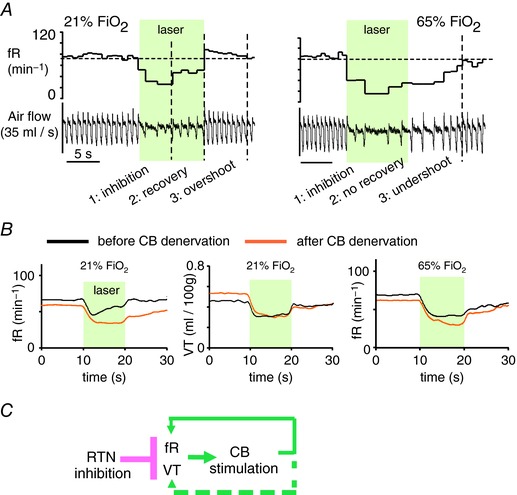
A, representative breathing response to instant bilateral optogenetic inhibition of RTN in a conscious rat. Under normoxia, the nadir occurs within 5 s and is followed by a gradual recovery. A brief overshoot occurs when the laser is switched off. Under hyperoxia, to silence the carotid bodies, the recovery phase is not observed and breathing inhibition persists for some time after the light is switched off. Recovery and overshoot of f R are therefore attributable to carotid body stimulation. Adapted from Basting et al. (2016). B, mean breathing response to bilateral opto‐inhibition of RTN in conscious rats (N = 6). Left panel: frequency response before and 7 days after bilateral CB denervation (same 6 rats). Middle panel: tidal volume (V T) response before and after CB denervation. Note that, contrary to f R, V T inhibition is unaffected by CB denervation. Right panel: under hyperoxia, the breathing response to RTN opto‐inhibition is no longer affected by CB denervation. From Basting et al. (2016). Confidence intervals removed for clarity. C, sequence of events (schematic): RTN inhibition reduces both f R and V T causing a decrease in alveolar ventilation and subsequent carotid body activation via the ensuing changes in blood gases. In rats, the carotid bodies stimulate breathing by increasing breathing frequency primarily; this effect quickly mitigates the hypoventilation elicited by RTN inhibition.
Parallel or opposite changes in RTN vs. carotid body activity enhance arterial stability; implication for diseases in which the carotid bodies are hyperactive
The activity of peripheral and central chemoreceptors usually varies in the same direction because the most common trigger is hypo‐ or hyperventilation, perturbations that are characterized by opposite changes in arterial and . In such situations the feedback exerted by carotid bodies and RTN is additive or even potentiative (Smith et al. 2015); its obvious function is to minimize arterial fluctuations.
During hypoxia, the activity of carotid bodies and RTN varies in opposite directions. By reducing the magnitude of the hyperventilation, RTN inhibition minimizes the respiratory alkalosis elicited by the increase in carotid body activity (Basting et al. 2015). Thus, in this situation, RTN inhibition also contributes to arterial stability, at least until reaches a level low enough that oxygen delivery takes precedence over CO2 homeostasis and ventilation increases substantially. In conscious rats, RTN inhibition elicits a rapid compensatory increase in carotid body activity that also largely and rapidly restores breathing (Basting et al. 2016). Thus, again under this circumstance, the activity of the carotid bodies and that of RTN change in opposite directions and the ultimate result of the increase in carotid body activity is to buffer the change in arterial (hypercapnia in this case) caused by a sudden decrease in central chemoreceptor activity that was not caused by reduced arterial . In other words, the network is built to favour homeostasis at the expense of stability until hypoxaemia becomes severe and oxygen delivery becomes the overriding priority.
Carotid body hyperactivity consisting of hyper‐sensitivity to hypoxia and aberrant ‘tonicity’ occurs in animal models of essential hypertension, heart failure and obstructive sleep apnoea (OSA) (Kumar & Prabhakar, 2012; Del Rio et al. 2013; Pijacka et al. 2016a,b). This hyperactivity is not driven by a change in arterial blood gases and contributes to the pathogenic increase in sympathetic nerve activity present in these human diseases and their animal models (Pijacka et al. 2016a,b).
In human essential hypertension, OSA, mild to moderate congestive heart failure, sympathetic tone and blood pressure are elevated but, unlike in some animal models of these diseases, resting ventilation under normoxia is unchanged (Narkiewicz et al. 1999a,b; Guardiola et al. 2004; Sinski et al. 2012; Del Rio et al. 2016). Based on our experiments on rodents, we suggest that in the waking state, a mild and sustained rise in carotid body activity may have little effect on breathing because a simultaneous reduction in central chemoreceptor (RTN) activity restores and breathing to control (Basting et al. 2015). By contrast sympathetic stimulation could be sustained because its main countervailing influence, the baroreflex, is reduced when the carotid bodies are activated (McBryde et al. 2013).
A change in central cardiorespiratory coupling has been repeatedly invoked to explain the increased SNA associated with OSA, hypertension and CHF (Zoccal et al. 2008; Molkov et al. 2011; Menuet et al. 2017). The evidence largely derives from semi‐in vitro preparations in which the intensity of the respiratory drive cannot be precisely compared between animals; without such quantification, it is near impossible to distinguish between normal coupling (increased phasic SNA caused by increased central respiratory drive in a particular preparation) and abnormal coupling (increased phasic SNA despite normal activity of the respiratory network). Since breathing is unchanged in OSA or hypertensive patients at rest, only an abnormally large coupling, evidence for which is very limited, could make a difference (Fatouleh & Macefield, 2011). Finally, a change in breathing or cardiorespiratory coupling is not the only way in which carotid body afferent activity could enhance sympathetic tone: carotid body stimulation increases the discharge of ventrolateral medullary presympathetic neurons via a direct excitatory pathway from the NTS (Koshiya & Guyenet, 1996; Dempsey et al. 2017).
The ability of central chemoreceptors to compensate for an increase in carotid body activity is evidently limited by the amount of tonic excitatory drive that RTN neurons, and possibly additional central chemoreceptors, contribute to the respiratory pattern generator at rest. Under more severe hypoxia in rodents, ventilation does rise. Based on experiments in rats breathing 12% , this rise occurs despite RTN being silenced. Under pathological conditions such as severe heart failure in humans, carotid body hyperactivity at rest may be high enough to overcome the loss of central chemoreceptor drive and hyperventilation is the result (Marcus et al. 2014). Chronic intermittent hypoxia (CIH), an animal model of OSA (Fletcher, 2001) sometimes produces a persistent increase in breathing rate and amplitude in rats (Reeves & Gozal, 2006) and may even elicit a permanent state of active expiration (Zoccal et al. 2008). CIH also modifies the discharge characteristics and hypoxic sensitivity of the preBötzinger complex in slices, denoting some persistent changes in cellular properties or a circuit configuration (Garcia et al. 2017). Animal models in which resting ventilation is elevated or disrupted at rest (CIH, heart failure) may present with a level of carotid body afferent activity considerably higher than in the human disease they are designed to mimic. These models may also experience brainstem abnormalities caused by direct effects of repeated cycles of hypoxia/re‐oxygenation on brainstem circuits.
Summary and concluding remarks
The retrotrapezoid nucleus is a small cluster of glutamatergic neurons that have a well‐defined developmental lineage (Egr‐2, Phox2b, Atoh‐1) and biochemical markers (e.g. Phox2b, NMB, VGlut2). These neurons selectively innervate the pontomedullary regions that harbour the respiratory pattern generator.
Most RTN neurons are CO2 activated and respond with high sensitivity to changes in arterial pH in vivo (0.5 Hz per 0.01 pH). Collectively, RTN neurons drive breathing in direct proportion to arterial [H+] with a pH recruitment threshold close to 7.5 in conscious rats. The pH sensitivity of RTN neurons is at least partly intrinsic and relies on proton receptors GPR4 or TASK‐2. This pH sensitivity is boosted by paracrine mechanisms mediated by pH‐sensitive astrocytes and by a regionally specific vasoconstrictive effect of CO2. RTN neurons also receive excitatory inputs from the carotid bodies. Finally, input from serotonergic and other classes of CNS neurons likely potentiate the response of RTN neurons response to CO2. An upcoming challenge will be to assess the relative contribution of these various mechanisms to the CO2 sensitivity of RTN neurons under different circumstances in vivo.
RTN neurons regulate alveolar ventilation and pulmonary CO2 excretion by facilitating multiple aspects of breathing (inspiratory amplitude, breathing rate, active expiration and, possibly, airway patency). The effect of RTN on these parameters is differentially gated or modulated according to the state of vigilance. Specifically, during REM sleep, the breathing rate is affected neither by hypercapnia not by RTN stimulation and the active expiration triggered by RTN stimulation during quiet waking disappears during slow‐wave sleep. Each of these breathing parameters could conceivably be regulated by a dedicated subset of RTN neurons. Whether RTN and the parafacial oscillator for active expiration (Huckstepp et al. 2016) are distinct or overlapping sets of neurons is unsettled.
At this time, there is no compelling reason to assume that the sole function of RTN is central respiratory chemoreception. RTN could also mediate the hyperpnoea of exercise as suggested by a Fos expression study (Barna et al. 2014); this possibility is consistent with the fact that these neurons stimulate active expiration (Marina et al. 2010; Abbott et al. 2011). Whether RTN also contributes to the emotional control of breathing and to diet‐ or obesity‐associated changes in ventilation has not been investigated.
The peripheral and the central respiratory chemoreflexes are highly interdependent. The breathing network is especially responsive to hypercapnic hypoxia, the consequence of hypoventilation, because RTN and the carotid bodies have cumulative excitatory effects on the respiratory pattern generator and RTN neurons receive an excitatory polysynaptic input from the carotid body afferents. The network is therefore poised to respond with extreme sensitivity to the slightest degree of hypo‐ or hyperventilation by triggering a breathing change of the opposite sign that restores arterial to normal.
Under other circumstances, a primary change in the activity of one type of chemoreceptor triggers a change of opposite sign in the other (e.g. RTN inhibition during hypoxia, or carotid body activation following a sudden reduction in RTN activity). These interactions are probably driven by changes in blood gases rather than by cross‐talk between brainstem pathways implicated in peripheral vs. central chemoreception but this point is not definitively established.
Chronically elevated carotid body activity underlies the increased sympathetic tone and hypertension associated with OSA and essential hypertension. In conscious humans, the rise in SNA is not accompanied by a change in resting breathing. As a tentative explanation we propose that when carotid body afferent activity is only moderately increased, breathing homeostasis is restored by a countervailing reduction in the activity of central chemoreceptors, notably RTN. As suggested previously (Pijacka et al. 2016a), SNA may remain chronically elevated because of a reduction in the baroreflex. Also, because resting breathing is unchanged in OSA and hypertension, we also suggest that the emphasis on cardiorespiratory coupling as the underlying cause of the increased SNA may be exaggerated and that the role of pathways that directly link carotid body afferents to the vasomotor presympathetic neurons of the RVLM should be further evaluated.
Finally, the theory that poikilocapnic hypoxia silences central respiratory chemoreceptors via alkalosis in conscious mammals has been axiomatic for decades but its cellular basis has long remained obscure. RTN is indeed switched off by poikilocapnic hypoxia despite the persistent or increased activation of the respiratory pattern generator (Basting et al. 2015). This evidence is consistent with the original theory regarding the effects of alkalosis on central chemoreceptors while, at the same time, providing additional evidence that RTN may indeed be the hub of the central respiratory chemoreflex.
Additional information
Competing interests
None declared.
Author contributions
All authors are current or very recent members of our laboratories (P.G.G. and D.A.B.). They have read, edited and contributed to and approved this review. P.G.G., D.A.B. and R.L.S. wrote the manuscript. P.G.G., D.A.B., R.L.S., R.K., Y.S., B.B.H., G.M.P.R.S., T.M.B., S.B.G.A. and I.C.W. designed and performed many of the experiments discussed in this review. All persons listed as authors qualify for authorship and all those who qualify are listed.
Funding
National Institute of Health grants HL074011, HL28785 (P.G.G.); HL108609 (D.A.B.)
Biographies
Patrice G. Guyenet was trained at Ecole Normale Supérieure (Paris), College de France (PhD) and Yale University (Postdoc). His principal mentors were Drs Jacques Glowinski and George K. Aghajanian. He is currently Professor of Pharmacology at the University of Virginia.

Douglas A. Bayliss received his PhD in physiology from the University of North Carolina. After a postdoctoral stint in physiology and biophysics at the University of Washington, he joined the Department of Pharmacology at the University of Virginia, where he is now Professor and Chair.
Ruth L. Stornetta is Professor of Pharmacology at the University of Virginia (UVA). She received a BA in psychology (UVA, 1975), an MA in psychology (College of William and Mary, 1978), a PhD in Neuroscience (UVA,1984), and postdoctoral training with D. J. Reis at Weill Cornell Medical College (1984–1987).
Edited by: Harold Schultz & Vsevolod Polotsky
This review was presented at the symposium ‘Advances in cellular and integrative control of oxygen and carbon dioxide homeostasis’, which took place at the XX ISAC meeting, Baltimore, MD, USA, 23–27 July 2017.
References
- Abbott SB, Stornetta RL, Coates MB & Guyenet PG (2011). Phox2b‐expressing neurons of the parafacial region regulate breathing rate, inspiration, and expiration in conscious rats. J Neurosci 31, 16410–16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC & Paton JF (2009). Abdominal expiratory activity in the rat brainstem–spinal cord in situ: patterns, origins, and implications for respiratory rhythm generation. J Physiol 587, 3539–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna BF, Takakura AC & Moreira TS (2014). Acute exercise‐induced activation of Phox2b‐expressing neurons of the retrotrapezoid nucleus in rats may involve the hypothalamus. Neuroscience 258, 355–363. [DOI] [PubMed] [Google Scholar]
- Basting TM, Abe C, Viar KE, Stornetta RL & Guyenet PG (2016). Is plasticity within the retrotrapezoid nucleus responsible for the recovery of the set‐point after carotid body denervation in rats? J Physiol 594, 3371–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basting TM, Burke PG, Kanbar R, Viar KE, Stornetta DS, Stornetta RL & Guyenet PG (2015). Hypoxia silences retrotrapezoid nucleus respiratory chemoreceptors via alkalosis. J Neurosci 35, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran‐Castillo S, Olivares MJ, Contreras RA, Zuniga G, Llona I, von Bernhardi R & Eugenin JL (2017). D‐serine released by astrocytes in brainstem regulates breathing response to CO2 levels. Nat Commun 8, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochorishvili G, Stornetta RL, Coates MB & Guyenet PG (2012). Pre‐Botzinger complex receives glutamatergic innervation from galaninergic and other retrotrapezoid nucleus neurons. J Comp Neurol 520, 1047–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke PG, Kanbar R, Basting TM, Hodges WM, Viar KE, Stornetta RL & Guyenet PG (2015). State‐dependent control of breathing by the retrotrapezoid nucleus. J Physiol 593, 2909–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coote JH (1982). Respiratory and circulatory control during sleep. J Exp Biol 100, 223–244. [DOI] [PubMed] [Google Scholar]
- Dahan A, Nieuwenhuijs D & Teppema L (2007). Plasticity of central chemoreceptors: effect of bilateral carotid body resection on central CO2 sensitivity. PLoS Med 4, e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daristotle L & Bisgard GE (1989). Central‐peripheral chemoreceptor ventilatory interaction in awake goats. Respir Physiol 76, 383–391. [DOI] [PubMed] [Google Scholar]
- de Britto AA & Moraes DJ (2017). Non‐chemosensitive parafacial neurons simultaneously regulate active expiration and airway patency under hypercapnia in rats. J Physiol 595, 2043–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Andrade DC, Lucero C, Arias P & Iturriaga R (2016). Carotid body ablation abrogates hypertension and autonomic alterations induced by intermittent hypoxia in rats. Hypertension 68, 436–445. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ & Schultz HD (2013). Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol 62, 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey B, Le S, Turner A, Bokiniec P, Ramadas R, Bjaalie JG, Menuet C, Neve R, Allen AM, Goodchild AK & McMullan S (2017). Mapping and analysis of the connectome of sympathetic premotor neurons in the rostral ventrolateral medulla of the rat using a volumetric brain atlas. Front Neural Circuits 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Ramanantsoa N, Trochet D, Vaubourg V, Amiel J, Gallego J, Brunet JF & Goridis C (2008). A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnea and specific loss of parafacial neurons. Proc Natl Acad Sci USA 105, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffin J & Mateika JH (2013). Cross‐Talk opposing view: peripheral and central chemoreflexes have additive effects on ventilation in humans. J Physiol 591, 4351–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlichman JS & Leiter JC (2010). Glia modulation of the extracellular milieu as a factor in central CO2 chemosensitivity and respiratory control. J Appl Physiol (1985) 108, 1803–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatouleh R & Macefield VG (2011). Respiratory modulation of muscle sympathetic nerve activity is not increased in essential hypertension or chronic obstructive pulmonary disease. J Physiol 589, 4997–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiamma MN, O'Connor ET, Roy A, Zuna I & Wilson RJ (2013). The essential role of peripheral respiratory chemoreceptor inputs in maintaining breathing revealed when CO2 stimulation of central chemoreceptors is diminished. J Physiol 591, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC ( 2001). Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol (1985) 90, 1600–1605. [DOI] [PubMed] [Google Scholar]
- Forsberg D, Ringstedt T & Herlenius E (2017). Astrocytes release prostaglandin E2 to modify respiratory network activity. eLife 6, e29566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AJ 3rd, Dashevskiy T, Khuu MA & Ramirez JM (2017). Chronic intermittent hypoxia differentially impacts different states of inspiratory activity at the level of the preBotzinger complex. Front Physiol 8, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Heitzmann D, Thomas J, Dubreuil V, Bandulik S, Reichold M, Bendahhou S, Pierson P, Sterner C, Peyronnet‐Roux J, Benfriha C, Tegtmeier I, Ehnes H, Georgieff M, Lesage F, Brunet JF, Goridis C, Warth R & Barhanin J (2010). Task2 potassium channels set central respiratory CO2 and O2 sensitivity. Proc Natl Acad Sci USA 107, 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K & Kasparov S (2010). Astrocytes control breathing through pH‐dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola J, Yu J, Hasan N & Fletcher EC (2004). Evening and morning blood gases in patients with obstructive sleep apnea. Sleep Med 5, 489–493. [DOI] [PubMed] [Google Scholar]
- Guyenet PG & Bayliss DA (2015). Neural control of breathing and CO2 homeostasis. Neuron 87, 946–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA, Stornetta RL, Ludwig MG, Kumar NN, Shi Y, Burke PG, Kanbar R, Basting TM, Holloway BB & Wenker IC (2016). Proton detection and breathing regulation by the retrotrapezoid nucleus. J Physiol 594, 1529–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins VE, Takakura AC, Trinh A, Malheiros‐Lima MR, Cleary CM, Wenker IC, Dubreuil T, Rodriguez EM, Nelson MT, Moreira TS & Mulkey DK (2017). Purinergic regulation of vascular tone in the retrotrapezoid nucleus is specialized to support the drive to breathe. eLife 6, e25232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BB, Viar KE, Stornetta RL & Guyenet PG (2015). The retrotrapezoid nucleus stimulates breathing by releasing glutamate in adult conscious mice. Eur J Neurosci 42, 2271–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner RL, Liu X, Gill H, Nolan P, Liu H & Sood S (2002). Effects of sleep‐wake state on the genioglossus vs. diaphragm muscle response to CO2 in rats. J Appl Physiol 92, 878–887. [DOI] [PubMed] [Google Scholar]
- Huckstepp RT, Cardoza KP, Henderson LE & Feldman JL (2015). Role of parafacial nuclei in control of breathing in adult rats. J Neurosci 35, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Henderson LE, Cardoza KP & Feldman JL (2016). Interactions between respiratory oscillators in adult rats. eLife 5, e14203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Id BR, Eason R, Spyer KM, Dicke N, Willecke K, Marina N, Gourine AV & Dale N (2010). Connexin hemichannel‐mediated CO2‐dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J Physiol 588, 3901–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski WA & Feldman JL (2006). Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol 570, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Worrell JW, Janczewski WA, Cui Y & Feldman JL (2013). Distinct inspiratory rhythm and pattern generating mechanisms in the preBotzinger complex. J Neurosci 33, 9235–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiya N & Guyenet PG (1996). Tonic sympathetic chemoreflex after blockade of respiratory rhythmogenesis in the rat. J Physiol 491, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NN, Velic A, Soliz J, Shi Y, Li K, Wang S, Weaver JL, Sen J, Abbott SB, Lazarenko RM, Ludwig MG, Perez‐Reyes E, Mohebbi N, Bettoni C, Gassmann M, Suply T, Seuwen K, Guyenet PG, Wagner CA & Bayliss DA (2015). Regulation of breathing by CO2 requires the proton‐activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 348, 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P & Prabhakar NR (2012). Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2, 141–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA & Paton JF (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 2395. [DOI] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH & Schultz HD (2014). Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol 592, 391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Trapp S, Li A, Nattie EE, Hewinson J, Smith JC, Paton JF & Gourine AV (2010). Essential role of Phox2b‐expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci 30, 12466–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuet C, Le S, Dempsey B, Connelly AA, Kamar JL, Jancovski N, Bassi JK, Walters K, Simms AE, Hammond A, Fong AY, Goodchild AK, McMullan S & Allen AM (2017). Excessive respiratory modulation of blood pressure triggers hypertension. Cell Metab 25, 739–748. [DOI] [PubMed] [Google Scholar]
- Molkov YI, Zoccal DB, Moraes DJ, Paton JF, Machado BH & Rybak IA (2011). Intermittent hypoxia‐induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J Neurophysiol 105, 3080–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA & Guyenet PG (2004). Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci 7, 1360–1369. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, van de Borne PJ, Kato M & Somers VK (1999a). Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation 100, 262–267. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N & Somers VK (1999b). Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 99, 1183–1189. [DOI] [PubMed] [Google Scholar]
- Nattie EE & Li A (2002). Substance P‐saporin lesion of neurons with NK1 receptors in one chemoreceptor site in rats decreases ventilation and chemosensitivity. J Physiol 544, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K, Mariho T & Kawakami K (2014). Cytoarchitecture and CO2 sensitivity of Phox2b‐positive parafacial neurons in the newborn rat medulla. Prog Brain Res 209, 57–71. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Janczewski WA, Tan W, Dickson CT, Deisseroth K & Feldman JL (2011). Active expiration induced by excitation of ventral medulla in adult anesthetized rats. J Neurosci 31, 2895–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan LG, Forster HV, Martino P, Strecker PJ, Beales J, Serra A, Lowry TF, Forster MM & Forster AL (1998). Important role of carotid afferents in control of breathing. J Appl Physiol 85, 1299–1306. [DOI] [PubMed] [Google Scholar]
- Pijacka W, McBryde FD, Marvar PJ, Lincevicius GS, Abdala AP, Woodward L, Li D, Paterson DJ & Paton JF (2016a). Carotid sinus denervation ameliorates renovascular hypertension in adult Wistar rats. J Physiol 594, 6255–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijacka W, Moraes DJ, Ratcliffe LE, Nightingale AK, Hart EC, da Silva MP, Machado BH, McBryde FD, Abdala AP, Ford AP & Paton JF (2016b). Purinergic receptors in the carotid body as a new drug target for controlling hypertension. Nat Med 22, 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR & Semenza GL (2015). Oxygen sensing and homeostasis. Physiology 30, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanantsoa N, Hirsch MR, Thoby‐Brisson M, Dubreuil V, Bouvier J, Ruffault PL, Matrot B, Fortin G, Brunet JF, Gallego J & Goridis C (2011). Breathing without CO2 chemosensitivity in conditional Phox2b mutants. J Neurosci 31, 12880–12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves SR & Gozal D (2006). Changes in ventilatory adaptations associated with long‐term intermittent hypoxia across the age spectrum in the rat. Resp Physiol Neurobiol 150, 135–143. [DOI] [PubMed] [Google Scholar]
- Richter DW & Smith JC (2014). Respiratory rhythm generation in vivo. Physiology 29, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffault PL, D'Autreaux F, Hayes JA, Nomaksteinsky M, Autran S, Fujiyama T, Hoshino M, Hagglund M, Kiehn O, Brunet JF, Fortin G & Goridis C (2015). The retrotrapezoid nucleus neurons expressing Phox2b and Atoh‐1 are essential for the respiratory response to CO2 . eLife 4, e07051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, Molkov YI, Jasinski PE, Shevtsova NA & Smith JC (2014). Rhythmic bursting in the pre‐Botzinger complex: mechanisms and models. Prog Brain Res 209, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Stornetta RL, Stornetta DS, Onengut‐Gumuscu S, Farber EA, Turner SD, Guyenet PG & Bayliss DA (2017). Neuromedin B expression defines the mouse retrotrapezoid nucleus. J Neurosci 37, 11744–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinski M, Lewandowski J, Przybylski J, Bidiuk J, Abramczyk P, Ciarka A & Gaciong Z (2012). Tonic activity of carotid body chemoreceptors contributes to the increased sympathetic drive in essential hypertension. Hypertens Res 35, 487–491. [DOI] [PubMed] [Google Scholar]
- Smith CA, Blain GM, Henderson KS & Dempsey JA (2015). Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO2: role of carotid body CO2 . J Physiol 593, 4225–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Saupe KW, Henderson KS & Dempsey JA (1995). Ventilatory effects of specific carotid body hypocapnia in dogs during wakefulness and sleep. J Appl Physiol (1985) 79, 689–699. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Barna BF, Cruz JC, Colombari E & Moreira TS (2014). Phox2b‐expressing retrotrapezoid neurons and the integration of central and peripheral chemosensory control of breathing in conscious rats. Exp Physiol 99, 571–585. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL & Guyenet PG (2006). Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2‐sensitive neurons in rats. J Physiol 572, 503–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ & Smith CA (2013). CrossTalk opposing view: peripheral and central chemoreceptors have hyperadditive effects on respiratory motor control. J Physiol 591, 4359–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby‐Brisson M, Karlen M, Wu N, Charnay P, Champagnat J & Fortin G (2009). Genetic identification of an embryonic parafacial oscillator coupling to the preBotzinger complex. Nat Neurosci 12, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Turovsky E, Theparambil SM, Kasymov V, Deitmer JW, Del Arroyo AG, Ackland GL, Corneveaux JJ, Allen AN, Huentelman MJ, Kasparov S, Marina N & Gourine AV (2016). Mechanisms of CO2/H+ sensitivity of astrocytes. J Neurosci 36, 10750–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakai J, Takamura D, Morinaga R, Nakamuta N & Yamamoto Y (2015). Differences in respiratory changes and Fos expression in the ventrolateral medulla of rats exposed to hypoxia, hypercapnia, and hypercapnic hypoxia. Respir Physiol Neurobiol 215, 64–72. [DOI] [PubMed] [Google Scholar]
- Wang S, Shi Y, Shu S, Guyenet PG & Bayliss DA (2013). Phox2b‐expressing retrotrapezoid neurons are intrinsically responsive to acidification and CO2 . J Neurosci 33, 7756–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Kreneisz O, Nishiyama A & Mulkey DK (2010). Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1‐Kir5.1‐like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol 104, 3042–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RJ & Day TA (2013). CrossTalk opposing view: peripheral and central chemoreceptors have hypoadditive effects on respiratory motor output. J Physiol 591, 4355–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Morgan BJ, Chenuel B, Khayat R, Reichmuth K, Lin J & Dempsey JA (2006). Influence of cerebrovascular function on the hypercapnic ventilatory response in healthy humans. J Physiol 577, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF & Machado BH (2008). Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol 586, 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]