

Abstract
Key points
Increasing blood flow (hyperaemia) to exercising muscle helps match oxygen delivery and metabolic demand. During exercise in hypoxia, there is a compensatory increase in muscle hyperaemia that maintains oxygen delivery and tissue oxygen consumption.
Nitric oxide (NO) and prostaglandins (PGs) contribute to around half of the augmented hyperaemia during hypoxic exercise, although the contributors to the remaining response are unknown.
In the present study, inhibiting NO, PGs, Na+/K+‐ATPase and inwardly rectifying potassium (KIR) channels did not blunt augmented hyperaemia during hypoxic exercise beyond previous observations with NO/PG block alone. Furthermore, although inhibition of only Na+/K+‐ATPase and KIR channels abolished hyperaemia during hypoxia at rest, it had no effect on augmented hyperaemia during hypoxic exercise.
This is the first study in humans to demonstrate that Na+/K+‐ATPase and KIR channel activation is required for augmented muscle hyperaemia during hypoxia at rest but not during hypoxic exercise, thus providing new insight into vascular control.
Abstract
Exercise hyperaemia in hypoxia is augmented relative to the same exercise intensity in normoxia. During moderate‐intensity handgrip exercise, endothelium‐derived nitric oxide (NO) and vasodilating prostaglandins (PGs) contribute to ∼50% of the augmented forearm blood flow (FBF) response to hypoxic exercise (HypEx), although the mechanism(s) underlying the remaining response are unclear. We hypothesized that combined inhibition of NO, PGs, Na+/K+‐ATPase and inwardly rectifying potassium (KIR) channels would abolish the augmented hyperaemic response in HypEx. In healthy young adults, FBF responses were measured (Doppler ultrasound) and forearm vascular conductance was calculated during 5 min of rhythmic handgrip exercise at 20% maximum voluntary contraction under regional sympathoadrenal inhibition in normoxia and isocapnic HypEx (O2 saturation ∼80%). Compared to control, combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels (l‐NMMA + ketorolac + ouabain + BaCl2; Protocol 1; n = 10) blunted the compensatory increase in FBF during HypEx by ∼50% (29 ± 6 mL min−1 vs. 62 ± 8 mL min−1, respectively, P < 0.05). By contrast, ouabain + BaCl2 alone (Protocol 2; n = 10) did not affect this augmented hyperaemic response (50 ± 11 mL min−1 vs. 60 ± 13 mL min−1, respectively, P > 0.05). However, the blocked condition in both protocols abolished the hyperaemic response to hypoxia at rest (P < 0.05). We conclude that activation of Na+/K+‐ATPase and KIR channels is involved in the hyperaemic response to hypoxia at rest, although it does not contribute to the augmented exercise hyperaemia during hypoxia in humans.
Keywords: hypoxia, blood flow, vasodilatation
Key points
Increasing blood flow (hyperaemia) to exercising muscle helps match oxygen delivery and metabolic demand. During exercise in hypoxia, there is a compensatory increase in muscle hyperaemia that maintains oxygen delivery and tissue oxygen consumption.
Nitric oxide (NO) and prostaglandins (PGs) contribute to around half of the augmented hyperaemia during hypoxic exercise, although the contributors to the remaining response are unknown.
In the present study, inhibiting NO, PGs, Na+/K+‐ATPase and inwardly rectifying potassium (KIR) channels did not blunt augmented hyperaemia during hypoxic exercise beyond previous observations with NO/PG block alone. Furthermore, although inhibition of only Na+/K+‐ATPase and KIR channels abolished hyperaemia during hypoxia at rest, it had no effect on augmented hyperaemia during hypoxic exercise.
This is the first study in humans to demonstrate that Na+/K+‐ATPase and KIR channel activation is required for augmented muscle hyperaemia during hypoxia at rest but not during hypoxic exercise, thus providing new insight into vascular control.
Introduction
During exercise, acute exposure to systemic hypoxia results in a compensatory increase in skeletal muscle blood flow relative to the same level of exercise under normoxic conditions (Hartley et al. 1973; Rowell et al. 1986; Wilkins et al. 2006, 2008, Casey et al. 2009, 2010; Crecelius et al. 2011b). This augmented hyperaemia compensates for the lower arterial oxygen content in hypoxia to maintain the matching of oxygen delivery with the oxygen demand of the contracting muscle (Koskolou et al. 1997; Roach et al. 1999; González‐Alonso et al. 2001; Crecelius et al. 2011b; Casey & Joyner, 2012). The response appears to be primarily the result of an increase in local vasodilatory factors rather than a decrease in or blunting of vasoconstrictor signals because hypoxia increases sympathetic nervous system activity (Hanada et al. 2003; Wilkins et al. 2008) but does not affect postjunctional α‐adrenergic receptor responsiveness (Dinenno et al. 2003; Wilkins et al. 2006; Dinenno, 2016). Although numerous studies have attempted to identify the contributing vasodilatory factors, our understanding of the response remains incomplete.
With sympathoadrenal signalling intact, adenosine and circulating epinephrine (and subsequent β‐adrenoceptor activation) do not contribute to the augmented vasodilatory response during moderate intensity exercise (Wilkins et al. 2008; Casey et al. 2009, 2011; Heinonen et al. 2010), whereas nitric oxide (NO) has been shown to play a significant role (Casey et al. 2010). Our laboratory has investigated the underlying mechanisms of the vascular response to hypoxia at rest and during exercise utilizing regional (forearm) sympathoadrenal blockade aiming to isolate local vasodilator mechanisms without concomitant α‐ and β‐adrenoceptor influences on vascular tone. At rest, we found an interaction between NO and prostaglandins (PGs), with inhibition of these pathways abolishing hypoxic vasodilatation (Markwald et al. 2011). However, during moderate intensity rhythmic handgrip exercise, combined inhibition of NO and PGs only blunts the hypoxic augmentation of blood flow by ∼50%, indicating that other vasodilator mechanisms are involved in the response (Crecelius et al. 2011b; Dinenno, 2016).
Recent investigations in animals and humans have demonstrated a role for Na+/K+‐ATPase and inwardly‐rectifying potassium (KIR) channels in the hyperaemic response to exercise and ischaemia (Edwards et al. 1998; Armstrong et al. 2007; Crecelius et al. 2013a, b , 2014; Ahn et al. 2017). Whether activation of these pathways contributes to vascular control during hypoxia at rest or in combination with exercise remains unknown. Both Na+/K+‐ATPase and KIR channels can be stimulated by extracellular K+ and there is also evidence that hypoxia can (i) increase venous plasma K+ concentrations (Marshall et al. 1993); (ii) stimulate K+ release from vascular smooth muscle (Park et al. 2005), the vascular endothelium or red blood cells (RBCs) (Mo & Ballard, 2005; Aziz et al. 2017); and (iii) increase local vasodilator responsiveness to K+ (Skinner & Costin, 1971). Alternatively, RBCs release ATP in response to haemoglobin deoxygenation, which can subsequently bind to purinergic (P2Y) receptors on the vascular endothelium to stimulate vasodilatation (Ellsworth et al. 2009, 2016; Ellsworth & Sprague, 2012). In this context, recent studies in humans have shown that venous plasma ATP concentrations are elevated in the circulation draining hypoxic and active skeletal muscle (González‐Alonso et al. 2002; Mortensen et al. 2011; Kirby et al. 2012). Once in circulation, ATP can evoke vasodilatation via stimulation of NO and PG production (Rongen et al. 1994; van Ginneken et al. 2004; Mortensen et al. 2009; Crecelius et al. 2011a, 2012) and, importantly, our laboratory has recently demonstrated that ∼40–50% of ATP‐mediated vasodilatation in the human forearm occurs via activation of KIR channels (Crecelius et al. 2012; Hearon et al. 2017).
Accordingly, the primary purpose of the present study was to test the hypothesis that combined inhibition of NO, PGs, and Na+/K+‐ATPase and KIR channel activation would abolish the augmented hyperaemic response during hypoxic exercise in humans. Further, we hypothesized that increased oxygen extraction would compensate for reductions in muscle blood flow (and thus oxygen delivery) to maintain oxygen consumption of both the resting and active skeletal muscle tissue.
Methods
Ethical approval and subjects
The present study conformed to the standards set by the Declaration of Helsinki, except for registration in a database. With approval from the Institutional Review Board at Colorado State University (protocol 14‐5392H), a total of 20 young healthy adults participated in the present study after providing their informed, written consent. Protocol 1 (combined inhibition of NO/PG synthesis and Na+/K+‐ATPase and KIR channel activation) comprised five men and five women; age 23 ± 2 years; weight 70.3 ± 5.2 kg; height 174 ± 3 cm; body mass index 22.9 ± 1.1 kg m−2. Protocol 2 (Na+/K+‐ATPase and KIR channel inhibition) comprised four men and six women; age 22 ± 2 years; weight 69.3 ± 3.9 kg; height 170 ± 2 cm; body mass index 23.9 ± 0.9 kg m−2. One subject participated in both protocols. All subjects were non‐smokers, non‐obese, normotensive (resting blood pressure <140/90 mmHg) and not taking any medications. Studies were performed in the Human Cardiovascular Physiology Laboratory at Colorado State University (altitude: ∼1500 m) after a 4 h fast and abstention from caffeine and exercise for 12 h, with subjects in the supine position, with the experimental arm abducted to 90° and slightly elevated above heart level upon a tilt‐adjustable table. Female subjects were studied during the early follicular phase of their menstrual cycle to minimize potential cardiovascular effects of sex‐specific hormones.
Arterial and venous catheterization
For both protocols, a 20‐gauge, 7.6 cm catheter was placed in the brachial artery of the non‐dominant arm under aseptic conditions after local anaesthesia (2% lidocaine) for local administration of study drugs and blood sampling. The catheter was connected to a three‐port connector and a pressure transducer for mean arterial pressure (MAP) measurement and was continuously flushed at 3 mL h−1 with heparinized saline, with the two side ports used for drug infusions (Markwald et al. 2011; Crecelius et al. 2011b). Additionally, in Protocol 1, a 20‐gauge, 5.1 cm catheter was inserted in retrograde fashion into an antecubital vein of the experimental arm for deep venous blood samples (Crecelius et al. 2011b). Saline was continuously infused through this catheter at a rate of ∼2 mL min−1 for the duration of the study to keep it patent.
Forearm blood flow and vascular conductance
A 12 MHz linear‐array ultrasound probe (Vivid 7; General Electric, Milwaukee, WI, USA) was used to determine brachial artery mean blood velocity (MBV) and diameter proximal to the catheter insertion site as described previously (Crecelius et al. 2011b; Hearon et al. 2017; Richards et al. 2017). Foam tape was used to mark the outline of the probe for consistent placement and measurement over the course of the experiments. For blood velocity measurements, the probe insonation angle was maintained at <60° and the frequency used was 5 MHz. The Doppler shift frequency spectrum was analysed via a Multigon 500M TCD (Multigon Industries, Mt. Vernon, NY, USA) spectral analyser from which MBV was determined as a weighted mean of the spectrum of Doppler shift frequencies. Brachial artery diameter measurements were made in duplex mode at end‐diastole and between contractions (in triplicate) during steady‐state conditions (Crecelius et al. 2011b). FBF was calculated as: FBF = MBV × π × (brachial artery diameter/2)2 × 60, where FBF is expressed as mL min−1, MBV as cm s−1, brachial diameter as cm, and 60 was used to convert from mL s−1 to mL min−1. Forearm vascular conductance (FVC) was calculated as (FBF/MAP) × 100, and expressed as mL min−1 100 mmHg−1 (Markwald et al. 2011; Crecelius et al. 2011b; Hearon et al. 2017; Richards et al. 2017). All studies were performed in a cool (20–22°C) temperature‐controlled environment with a fan directed toward the forearm to minimize the contribution of skin blood flow to forearm haemodynamics.
Rhythmic handgrip exercise
Maximum voluntary contraction (MVC) was determined for the experimental arm as the average of three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL, USA) that were within 3% of each other (Protocol 1: mean 33.8 ± 2.8 kg, range 24.0–50.0 kg; Protocol 2: mean 31.2 ± 2.4 kg, range 22.0–47.0 kg). Rhythmic handgrip exercise during the trials was performed with weight corresponding to 20% MVC (Protocol 1: mean 6.8 ± 0.6 kg, range 4.8–10.0 kg; Protocol 2: mean 6.2 ± 0.5 kg, range 4.4–9.4 kg) attached to a pulley system and lifted 4–5 cm over the pulley at a duty cycle of 1 s contraction/2 s relaxation (20 contractions min–1) using both visual and auditory feedback to ensure the correct timing (Dinenno & Joyner, 2003, 2004). We chose this workload based on recent data suggesting that a greater potential role for local vasodilators in muscle blood flow regulation during hypoxia at this relative intensity (Wilkins et al. 2008) and also to minimize sympathetically‐mediated increases in heart rate and mean arterial blood pressure as a result of exercise (Victor & Seals, 1989). Previous studies in our laboratory have determined that MVC is not affected by any of the vasoactive substances administered in the present study, particularly ouabain and BaCl2 (Crecelius et al. 2013a).
Systemic isocapnic hypoxia
To isolate the effects of systemic isocapnic hypoxia, we used a self‐regulating partial rebreathe system developed by Banzett et al. (2000) and more recently described by our laboratory (Markwald et al. 2011; Crecelius et al. 2011b; Richards et al. 2017). This system allows for constant alveolar fresh air ventilation independent of changes in breathing frequency or tidal volume (Banzett et al. 2000; Dinenno et al. 2003; Wilkins et al. 2008). Using this system, we were able to clamp end‐tidal CO2 levels despite the hypoxia‐induced increases in ventilation. The level of oxygen was manipulated by mixing nitrogen with medical air via an anaesthesia gas blender. For the hypoxic trials, inspired oxygen was titrated to achieve arterial oxygen saturations () of ∼80% as assessed via pulse oximetry. For normoxic trials, subjects were placed on the rebreathe system but inspired ambient air. Subjects breathed through a scuba mouthpiece with a nose‐clip to prevent nasal breathing. An anaesthesia monitor (Cardiocap/5; Datex‐Ohmeda, Louisville, CO, USA) was used to determine heart rate (HR) (3‐lead ECG) and expired CO2 sampled at the mouthpiece. Ventilation was measured via a turbine pneumotachograph (model 17125 UVM; Vacu‐Med, Ventura, CA, USA).
Pharmacological infusions
Regional sympathoadrenal inhibition (Protocols 1 and 2)
To isolate local vascular control mechanisms, sympathoadrenal influences on forearm vascular tone were blocked using brachial artery catheter administration of phentolamine mesylate (non‐selective α‐adrenoceptor antagonist; Bedford Laboratories, Bedford, OH, USA) and propranolol hydrochloride (non‐selective β‐adrenoceptor antagonist; Baxter, Deerfield, IL, USA) as described previously by our laboratory (Markwald et al. 2011; Crecelius et al. 2011b; Richards et al. 2014, 2017). A loading dose totalling 1000 μg (200 μg min−1 for 5 min) of each drug was infused prior to initiating experimental trials and a maintenance dose (50 μg min−1) was infused throughout the entire study to ensure continuous blockade. The dose of phentolamine used was twice as great as those previously reported to effectively block α‐adrenoceptors (Eklund & Kaijser, 1976; Dietz et al. 1997; Halliwill et al. 1997) and maintains effective α‐blockade for several hours (Markwald et al. 2011). The dose of propranolol used has been shown to inhibit forearm vasodilatation in response to the non‐selective β‐adrenoceptor agonist isoproterenol (Johnsson, 1967; Richards et al. 2017), as well as reduce vasodilatation in the resting forearm during contralateral isometric handgrip exercise (Eklund & Kaijser, 1976).
Regional NO synthase (NOS) inhibition and cyclooxygenase (COX) inhibition (Protocol 1)
Intra‐arterial administration of N G‐monomethyl‐l‐arginine (l‐NMMA; non‐selective NOS inhibitor; Bachem, Bubendorf, Switzerland) was used to inhibit the production of NO. A 5 min loading dose (5 mg min−1, 25 mg total) was infused initially, followed by a maintenance dose (1.25 mg min−1) for the duration of the study to ensure continuous blockade. This dose of l‐NMMA has been shown to significantly reduce basal tone and also the vasodilatory effects of ACh (Dietz et al. 1994; Eisenach et al. 2002; Dinenno & Joyner, 2004), consistent with effective NOS inhibition (Vallance et al. 1989). Additionally, intra‐arterial administration of ketorolac (non‐selective COX inhibitor; trade name Toradol; Hospira, Lake Forest, IL, USA) was used to inhibit the production of PGs (Markwald et al. 2011; Crecelius et al. 2011b). A 10 min loading dose (600 μg min−1, 6 mg total) was infused initially, followed by a maintenance dose (150 μg min−1) for the duration of the study to ensure continuous blockade. This dose of ketorolac is twice that previously demonstrated to reduce forearm blood flow during exercise (Schrage et al. 2004) and reduce circulating PGF1α (a stable breakdown product of PGs) at rest and during handgrip exercise (Dinenno & Joyner, 2004).
Regional Na+/K+‐ATPase and and KIR channel inhibition (Protocols 1 and 2)
Intra‐arterial administration of ouabain octahydrate (Na+/K+‐ATPase inhibitor, Sigma‐O3125; Sigma‐Aldrich, St Louis, MO, USA) was infused at 2.7 nmol min−1 in combination with barium chloride (BaCl2; KIR channel inhibitor; 10% w/v BDH3238; EMD Chemicals, Gibbstown, NJ, USA) at 0.9 μmol (dL forearm volume)−1 min−1 with an absolute dose of 8–10 μmol min−1 (Nelson & Quayle, 1995; Edwards et al. 1998; Crecelius et al. 2012, 2013a, 2014, 2015). This approach abolishes vasodilatation to increasing doses of intra‐arterial potassium chloride (Crecelius et al. 2012). Ouabain and BaCl2 were loaded for 15 and 3 min, respectively, prior to the first trial of the blocked condition and were continued at these same doses for the duration of the trial. In the second trial of the blocked condition, infusion of all inhibitors began 3 min before the start of muscle contractions. Ouabain and BaCl2 were prepared in saline and confirmed sterile and free of fungus/endotoxin and particulate matter with a standard microbiology report (JCB‐Analytical Research Labs, Wichita, KS, USA) prior to use. The forearm volume used for normalization of BaCl2 was determined from regional analysis of whole‐body dual‐energy X‐ray absorptiometry scans (QDR series software; Hologic, Inc., Marlborough, MA, USA).
Experimental protocol
A timeline for the overall study and each trial is depicted in Fig. 1. Following catheter placement and experimental set‐up in both protocols, resting haemodynamics were determined before and after regional sympathoadrenal inhibition. Normoxic trials consisted of a 3 min baseline period and 5 min of 20% MVC rhythmic handgrip exercise. Hypoxic trials consisted of a 3 min baseline period, 3 min of steady‐state hypoxia at ∼80% (plus ∼2 min for the normoxia to hypoxia transition; monitored via pulse oximeter) and 5 min of hypoxia plus 20% MVC rhythmic handgrip exercise.
Figure 1. Timeline and experimental protocol.
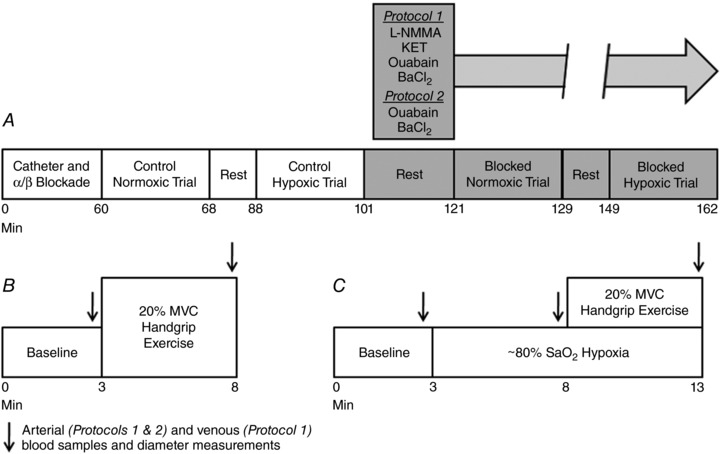
A, overall experimental protocol; after placement of a brachial artery catheter (Protocols 1 and 2) and a deep venous catheter (Protocol 1), phentolamine and propranolol were administered intra‐arterially to block α‐ and β‐adrenoceptors, respectively, and to isolate local vascular control mechanisms. Normoxic and hypoxic exercise trials were performed under the control (saline) condition, and then repeated following intra‐arterial administration of pharmacological antagonists to inhibit NO/PG synthesis and Na+/K+‐ATPase and KIR channel activation (Protocol 1) or only Na+/K+‐ATPase and KIR channel activation (Protocol 2). B, normoxic trial timeline; baseline measurements were made for 3 min, followed by 5 min of 20% MVC rhythmic handgrip exercise. C, hypoxic trial timeline; baseline measurements were made for 3 min and oxygen saturations were then lowered to ∼80% within the first 2 min and maintained for the duration of the trial. After 3 min of steady‐state hypoxia, subjects performed 5 min of 20% MVC rhythmic handgrip exercise. l‐NMMA, N G‐monomethyl‐l‐arginine; KET, ketorolac; BaCl2, barium chloride.
In Protocol 1, we aimed to determine whether combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels (l‐NMMA, ketorolac, ouabain, and BaCl2, respectively) would abolish the augmented vasodilatation and hyperaemia during hypoxic exercise. Thus, subjects performed exercise in normoxia and hypoxia under control (saline) and blocked (aforementioned inhibitors) conditions. In total, four experimental trials each separated by 20 min of rest were performed, and the order of normoxic and hypoxic trials before and after blockade was counterbalanced between subjects. Given the lack of an additional effect of Na+/K+‐ATPase and KIR channel inhibition (see Results) compared to our previous findings with NO/PG inhibition alone (Crecelius et al. 2011b) and the possibility that redundant vasodilator signalling could mask an effect of inhibiting these pathways (Schrage et al. 2004; Markwald et al. 2011; Joyner & Casey, 2014; Lamb & Murrant, 2015), we performed a follow‐up protocol (Protocol 2) to test the contribution of Na+/K+‐ATPase and KIR channel activation alone. The experimental approach in Protocol 2 was identical to Protocol 1, with the exception being that only Na+/K+‐ATPase and KIR channels were inhibited (ouabain and BaCl2, respectively) in the blocked condition.
Data acquisition and analysis
Data were collected and stored on a computer at 250 Hz and were analysed offline with signal‐processing software (WinDaq; DATAQ Instruments, Akron, OH, USA). MAP was determined from the brachial artery pressure waveform. FBF, HR, MAP and oxygen saturations (pulse oximetry) represent an average of the last 30 s of the appropriate time period. Minute ventilation and end‐tidal CO2 were determined from an average of the data over the last minute of each time period. The sampling timeframe used for averaging was greater for respiratory variables than hemodynamic variables to ensure an adequate number of sampling points.
In both protocols, blood samples were taken at the end of rest, normoxic exercise, steady‐state hypoxia, and hypoxic exercise under control and blocked conditions (Fig. 1). In Protocol 1, brachial artery and deep venous blood samples (∼2 mL) were drawn and immediately (<1 min) analysed with a clinical blood gas analyser (Rapid Point 400 Series Automatic Blood Gas System; Siemens Healthcare Diagnostics, Deerfield, IL, USA) for partial pressures of oxygen and carbon dioxide ( and ), fraction of oxygenated haemoglobin (FO2Hb), oxygen content and pH. In Protocol 2, only brachial artery blood samples were drawn and analysed as described for Protocol 1. From the blood gas data in Protocol 1, arteriovenous oxygen difference (a − ) was calculated as the difference between arterial () and venous () oxygen content. Oxygen delivery was calculated as ( × FBF × 0.001) and expressed in mL min−1. Oxygen extraction, reported as a percent, was calculated as [( – ) × −1 × 100]. Oxygen consumption across the forearm () was calculated as [( – ) × FBF × 0.001] and expressed in mL min−1.
We also have expressed the FBF data in two alternative forms. First, we calculated ‘hypoxic augmentation’ as the difference between absolute FBF during hypoxic exercise and during normoxic exercise under control (saline), combined NO/PG/Na+/K+‐ATPase/KIR channel inhibition (Protocol 1) or Na+/K+‐ATPase and KIR channel inhibition alone (Protocol 2) conditions, as described previously (Crecelius et al. 2011b). We consider that this accurately reflects the underlying physiological responses of the primary research question addressed in the present study. In this context, we are interested in the substances or pathways involved in augmenting FBF during hypoxic compared to normoxic exercise because this difference in blood flow is what drives the maintenance of oxygen delivery during systemic hypoxia when arterial oxygen content is reduced. Second, we calculated the change in FBF from rest under a given physiological (normoxic or hypoxic) and drug condition because there is interest in this means of data expression by investigators in the field. We also performed this calculation with the data obtained in our original study on this topic (Crecelius et al. 2011b) and have addressed these findings in the Discussion.
Statistical analysis
Data are presented as the mean ± SEM. Differences within and between trials and conditions were determined via two‐way repeated measures ANOVA. As a result of large differences in the magnitude of values of exercise and rest, for comparisons of FBF, FVC, a − , oxygen extraction, oxygen delivery and oxygen consumption, two ANOVAs were completed, one including both exercise conditions (normoxic and hypoxic) and one including both rest periods and hypoxia, as described previously (Crecelius et al. 2011b). For all other variables collected, all time points were included in the ANOVA analysis. Specific hypothesis testing was performed using a two‐tailed Student's t test for paired data and post hoc comparisons were made with Tukey's honestly significant test when appropriate. Significance was set a priori at P < 0.05.
Results
Effect of regional α‐ and β‐adrenoceptor blockade
Prior to all experimental trials, combined blockade of α‐ and β‐adrenoceptors significantly increased resting FBF (Protocol 1: 44 ± 8 vs. 81 ± 12 mL min−1; Protocol 2: 35 ± 4 vs. 70 ± 7 mL min−1) and FVC (Protocol 1: 50 ± 10 vs. 91 ± 15 mL min−1 100 mmHg−1; Protocol 2: 38 ± 4 vs. 74 ± 8 mL min−1 100 mmHg−1) by ∼100% (P < 0.05). MAP (Protocol 1: 90 ± 2 vs. 90 ± 2 mmHg; Protocol 2: 93 ± 2 vs. 91 ± 2 mmHg) and HR (Protocol 1: 61 ± 4 vs. 59 ± 4 beats min−1; Protocol 2: 67 ± 3 vs. 63 ± 3 beats min−1) did not change as a result of local α‐and β‐receptor blockade. These effects are comparable to our previous observations (Crecelius et al. 2011b).
Systemic haemodynamic and respiratory responses
Systemic haemodynamic and respiratory responses are presented for each trial and condition in Protocol 1 (Table 1) and Protocol 2 (Table 2). Briefly, the targeted of ∼80% was achieved under all hypoxic conditions and was similar across trials in both protocols. MAP was increased during normoxic and hypoxic exercise relative to rest and was significantly greater in the blocked vs. control condition at all time points in both protocols (P < 0.05). As expected, hypoxia at rest and during exercise significantly increased HR and ventilation under all conditions (P < 0.05).
Table 1.
Protocol 1 systemic haemodynamic and ventilatory responses
| Rest 1 | Normoxic exercise | Rest 2 | Steady‐state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| MAP (mmHg) | 90 ± 2 | 95 ± 1† | 91 ± 2 | 94 ± 2 | 97 ± 2† |
| HR (beats min−1) | 59 ± 4 | 69 ± 6† | 59 ± 3 | 75 ± 4† | 79 ± 4†‡ |
| FVC (mL min−1 100 mmHg−1) | 91.4 ± 14.9 | 357.3 ± 40.1† | 91.2 ± 17.1 | 117.0 ± 24.8† | 415.9 ± 44.8†‡ |
| (%) | 99 ± 0 | 98 ± 1 | 99 ± 0 | 82 ± 2†‡ | 82 ± 1†‡ |
| Minute vent. (L min−1; BTPS) | 8.4 ± 0.4 | 10.5 ± 0.6 | 9.1 ± 0.4 | 17.0 ± 1.4†‡ | 23.1 ± 2.3†‡# |
| End‐tidal CO2 (mmHg) | 40.2 ± 0.7 | 38.7 ± 0.9 | 39.1 ± 1.4 | 38.6 ± 0.9 | 39.5 ± 1.0 |
| NO, PG, Na+/K+‐ATPase, KIR channel block | |||||
| MAP (mmHg) | 98 ± 2* | 105 ± 2*† | 97 ± 3* | 103 ± 3*† | 106 ± 2*† |
| HR (beats min−1) | 57 ± 3 | 63 ± 4* | 57 ± 3 | 75 ± 3†‡ | 79 ± 4†‡ |
| FVC (mL min−1 100 mmHg−1) | 41.0 ± 6.1* | 244.4 ± 31.1*† | 41.5 ± 7.4* | 36.9 ± 5.0* | 270.3 ± 31.2*†‡ |
| (%) | 99 ± 0 | 98 ± 1 | 99 ± 0 | 84 ± 1*†‡ | 82 ± 1†‡ |
| Minute vent. (L min−1; BTPS) | 9.5 ± 0.7 | 11.4 ± 0.6 | 8.8 ± 0.7 | 22.1 ± 1.6*†‡ | 28.8 ± 2.7*†‡# |
| End‐tidal CO2 (mmHg) | 37.3 ± 1.3* | 36.5 ± 1.1* | 38.1 ± 0.8 | 38.0 ± 0.9 | 38.5 ± 1.0‡ |
BTPS, body temperature and pressure, saturated.
* P < 0.05 vs. control; † P < 0.05 vs. rest (within trial); ‡ P < 0.05 vs. normoxic exercise; # P < 0.05 vs. hypoxia; all comparisons within protocol; , oxygen saturation via pulse oximetry.
Table 2.
Protocol 2 systemic haemodynamic and ventilatory responses
| Rest 1 | Normoxic exercise | Rest 2 | Steady‐state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| MAP (mmHg) | 91 ± 2 | 96 ± 3 | 91 ± 2 | 94 ± 3 | 96 ± 2 |
| HR (beats min−1) | 63 ± 3 | 69 ± 3† | 62 ± 2 | 78 ± 2†‡ | 79 ± 2†‡ |
| FVC (mL min−1 100 mmHg−1) | 74.0 ± 7.8 | 311.0 ± 31.5† | 64.0 ± 6.6 | 87.0 ± 11.3† | 370.9 ± 38.1†‡ |
| (%) | 99 ± 0 | 98 ± 1 | 98 ± 0 | 81 ± 1†‡ | 79 ± 0†‡ |
| Minute vent. (L min−1; BTPS) | 8.7 ± 1.0 | 10.4 ± 0.9 | 8.7 ± 0.8 | 16.2 ± 2.3† | 18.9 ± 2.4†‡ |
| End‐tidal CO2 (mmHg) | 35.3 ± 1.4 | 34.7 ± 1.7 | 35.2 ± 0.9 | 35.2 ± 1.0 | 35.8 ± 1.1 |
| Na+/K+‐ATPase, KIR channel block | |||||
| MAP (mmHg) | 98 ± 3* | 105 ± 3*† | 98 ± 2* | 102 ± 3* | 106 ± 3*† |
| HR (beats min−1) | 61 ± 3 | 65 ± 3* | 62 ± 2 | 79 ± 3†‡ | 79 ± 2†‡ |
| FVC (mL min−1 100 mmHg−1) | 35.8 ± 2.1* | 215.8 ± 19.9*† | 36.6 ± 2.1* | 33.7 ± 1.8* | 259.1 ± 24.7*†‡ |
| (%) | 98 ± 0 | 98 ± 0 | 98 ± 0 | 80 ± 1†‡ | 81 ± 1*†‡ |
| Minute vent. (L min−1; BTPS) | 9.3 ± 0.4 | 10.6 ± 0.9 | 9.5 ± 0.8 | 21.5 ± 3.3*†‡ | 23.4 ± 2.9*†‡ |
| End‐tidal CO2 (mmHg) | 34.0 ± 1.1* | 33.2 ± 1.3* | 35.2 ± 0.7 | 34.5 ± 1.2 | 34.8 ± 1.0 |
BTPS, body temperature and pressure, saturated.
* P < 0.05 vs. control; † P < 0.05 vs. rest (within trial); ‡ P < 0.05 vs. normoxic exercise; all comparisons within protocol; , oxygen saturation via pulse oximetry.
Forearm blood flow and vascular conductance responses
Absolute values of FBF for each trial in both protocols are presented in Fig. 2 and all main effects are similar when the data are presented as FVC (Tables 1 and 2). In Protocol 1 (Fig. 2 A), resting FBF was not different between normoxic (Rest 1) and hypoxic (Rest 2) trials. As expected, FBF during control hypoxic exercise was significantly augmented compared to normoxia (401 ± 40 vs. 339 ± 37 mL min−1; P < 0.05). Combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels significantly reduced resting FBF and, furthermore, significantly reduced steady‐state FBF during normoxic exercise (256 ± 31 mL min−1 vs. 339 ± 37 mL min−1; P < 0.05). Blockade of these pathways abolished the FBF response to hypoxia at rest (Fig. 3) and also reduced steady‐state FBF during hypoxic exercise (285 ± 32 vs. 401 ± 40 mL min−1; P < 0.05). The augmentation of FBF during hypoxic exercise was reduced by ∼50% during blockade conditions (∆FBF: control = 62 ± 8 vs. blockade = 29 ± 6 mL min−1; P < 0.05) (Fig. 4), although FBF was still greater than normoxic exercise (P < 0.05) (Fig. 2 A). The change in FBF from rest to steady‐state exercise was reduced by ∼20% under both normoxic and hypoxic conditions (Fig. 5 B). Furthermore, the change in FBF was greater during hypoxic compared to normoxic exercise under both control and blockade conditions (Fig. 5 B).
Figure 2. Forearm haemodynamics at rest, during normoxic exercise, hypoxia and hypoxic exercise.
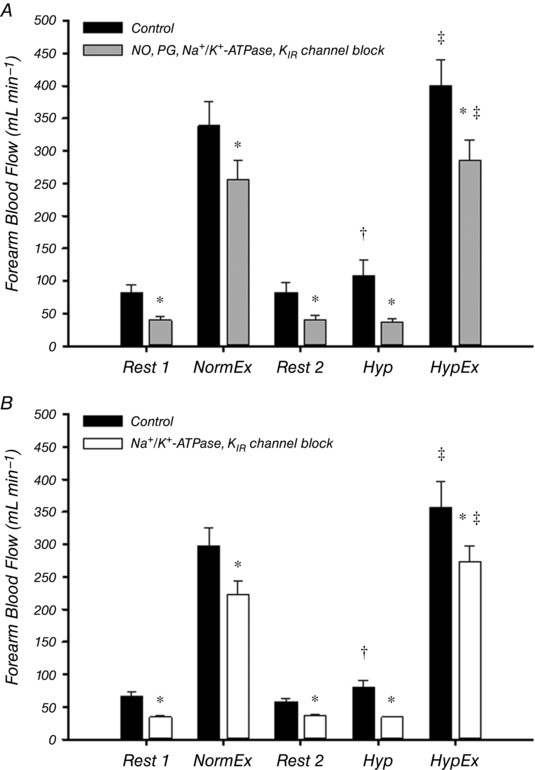
Forearm blood flow across all trials in Protocol 1 (A) and Protocol 2 (B). A, combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels significantly reduced FBF (and FVC) (Table 1) at all time points. Under control conditions, hypoxic exercise (HypEx) FBF was significantly greater than normoxic exercise (NormEx) FBF and this augmentation remained in the blocked condition. FBF during hypoxia (Hyp) under resting conditions was significantly greater than rest (Rest 2) under control conditions but not after blockade. B, with combined inhibition of Na+/K+‐ATPase and KIR channels, the trends in the data are not different from that observed in Protocol 1 (A). * P < 0.05 vs. control; † P < 0.05 vs. Rest 2; ‡ P < 0.05 vs. normoxic exercise (within condition).
Figure 3. Effect of blockade on hypoxia‐induced hyperaemia.
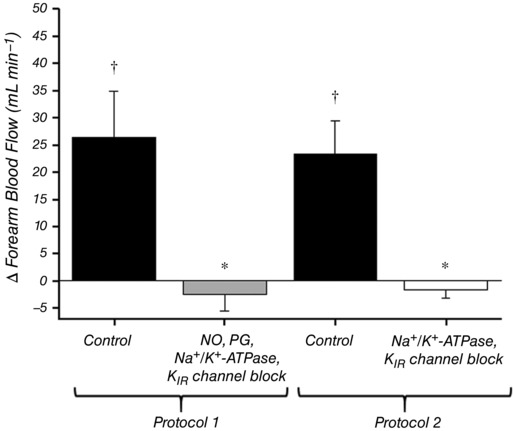
The effect of hypoxia on forearm blood flow at rest was calculated for each condition as the difference in absolute FBF between hypoxia (Hyp) and rest (Rest 2). In Protocol 1, combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels abolished the hypoxia‐induced increase in FBF at rest. In Protocol 2, combined inhibition of Na+/K+‐ATPase and KIR channels also abolished the hypoxia‐induced increase in FBF at rest. * P < 0.05 vs. control; † P < 0.05 vs. zero.
Figure 4. Effect of blockade on augmented hyperaemia during hypoxic exercise.
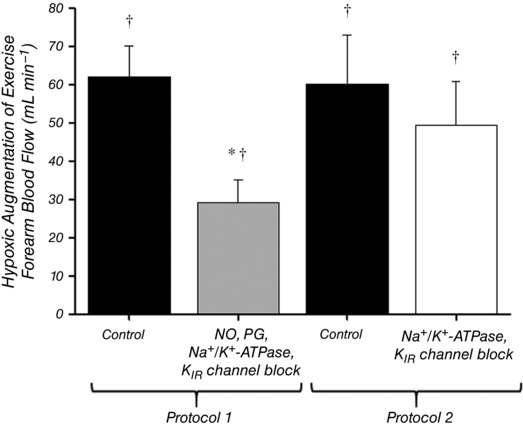
Hypoxic augmentation of forearm blood flow during exercise was calculated for each condition as the difference in absolute FBF between hypoxic exercise (HypEx) and normoxic exercise (NormEx). In Protocol 1, combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels blunted the hypoxic augmentation by ∼50%. In Protocol 2, combined inhibition of Na+/K+‐ATPase and KIR channels did not significantly affect hypoxic augmentation. * P < 0.05 vs. control; † P < 0.05 vs. zero.
Figure 5. Effect of blockade on change in forearm blood flow from rest to steady‐state exercise in normoxia and hypoxia.
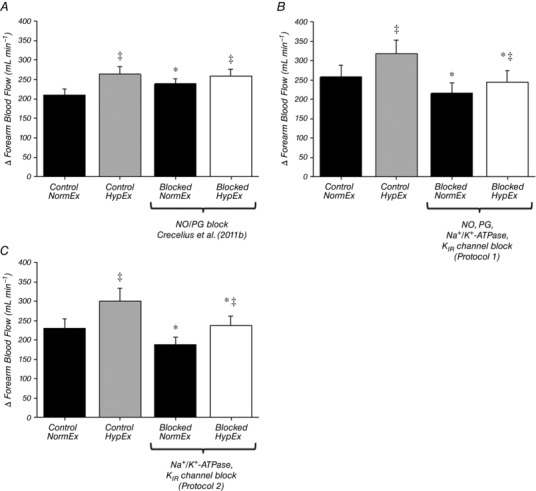
The absolute change in forearm blood flow from rest to exercise at 20% MVC in normoxia and hypoxia in each condition was calculated using data from our previous study (A) (Crecelius et al. 2011b) and also for Protocol 1 (B) and Protocol 2 (C) of the present study. The increase in FBF during hypoxic exercise was not reduced by combined NO/PG blockade (A). Combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels significantly reduced the change in FBF from rest to exercise in both normoxia and hypoxia by ∼20% (B), as did inhibition of Na+/K+‐ATPase and KIR channels alone (C). * P < 0.05 vs. control (within normoxic/hypoxic condition); ‡ P < 0.05 vs. normoxic exercise (within drug condition).
In Protocol 2 (Fig. 2 B), resting FBF was not different between normoxic (Rest 1) and hypoxic (Rest 2) trials. As expected, exercise during hypoxia under control conditions significantly augmented FBF compared to normoxia (358 ± 39 vs. 297 ± 29 mL min−1; P < 0.05). Inhibition of Na+/K+‐ATPase and KIR channels significantly reduced resting FBF and, furthermore, significantly reduced steady‐state FBF during normoxic exercise (224 ± 19 mL min−1 vs. 297 ± 29 mL min−1; P < 0.05). Blockade of these pathways abolished the FBF response to hypoxia at rest (Fig. 3) and also reduced steady‐state FBF during hypoxic exercise (273 ± 32 vs. 358 ± 39 mL min−1; P < 0.05). By contrast to Protocol 1, the augmentation of FBF during hypoxic exercise was not significantly altered by blockade of Na+/K+‐ATPase and KIR channels (∆FBF: control = 60 ± 13 vs. blockade = 50 ± 11 mL min−1; P = 0.45) (Fig. 4). The change in FBF from rest to steady‐state exercise was reduced by ∼20% under both normoxic and hypoxic conditions (Fig. 5 C). Furthermore, the change in FBF was greater during hypoxic compared to normoxic exercise under both control and blockade conditions (Fig. 5 C).
Blood gases and forearm oxygen delivery, extraction and consumption
Blood gas data for Protocols 1 and 2 are presented in Tables 3 and 4, respectively. As expected, hypoxia reduced arterial oxygen saturation (), arterial partial pressure of oxygen () and compared to normoxia in both protocols (P < 0.05). Under control and blocked conditions, there was no significant difference in arterial partial pressure of CO2 () between normoxic and hypoxic trials. In Protocol 1, both normoxic and hypoxic exercise significantly reduced venous pH (pHv), venous () and compared to rest (P < 0.05). Venous catheters were not placed in Protocol 2.
Table 3.
Protocol 1 blood gases
| Rest 1 | Normoxic exercise | Rest 2 | Steady‐state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| pHa | 7.40 ± 0.01 | 7.40 ± 0.01 | 7.42 ± 0.01 | 7.42 ± 0.01 | 7.41 ± 0.01 |
| pHv | 7.39 ± 0.01 | 7.30 ± 0.01† | 7.40 ± 0.01 | 7.41 ± 0.01‡ | 7.31 ± 0.01†# |
| (mmHg) | 37.2 ± 1.1 | 37.2 ± 1.0 | 35.4 ± 1.5 | 35.9 ± 1.1 | 36.0 ± 1.2 |
| (mmHg) | 40.6 ± 0.8 | 54.1 ± 2.5† | 38.8 ± 1.6 | 37.8 ± 1.5‡ | 52.7 ± 2.7†# |
| [Hb]a (g dL−1) | 15.0 ± 0.4 | 15.1 ± 0.4 | 15.0 ± 0.4 | 14.9 ± 0.4 | 14.9 ± 0.4 |
| (%) | 95.2 ± 0.1 | 95.7 ± 0.2 | 95.7 ± 0.3 | 81.8 ± 1.4†‡ | 81.6 ± 1.4†‡ |
| (%) | 82.9 ± 3.1 | 46.4 ± 2.5† | 78.7 ± 1.6 | 71.9 ± 1.1† | 35.3 ± 2.3†# |
| (mmHg) | 80.0 ± 1.0 | 83.2 ± 1.6 | 83.3 ± 2.2 | 46.5 ± 1.6†‡ | 47.8 ± 2.0†‡ |
| PvO2 (mmHg) | 53.2 ± 5.2 | 29.3 ± 0.8† | 44.7 ± 2.2 | 38.6 ± 1.0 | 23.9 ± 0.5†# |
| (mL L−1) | 199 ± 5 | 200 ± 5 | 200 ± 6 | 167 ± 5†‡ | 168 ± 7†‡ |
| (mL L−1) | 167 ± 7 | 95 ± 7† | 163 ± 7 | 148 ± 5‡ | 72 ± 6†‡# |
| Extraction (%) | 17 ± 3 | 53 ± 3† | 20 ± 2 | 13 ± 2‡ | 58 ± 3†# |
| NO, PG, Na+/K+‐ATPase, KIR channel block | |||||
| pHa | 7.43 ± 0.01* | 7.42 ± 0.01* | 7.42 ± 0.01 | 7.43 ± 0.01 | 7.42 ± 0.01 |
| pHv | 7.36 ± 0.01* | 7.28 ± 0.02† | 7.36 ± 0.01* | 7.36 ± 0.01*‡ | 7.29 ± 0.02†# |
| (mmHg) | 33.1 ± 1.7* | 32.6 ± 1.2* | 35.1 ± 1.0 | 34.3 ± 1.1 | 34.9 ± 1.1 |
| (mmHg) | 40.6 ± 1.2 | 58.1 ± 5.0† | 41.1 ± 2.0 | 41.8 ± 1.5‡ | 55.7 ± 3.9†# |
| [Hb]a (g dL−1) | 15.0 ± 0.4 | 14.8 ± 0.4* | 15.0 ± 0.4 | 14.8 ± 0.4† | 14.8 ± 0.4 |
| (%) | 95.8 ± 0.3 | 96.4 ± 0.2 | 95.7 ± 0.3 | 82.6 ± 1.3*†‡ | 82.8 ± 1.1*†‡ |
| (%) | 65.6 ± 3.9* | 33.6 ± 2.7*† | 65.7 ± 8.7 | 46.7 ± 4.5*† | 26.1 ± 3.0*†# |
| (mmHg) | 83.8 ± 2.7* | 87.3 ± 1.1* | 82.8 ± 2.0 | 47.0 ± 1.6†‡ | 47.8 ± 1.4†‡ |
| (mmHg) | 36.7 ± 2.3 | 21.4 ± 1.6† | 39.3 ± 5.1 | 26.7 ± 1.6† | 18.3 ± 1.3† |
| (mL L−1) | 200 ± 5 | 201 ± 5 | 198 ± 6 | 168 ± 6†‡ | 170 ± 6†‡ |
| (mL L−1) | 126 ± 9* | 60 ± 9*† | 124 ± 17* | 84 ± 8*†‡ | 47 ± 8*†# |
| Extraction (%) | 38 ± 4* | 71 ± 4*† | 37 ± 9* | 50 ± 5*†‡ | 73 ± 4*†# |
* P < 0.05 vs. control; † P < 0.05 vs. rest (within trial); ‡ P < 0.05 vs. normoxic exercise; # P < 0.05 vs. hypoxia; all comparisons within protocol.
Table 4.
Protocol 2 blood gases
| Rest 1 | Normoxic exercise | Rest 2 | Steady‐state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| pHa | 7.45 ± 0.01 | 7.44 ± 0.01 | 7.44 ± 0.01 | 7.45 ± 0.01 | 7.43 ± 0.01 |
| (mmHg) | 31.2 ± 1.5 | 32.0 ± 1.7 | 32.7 ± 1.0 | 32.1 ± 1.1 | 32.0 ± 1 |
| [Hb]a (g dL−1) | 14.7 ± 0.4 | 14.7 ± 0.4 | 14.5 ± 0.4 | 14.5 ± 0.4 | 14.4 ± 0.3 |
| (%) | 96.0 ± 0.5 | 96.3 ± 0.3 | 96.1 ± 0.3 | 79.6 ± 0.8†‡ | 81.6 ± 0.7†‡ |
| (mmHg) | 85.9 ± 3.0 | 88.4 ± 2.8 | 84.5 ± 1.5 | 43.1 ± 0.7†‡ | 46.0 ± 0.6†‡ |
| (mL L−1) | 197 ± 5 | 197 ± 5 | 195 ± 5 | 159 ± 5†‡ | 164 ± 5†‡ |
| Na+/K+‐ATPase, KIR channel block | |||||
| pHa | 7.45 ± 0.01 | 7.44 ± 0.01 | 7.44 ± 0.01 | 7.45 ± 0.01 | 7.44 ± 0.01 |
| (mmHg) | 29.7 ± 1.0* | 29.2 ± 1.1* | 31.7 ± 1.1 | 30.0 ± 1.2* | 31.2 ± 1.1 |
| [Hb]a (g dL−1) | 14.9 ± 0.4 | 14.4 ± 0.4† | 14.9 ± 0.4* | 14.8 ± 0.4 | 15.0 ± 0.4* |
| (%) | 96.4 ± 0.2 | 96.2 ± 0.2 | 96.1 ± 0.2 | 80.5 ± 0.9†‡ | 80.4 ± 0.7†‡ |
| (mmHg) | 87.0 ± 2.1 | 86.9 ± 2.1 | 84.3 ± 1.7 | 43.9 ± 0.8†‡ | 44.4 ± 0.9†‡ |
| (mL L−1) | 201 ± 6 | 194 ± 6 | 200 ± 6 | 165 ± 6†‡ | 168 ± 5†‡ |
* P < 0.05 vs. control; † P < 0.05 vs. rest (within trial); ‡ P < 0.05 vs. normoxic exercise; all comparisons within protocol.
In Protocol 1, oxygen delivery was significantly reduced at all time points under the blocked condition compared to control (P < 0.05) (Fig. 6 A) primarily as a result of the decrease in FBF because was unaffected by the blockade (Table 3). Oxygen extraction was significantly greater during the hypoxic vs. normoxic exercise trial under the control condition and was also greater at all time points under the blocked condition compared to control (P < 0.05) (Fig. 6 B). Forearm oxygen consumption () was not significantly different at any time point under blocked vs. control conditions (P = 0.08–0.86) (Fig. 6 C).
Figure 6. Forearm oxygen delivery, extraction and consumption before and after combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels.
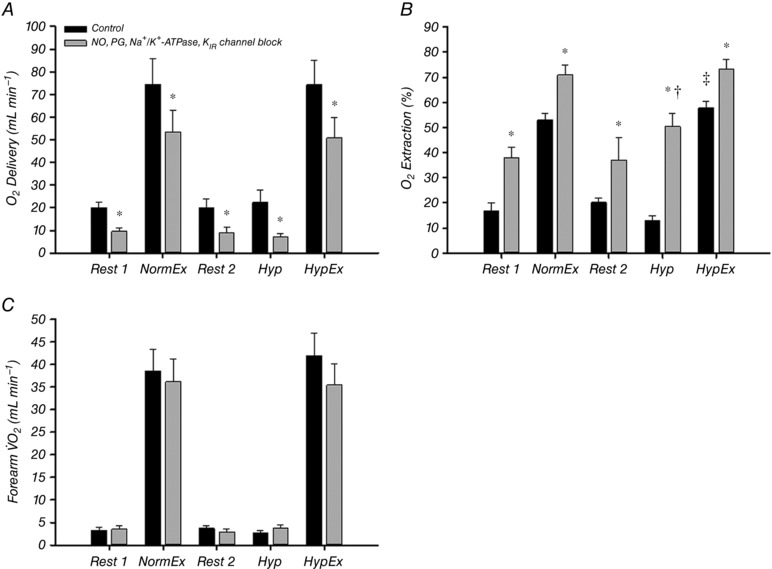
Combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels significantly attenuated oxygen delivery in all trials (A). Subsequently, oxygen extraction was significantly increased after blockade in all trials (B) and thus oxygen consumption (forearm ) was maintained after blockade in all trials (C). All values for NormEx and HypEx under both conditions are significantly greater than rest and hypoxia (P < 0.05 within condition). * P < 0.05 vs. control condition; † P < 0.05 vs. rest (within trial and condition); ‡ P < 0.05 vs. normoxic exercise (within condition).
Discussion
The present study provides several primary new findings. First, under conditions of forearm sympathoadrenal blockade to isolate local vasodilatory mechanisms, combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels blunts the augmented skeletal muscle hyperaemia during hypoxic exercise ∼50% (Protocol 1) (Figs 2 A and 4). These data are almost identical to our recent findings showing that inhibition of NO and PGs reduces the response to a similar extent (Crecelius et al. 2011b). As a result of concerns regarding redundant pathways masking an effect of inhibiting Na+/K+‐ATPase and KIR channels during concomitant NO and PG inhibition, we performed additional experiments with Na+/K+‐ATPase and KIR channel inhibition alone (Protocol 2). Thus, our second primary finding is that activation of Na+/K+‐ATPase and KIR channels does not contribute to the augmented skeletal muscle hyperaemia during hypoxic exercise (Figs 2 B and 4) and thus FBF remains elevated compared to normoxic exercise. Third, despite the lack of contribution of Na+/K+‐ATPase and KIR channel activation to hypoxic augmentation during exercise, these hyperpolarizing pathways are an essential component of the hyperaemic response to hypoxia at rest (Fig. 3). Finally, the decline in blood flow and therefore oxygen delivery in all experimental trials following combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels does not significantly impact oxygen consumption as a result of significant increases in oxygen extraction (Fig. 6).
Na+/K+‐ATPase and KIR channels in vasodilatation and hyperaemia during hypoxia at rest
Under similar experimental conditions, we have demonstrated previously that combined blockade of NO and PGs decreases resting forearm blood flow and abolishes the vasodilator response to hypoxia in quiescent skeletal muscle (Markwald et al. 2011; Crecelius et al. 2011b). Consistent with this, combined NO, PG, Na+/K+‐ATPase and KIR channel blockade in Protocol 1 of the present study also abolished the response. Interestingly, blockade of only Na+/K+‐ATPase and KIR channels in Protocol 2 both decreased resting blood flow and abolished the hyperaemic response to hypoxia in quiescent skeletal muscle as well (Fig. 3). Although determining the exact mechanisms by which these disparate pathways contribute to hypoxic vasodilatation at rest is beyond the scope of the present study, this merits a brief discussion. First, any effect of our pharmacological inhibitors is not simply the result of reducing forearm blood flow at rest (i.e. vasoconstriction) because studies have shown that infusion of phenylephrine (α1‐adrenergic agonist) to reduce blood flow prior to systemic hypoxia does not reduce the dilatory response (Blitzer et al. 1996). Second, this could suggest that NO and PGs and Na+/K+‐ATPase and KIR channels operate in a redundant fashion. However, if these pathways were operating in a redundant manner, we would expect that NO/PG inhibition alone, or Na+/K+‐ATPase and KIR channel inhibition alone, would not impact hypoxic vasodilatation and that the response would be abolished only when all four pathways are inhibited (Markwald et al. 2011; Lamb & Murrant, 2015). Finally, it is possible that multiple pathways are involved in the regulation of vascular tone during hypoxia and perhaps, depending on the study, inhibition of a given pathway may significantly reduce or almost abolish the response. In this context, adenosine receptor antagonism (∼85%: Leuenberger et al. 1999), NO inhibition alone (∼60%: Blitzer et al. 1996; ∼75%: Casey et al. 2010) or in combination with PG inhibition (∼85–90%: Markwald et al. 2011; ∼90%: Crecelius et al. 2011b) and cytochrome P‐450 inhibition (∼50%: Spilk et al. 2013), have all been demonstrated to significantly reduce forearm vasodilatation during hypoxia. We propose that during systemic hypoxia when is reduced to ∼80%, inhibiting the normal vasodilator and hyperaemic response (typically by the order of ∼20–30%) results in a fairly small absolute change in forearm blood flow (Fig. 2) and oxygen delivery (Fig. 6 A). Given that blood flow and oxygen demand in resting skeletal muscle is low, pharmacological inhibition of a dilatory signal or pathway may not evoke much of an ‘error signal’ to initiate a redundant response.
Na+/K+‐ATPase and KIR channels in vasodilatation and hyperaemia during hypoxic exercise
The primary purpose of the present study was to determine whether Na+/K+‐ATPase and KIR channels are involved in the augmented hyperaemia during hypoxic exercise in humans. In Protocol 1, we combined Na+/K+‐ATPase and KIR channel inhibition with the same NO/PG blockade approach that decreased the compensatory increase in blood flow during hypoxic exercise by ∼50% in our previous study (Crecelius et al. 2011b). Although this approach significantly reduced steady‐state hyperaemia during exercise in normoxia and hypoxia (Fig. 2), which is most probably primarily a result of KIR channel inhibition (Crecelius et al. 2014), it did not result in a further decrease in the augmented hyperaemia during hypoxic exercise beyond ∼50% (Fig. 4). Given that exercise hyperaemia is regulated by many redundant factors (Joyner & Wilkins, 2007) and to test that this lack of an additional effect was not influenced by NO/PG inhibition, we repeated the experiment with combined ouabain and BaCl2 infusion alone (Protocol 2). Consistent with our observations in Protocol 1, inhibition of Na+/K+‐ATPase and KIR channels alone significantly reduced exercise hyperaemia in normoxia and hypoxia by ∼25%, although it did not impact the augmented hyperaemia during hypoxic exercise (Figs 2 and 4). Collectively, these data indicate that activation of Na+/K+‐ATPase and KIR channels does not contribute to the augmented skeletal muscle hyperaemia during hypoxic exercise in humans (Fig. 4).
Possible mechanisms of the unexplained augmented exercise hyperaemia during hypoxia
Our data clearly indicate that distinct mechanisms beyond the synthesis of the endothelial autocoids NO and PGs and activation of Na+/K+‐ATPase and KIR channels must be involved in the augmented hyperaemic response during hypoxic exercise. Although there are many potential explanations, it is of interest that skeletal muscle blood flow responses are more closely associated with reductions in than (Roach et al. 1999; González‐Alonso et al. 2001). Given that changes in are directly related to reductions in the oxygenation of haemoglobin, RBCs may be implicated as a potential sensor of the hypoxic stimulus and modulator of vascular tone, blood flow and oxygen delivery through the release of vasodilatory molecules such as ATP (González‐Alonso et al. 2002; Ellsworth et al. 2009, 2016; Dufour et al. 2010; Ellsworth & Sprague, 2012; Dinenno, 2016). Circulating ATP can then bind to purinergic P2Y receptors on the endothelium and evoke vasodilatation, in part through the activation of small‐ and intermediate‐conductance calcium‐activated potassium channels (SKCa and IKCa, respectively) (Winter & Dora, 2007) and subsequent hyperpolarization that conducts along and between the endothelium and vascular smooth muscle through gap junctions (Bagher & Segal, 2011; Garland et al. 2011; Straub et al. 2014; Dora, 2017). Unfortunately, we were unable to inhibit P2Y receptors, SKCa/IKCa channels or gap junctions in the present study as a result of specific inhibitors being unavailable for use in humans. As such, the vasodilatory response to intravascular ATP that persists following combined inhibition of NO, PGs, Na+/K+‐ATPase and KIR channels (∼40% based on Hearon et al. 2017) may have contributed to the remaining augmented hyperaemia during hypoxic exercise. RBCs can also act as an endothelium‐independent source of NO through the formation of S‐nitrosohemoglobin in the lung (Jia et al. 1996; Stamler et al. 1997), hypoxia‐induced increases in the conversion of nitrite to NO via the nitrite reductase activity of deoxyhaemoglobin, as well as synthesis via RBC eNOS (Kleinbongard et al. 2006; Halpin & Spence, 2010; Cortese‐Krott et al. 2012; Wood et al. 2013), all of which would be unaffected by NOS inhibition in the present study.
Beyond the RBC, other pathways could potentially be involved in the remaining hypoxic response during exercise. For example, adenosine has been shown to contribute to the vasodilatory response to hypoxia alone in both humans and experimental animals (Bryan & Marshall, 1999; Leuenberger et al. 1999; Ray et al. 2002), as well as to exercise hyperaemia (Marshall, 2007). However, an obligatory role of adenosine in the augmented hyperaemic response to hypoxic exercise in humans has not been demonstrated (Casey et al. 2009; Heinonen et al. 2010). It is important to note that these findings may be influenced by experimental limitations associated with the use of adenosine receptor antagonists such as aminophylline in humans, which include non‐selective targeting of multiple adenosine receptor subtypes, incomplete inhibition of adenosine‐mediated dilatation (∼50% inhibition of the vasodilatory response to intra‐brachial adenosine infusion (Kirby et al. 2010)) and non‐selective inhibition of phosphodiesterases, which can increase resting muscle blood flow (Rådegran & Hellsten, 2000; Casey et al. 2009; Lomas & Zaccolo, 2014; Dinenno, 2016). Furthermore, given the reduction in steady‐state exercise hyperaemia with Na+/K+‐ATPase and KIR channel inhibition, it is possible that adenosine could play a role under these conditions of reduced tissue perfusion.
Cytochrome P450 metabolites of arachidonic acid are another group of vasoactive molecules that could contribute to the augmented hyperaemic response to hypoxic exercise. These metabolites are comprised of two groups, hydroxyeicosatetraenoic acids (HETEs) and epoxyeicosatrienoic acids (EETs), with HETEs generally evoking vasoconstriction through inhibition of KCa channels and activation of PKC in the vascular smooth muscle and EETs generally evoking vasodilatation through activation of KCa channels (Clifford & Hellsten, 2004; Miyata & Roman, 2005; Félétou & Vanhoutte, 2009; Spector, 2009). 20‐HETE is the primary vasoconstricting HETE that has been studied in the context of vascular control, whereas there are multiple EET isoforms and it is less clear which of these contribute to regulation of vascular tone (Félétou & Vanhoutte, 2009; Spector, 2009). The production and release of 20‐HETE is oxygen‐dependent, such that declines in decrease [20‐HETE]; thus, the contribution of 20‐HETE to hypoxic vasodilatation has been proposed to result from less vasoconstrictor signalling through this pathway (Frisbee et al. 2001, 2002; Jackson, 2016). By contrast, an increase in EETs in hypoxia would contribute to vasodilatation and, although studies in isolated rat arteries suggest that EETs are not involved in hypoxic vasodilatation (Frisbee et al. 2001, 2002), Spilk et al. (2013) found that regional infusion of fluconazole (an inhibitor of cytochrome P450 2C9) in the human forearm significantly attenuates hypoxia‐induced dilatation. There is also evidence in humans that EETs can compensate for the loss of NO at rest and during exercise in humans, thus indicating potential cross‐talk between these two pathways (Hillig et al. 2003; Ozkor & Quyyumi, 2011; Ozkor et al. 2011). Additionally, given that both vasodilating PGs and EETs are derived from the metabolism of arachidonic acid, it is important to consider that COX inhibition (as in Protocol 1 of the present study) could conceivably shunt arachidonic acid metabolism through the cytochrome P450 pathway, thereby resulting in a compensatory increase in EETs. Thus, it is possible that reduced 20‐HETE and/or increased EET production contributed to the remaining augmented exercise hyperaemia during hypoxia in the present study.
Finally, activation of other K+ channels could be involved in the response. In isolated vessels and in situ preparations, ATP‐sensitive K+ (KATP) channels have been shown to be involved in the vasodilatory response to hypoxia (Gasser et al. 1993; Marshall et al. 1993; Frisbee et al. 2002; Ngo et al. 2010), although other studies have shown they are not involved (Smani et al. 2002; Lynch et al. 2006). Similarly, studies in experimental animals suggest that KATP channels mediate arteriolar vasodilatation during muscle contractions, (Murrant & Sarelius, 2002; Dua et al. 2009), yet studies in humans do not support this (Keller et al. 2004; Schrage et al. 2006). KCa channels have also been shown to be activated by hypoxia (Gebremedhin et al. 1994; Frisbee et al. 2002) and can be stimulated by factors that were not blocked in the present study including EETs and ATP (Gebremedhin et al. 1992; Campbell et al. 1996; Zou et al. 1996; Eckman et al. 1998; Hayabuchi et al. 1998; Miura & Gutterman, 1998; Fisslthaler et al. 1999; Earley et al. 2005; Huang et al. 2005; Weston et al. 2005; Winter & Dora, 2007). However, confirmation of these findings is not possible as a result of the lack of specific inhibitors available for use in humans. Future studies will be needed to determine whether these channels or other mechanisms contribute significantly to the augmented hyperaemic response during hypoxic exercise.
Experimental considerations and limitations
A key issue for studies on blood flow and vascular control in response to pharmacological or physiological perturbations is the proper quantification of the vasomotor responses. With respect to the research question addressed in the present study, the augmented hyperaemia observed during hypoxic exercise occurs as a means to maintain oxygen delivery for a given oxygen demand. Many studies have demonstrated this, including our own (Koskolou et al. 1997; Roach et al. 1999; González‐Alonso et al. 2001; Crecelius et al. 2011b; Casey & Joyner, 2012) (Fig. 6 A). Thus, when attempting to understand the signalling mechanisms underlying this response, we consider that quantifying the ‘hypoxic augmentation of exercise forearm blood flow’ is most appropriate because this difference in blood flow is what drives oxygen delivery to be maintained during systemic hypoxia when arterial oxygen content is reduced. In an effort to be comprehensive, we have also presented the data as a change from the respective baseline condition from both our previous study on this topic (Crecelius et al. 2011b) and the present study (Fig. 5). When this analysis is performed, the collective data indicate that (i) NO and PGs do not play a role in the change in forearm blood flow from rest to exercise in either normoxia or hypoxia (Fig. 5 A) (Crecelius et al. 2011b); (ii) Na+/K+‐ATPase and KIR channels contribute significantly to this change from rest to exercise in both normoxia and hypoxia (Fig. 5 B and C); and (iii) additional vasodilator mechanisms are involved in this change from rest to exercise in hypoxia (Fig. 5 B and C).
Given the specific research question of the present study as it relates to blood flow and oxygen delivery during hypoxic exercise, we do not consider that quantifying the change in blood flow from rest to exercise (within a given normoxia/hypoxia or drug condition) is most reflective of the underlying physiology. For example, despite the appearance of a ‘normal’ blood flow response from rest to exercise in our prior study with combined NO/PG inhibition (Fig. 5 A), muscle blood flow and oxygen delivery (Crecelius et al. 2011b) were clearly reduced during hypoxic exercise compared with normoxic exercise by inhibiting these autocoids, strongly implicating their role in blood flow regulation under hypoxic conditions. In the present study, inhibition of Na+/K+‐ATPase and KIR channels (in both protocols) reduced steady‐state blood flow during both normoxic and hypoxic exercise (Fig. 2). However, blood flow during hypoxic exercise was greater than normoxic exercise within the blocked condition and, as such, the ‘hypoxic augmentation’ was largely unaffected (Fig. 4). Thus, we consider this to indicate that Na+/K+‐ATPase and KIR channels are not involved in the augmented hyperaemia during hypoxic exercise in humans. However, in an effort to be comprehensive, we present these data because there is interest in this means of data expression by investigators in the field.
One potential limitation of the present study is that we did not test the individual effects of ouabain and BaCl2 and are therefore unable to determine the respective contributions of Na+/K+‐ATPase and KIR channels to the hyperaemic response to hypoxia at rest. However, in previous studies, we have found little role for Na+/K+‐ATPase activation in the vascular responses to intra‐arterial ATP infusion or exercise, whereas activation of KIR channels does contribute significantly (Crecelius et al. 2012, 2014). Thus, it is possible that KIR channels were the primary contributor to the hypoxic vasodilatory response at rest. We also did not directly test the efficacy of the pharmacological inhibitors used in the present study, although our laboratory and others have demonstrated these doses are effective in humans (see Methods). Furthermore, in the present study, the combined infusion of ouabain and BaCl2 significantly attenuated blood flow at rest and during exercise in both normoxia and hypoxia in both protocols (Figs 2 and 3). Despite this effect, the augmented hyperaemia during hypoxic exercise was not different from NO/PG block alone in Protocol 1 and unaffected in Protocol 2 (Fig. 4).
Finally, the addition of ouabain and BaCl2 in both protocols significantly increased MAP (∼6–10 mmHg) and decreased FBF under all experimental conditions (Fig. 2 and Tables 1 and 2); in contrast, the combined NO/PG blockade from our previous investigation had no effect on MAP and only decreased FBF at rest and during hypoxic exercise (Crecelius et al. 2011b). Despite these differences, this probably does not influence our primary conclusion regarding the role of the Na+/K+‐ATPase and KIR channels in hypoxic augmentation of blood flow during exercise for two key reasons. First, all experiments were performed during local sympathoadrenal blockade, which would minimize any impact of baroreflex‐mediated changes in sympathetic outflow on forearm vascular tone. Second, the blockade‐induced change in MAP and FBF during exercise was the same between protocols and between normoxic/hypoxic conditions within each protocol; however, the effect on forearm haemodynamics during hypoxic exercise was different. Specifically, hypoxic augmentation of exercise hyperaemia was blunted only when NO and PGs were included in the blockade conditions, which supports our previous findings (Crecelius et al. 2011b).
Conclusions
Skeletal muscle blood flow is augmented during hypoxia to maintain oxygen delivery to the tissue. In combination with our previous work (Markwald et al. 2011; Crecelius et al. 2011b), the results from the present study indicate that combined inhibition of NO and PGs, or inhibition of Na+/K+‐ATPase and KIR channels, abolishes the hyperaemic response to systemic isocapnic hypoxia at rest. How these pathways are interrelated under these conditions is presently unknown. By contrast, NO and PGs appear to mediate ∼50% of the augmented hyperaemia during hypoxic exercise, whereas the present study provides the first experimental evidence that Na+/K+‐ATPase and KIR channels do not contribute to this response. The factors or signalling pathways underlying the remaining augmented hyperaemia during hypoxic exercise remain to be determined.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the Human Cardiovascular Physiology Laboratory, Colorado State University, Fort Collins, CO, USA. MLR and FAD contributed to the conception and design of the experiments, collection, analysis and interpretation of the data, as well as the writing of the manuscript. ARC contributed to the conception and design of the experiments, collection, analysis and interpretation of the data, and provided critical revision of the manuscript. GJL and DGL contributed to the experimental design, provided invasive methodology for data collection, and contributed to the critical revision of the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This research was supported by the National Institutes of Health award HL095573 (FAD).
Acknowledgements
We thank the subjects who volunteered to participate, as well as Jennifer Richards, Hannah Scott and Devin V. Dinenno for their assistance with conducting these studies and the preparation of the manuscript. Anne Crecelius is currently an Assistant Professor in the Department of Health and Sport Science at the University of Dayton.
Biography
Matthew L. Racine received his undergraduate degree from the University of Idaho (2009) and his Master's degree from the University of Colorado Boulder (2012). He is presently a PhD candidate and Predoctoral Fellow training under Dr Frank Dinenno in the Human Cardiovascular Physiology Laboratory at Colorado State University. His research focuses on the mechanisms of skeletal muscle blood flow regulation during physiological stressors such as exercise and hypoxia in young and older adults, with more recent work on the contribution of red blood cells to this process through the release of ATP.

Edited by: Harold Schultz & Giovanni Mann
References
- Ahn SJ, Fancher IS, Bian J, Zhang CX, Schwab S, Gaffin R, Phillips SA & Levitan I (2017). Inwardly‐rectifying K+ channels are major contributors to flow‐induced vasodilation in resistance arteries. J Physiol 595, 2339–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz Q, Li Y, Anderson N, Ojake L, Tsisanova E & Tinker A (2017). Molecular and functional characterization of the endothelial ATP‐sensitive potassium channel. J Biol Chem 292, 17587–17597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P & Segal S (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol 202, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzett RB, Garcia RT & Moosavi SH (2000). Simple contrivance ‘clamps’ end‐tidal PCO(2) and PO(2) despite rapid changes in ventilation. J Appl Physiol 88, 1597–1600. [DOI] [PubMed] [Google Scholar]
- Blitzer ML, Lee SD & Creager MA (1996). Endothelium‐derived nitric oxide mediates hypoxic vasodilation of resistance vessels in humans. Am J Physiol Heart Circ Physiol 271, H1182–H1185. [DOI] [PubMed] [Google Scholar]
- Bryan PT & Marshall JM (1999). Adenosine receptor subtypes and vasodilatation in rat skeletal muscle during systemic hypoxia: a role for A1 receptors. J Physiol 514, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF & Harder DR (1996). Identification of epoxyeicosatrienoic acid as endothelium‐derived hyperpolarizing factors. Circ Res 78, 415–423. [DOI] [PubMed] [Google Scholar]
- Casey DP, Curry TB, Wilkins BW & Joyner MJ (2011). Nitric oxide‐mediated vasodilation becomes independent of beta‐adrenergic receptor activation with increased intensity of hypoxic exercise. J Appl Physiol 110, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP & Joyner MJ (2012). Compensatory vasodilatation during hypoxic exercise: mechanisms responsible for matching oxygen supply to demand. J Physiol 24, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Curry TB, Eisenach JH, Wilkins BW & Joyner MJ (2010). Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J Physiol 588, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Pike TL, Eisenach JH, Dietz NM, Joyner MJ & Wilkins BW (2009). Adenosine receptor antagonist and augmented vasodilation during hypoxic exercise. J Appl Physiol 107, 1128–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS & Hellsten Y (2004). Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97, 393–403. [DOI] [PubMed] [Google Scholar]
- Cortese‐Krott MM, Rodriguez‐Mateos A, Sansone R, Kuhnle GGC, Thasian‐Sivarajah S, Krenz T, Horn P, Krisp C, Wolters D, Heiß C, Kröncke K‐D, Hogg N, Feelisch M & Kelm M (2012). Human red blood cells at work: identification and visualization of erythrocytic eNOS activity in health and disease. Blood 120, 4229–4237. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Hearon CM Jr, Luckasen GJ, Larson DG & Dinenno FA (2015). Contracting human skeletal muscle maintains the ability to blunt α1‐adrenergic vasoconstriction during KIR channel and Na+/K+‐ATPase inhibition. J Physiol 593, 2735–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2012). ATP‐mediated vasodilatation occurs via activation of inwardly rectifying potassium channels in humans. J Physiol 590, 5349–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2013a). Mechanisms of rapid vasodilation following a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305, H29–H40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Richards JC, Garcia LJ, Voyles WF, Larson DG, Luckasen GJ & Dinenno FA (2011a). Mechanisms of ATP‐mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol 301, H1302–H1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Voyles WF & Dinenno FA (2011b). Augmented skeletal muscle hyperaemia during hypoxic exercise in humans is blunted by combined inhibition of nitric oxide and vasodilating prostaglandins. J Physiol 589, 3671–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2014). KIR channel activation contributes to onset and steady‐state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol 307, H782–H791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Richards JC, Luckasen GJ, Larson DG & Dinenno FA (2013b). Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+‐ATPase in humans. Circ Res 113, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz NM, Halliwill JR & Joyner MJ (1994). Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol 480, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz NM, Halliwill JR, Spielmann JM, Lawler LA, Papouchado BG, Eickhoff TJ & Joyner MJ (1997). Sympathetic withdrawal and forearm vasodilation during vasovagal syncope in humans. J Appl Physiol 82, 1785–1793. [DOI] [PubMed] [Google Scholar]
- Dinenno FA (2016). Skeletal muscle vasodilation during systemic hypoxia in humans. J Appl Physiol 120, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA & Joyner MJ (2003). Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol 553, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA & Joyner MJ (2004). Combined NO and PG inhibition augments alpha‐adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Hear Circ Physiol 287, H2576–H2584. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ & Halliwill JR (2003). Failure of systemic hypoxia to blunt alpha‐adrenergic vasoconstriction in the human forearm. J Physiol 549, 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA (2017). Conducted dilatation to ATP and K+ and in rat skeletal muscle arterioles. Acta Physiol 219, 202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua A, Dua N & Murrant C (2009). Skeletal muscle contraction‐induced vasodilator complement production is dependent on stimulus and contraction frequency. Am J Physiol Heart Circ Physiol 297, H433–H442. [DOI] [PubMed] [Google Scholar]
- Dufour SP, Patel RP, Brandon A, Teng X, Pearson J, Barker H, Ali L, Yuen AHY, Smolenski RT & González‐Alonso J (2010). Erythrocyte‐dependent regulation of human skeletal muscle blood flow: role of varied oxyhemoglobin and exercise on nitrite, S‐nitrosohemoglobin, and ATP. Am J Physiol 299, H1936–H1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT & Brayden JE (2005). TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97, 1270–1279. [DOI] [PubMed] [Google Scholar]
- Eckman DM, Hopkins N, McBride C & Keef KD (1998). Endothelium‐dependent relaxation and hyperpolarization in guinea‐pig coronary artery: Role of epoxyeicosatrienoic acid. Br J Pharmacol 124, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Eisenach J, Clark E, Charkoudian N, Dinenno F, Atkinson J, Fealey R, Dietz N & Joyner M (2002). Effects of chronic sympathectomy on vascular function in the human forearm. J Appl Physiol 92, 2019–2025. [DOI] [PubMed] [Google Scholar]
- Eklund B & Kaijser L (1976). Effect of regional alpha‐ and beta‐adrenergic blockade on blood flow in the resting forearm during contralateral isometric handgrip. J Physiol 262, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH & Sprague RS (2009). Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology 24, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG & Sprague RS (2016). Role of erythrocyte‐released ATP in the regulation of microvascular oxygen supply in skeletal muscle. Acta Physiol 216, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML & Sprague RS (2012). Regulation of blood flow distribution in skeletal muscle: role of erythrocyte‐released ATP. J Physiol 590, 4985–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M & Vanhoutte P (2009). EDHF: an update. Clin Sci 117, 139–155. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I & Busse R (1999). Cytochrome P4502C is an EDHF synthase in coronary arteries. Nature 401, 493–497. [DOI] [PubMed] [Google Scholar]
- Frisbee J, Maier K, Falck J, Roman R & Lombard J (2002). Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 283, R309–R319. [DOI] [PubMed] [Google Scholar]
- Frisbee J, Roman R, Krishna U, Falck J & Lombard J (2001). Relative contributions of cyclooxygenase‐ and cytochrome P450 omega‐hydroxylase‐dependent pathways to hypoxic dilation of skeletal muscle resistance arteries. J Vasc Res 38, 305–314. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: Spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser R, Klein W & Kickenweiz E (1993). Vasodilative response to hypoxia and simulated ischemia is mediated by ATP‐sensitive K+ channels in guinea pig thoracic aorta. Angiology 44, 228–243. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH & Harder DR (1994). Hypoxia increases the activity of Ca(2+)‐sensitive K+ channels in cat cerebral arterial muscle cell membranes. Pflügers Arch Eur J Physiol 428, 621–630. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M & Harder DR (1992). Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol Heart Circ Physiol 263, H519–H525. [DOI] [PubMed] [Google Scholar]
- van Ginneken E, Meijer P, Verkaik N, Smits P & Rongen G (2004). ATP‐induced vasodilation in human skeletal muscle. Br J Pharmacol 141, 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Alonso J, Olsen DB & Saltin B (2002). Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91, 1046–1055. [DOI] [PubMed] [Google Scholar]
- González‐Alonso J, Richardson RS & Saltin B (2001). Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol 530, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwill JR, Lawler LA, Eickhoff TJ, Dietz NM, Nauss LA & Joyner MJ (1997). Forearm sympathetic withdrawal and vasodilatation during mental stress in humans. J Physiol 504, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin ST & Spence DM (2010). Direct plate‐reader measurement of nitric oxide released from hypoxic erythrocytes flowing through a microfluidic device. Anal Chem 82, 7492–7497. [DOI] [PubMed] [Google Scholar]
- Hanada A, Sander M & González‐Alonso J (2003). Human skeletal muscle sympathetic nerve activity, heart rate and limb haemodynamics with reduced blood oxygenation and exercise. J Physiol 551, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley L, Vogel J & Landowne M (1973). Central, femoral, and brachial circulation during exercise in hypoxia. J Appl Physiol 34, 87–90. [DOI] [PubMed] [Google Scholar]
- Hayabuchi Y, Nakaya Y, Matsuoka S & Kuroda Y (1998). Endothelium‐derived hyperpolarizing factor activates Ca2+‐activated K+ channels in porcine coronary artery smooth muscle cells. J Cardiovasc Pharmacol 32, 642–649. [DOI] [PubMed] [Google Scholar]
- Hearon CM Jr, Richards JC, Racine ML, Luckasen GJ, Larson DG, Joyner MJ & FA Dinenno (2017). Sympatholytic effect of intravascular ATP is independent of nitric oxide, prostaglandins, Na+/K+‐ATPase and KIR channels in humans. J Physiol 15, 5175–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen IH, Kemppainen J, Kaskinoro K, Peltonen JE, Borra R, Lindroos M, Oikonen V, Nuutila P, Knuuti J, Boushel R & Kalliokoski KK (2010). Regulation of human skeletal muscle perfusion and its heterogeneity during exercise in moderate hypoxia. Am J Physiol Regul Integr Comp Physiol 299, R72–R79. [DOI] [PubMed] [Google Scholar]
- Hillig T, Krustrup P, Fleming I, Osada T, Saltin B & Hellsten Y (2003). Cytochrome P450 2C9 plays an important role in the regulation of exercise‐induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol 546, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Sun D, Jacobson A, Carroll MA, Falck JR & Kaley G (2005). Epoxyeicosatrienoic acids are released to mediate shear stress‐dependent hyperpolarization of arteriolar smooth muscle. Circ Res 96, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WF (2016). Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action. J Physiol 594, 5055–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Bonaventura C, Bonaventura J & Stamler JS (1996). S‐nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380, 221–226. [DOI] [PubMed] [Google Scholar]
- Johnsson G (1967). The effects of intra‐arterially administered propranolol and H 56/28 on blood flow in the forearm – a comparative study of two β‐adrenergic receptor antagonists. Acta Pharmacol Toxicol 25, 63–74. [DOI] [PubMed] [Google Scholar]
- Joyner MJ & Casey DP (2014). Muscle blood flow, hypoxia and hypoperfusion. J Appl Physiol 116, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ & Wilkins BW (2007). Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol 583, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DM, Ogoh S, Greene S, Olivencia‐Yurvati A & Raven PB (2004). Inhibition of KATP channel activity augments baroreflex‐mediated vasoconstriction in exercising human skeletal muscle. J Physiol 561, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2010). Vasodilatory responsiveness to adenosine triphosphate in ageing humans. J Physiol 588, 4017–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2012). Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, Kumara I, Gharini P, Kabanova S, Ozüyaman B, Schnürch H‐G, Gödecke A, Weber A‐A, Robenek M, Robenek H, Bloch W, Rösen P & Kelm M (2006). Red blood cells express a functional endothelial nitric oxide synthase. Blood 107, 2943–2951. [DOI] [PubMed] [Google Scholar]
- Koskolou MD, Calbet JAL, Rådegran G & Roach RC (1997). Hypoxia and the cardiovascular response to dynamic knee‐extensor exercise. Am J Physiol Heart Circ Physiol 272, H2655–H2663. [DOI] [PubMed] [Google Scholar]
- Lamb IR & Murrant CL (2015). Potassium inhibits nitric oxide and adenosine arteriolar vasodilatation via KIR and Na+/K+ATPase: Implications for redundancy in active hyperaemia. J Physiol 593, 5111–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuenberger U, Gray K & Herr M (1999). Adenosine contributes to hypoxia‐induced forearm vasodilation in humans. J Appl Physiol 87, 2218–2224. [DOI] [PubMed] [Google Scholar]
- Lomas O & Zaccolo M (2014). Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology 29, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch FM, Austin C, Heagerty AM & Izzard AS (2006). Adenosine and hypoxic dilation of rat coronary small arteries: roles of the ATP‐sensitive potassium channel, endothelium, and nitric oxide. Am J Physiol Heart Circ Physiol 290, H1145–H1150. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Kirby BS, Crecelius AR, Carlson RE, Voyles WF & Dinenno FA (2011). Combined inhibition of nitric oxide and vasodilating prostaglandins abolishes forearm vasodilatation to systemic hypoxia in healthy humans. Am J Physiol Heart Circ Physiol 589, H1979–H1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM (2007). The roles of adenosine and related substances in exercise hyperaemia. J Physiol 583, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM, Thomas T & Turner L (1993). A link between adenosine, ATP‐sensitive K+ channels, potassium and muscle vasodilatation in the rat in systemic hypoxia. J Physiol 472, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H & Gutterman DD (1998). Human coronary arteriolar dilation to arachidonic acid. Circ Res 83, 501–507. [DOI] [PubMed] [Google Scholar]
- Miyata N & Roman RJ (2005). Role of 20‐hydroxyeicosatetraenoic acid (20‐HETE) in vascular system. J Smooth Muscle Res 41, 175–193. [DOI] [PubMed] [Google Scholar]
- Mo FM & Ballard HJ (2005). Acute systemic hypoxia elevates venous but not interstitial potassium of dog skeletal muscle. Am J Physiol Hear Circ Physiol 289, H1710–H1718. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, González‐Alonso J, Bune LT, Saltin B, Pilegaard H & Hellsten Y (2009). ATP‐induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol 296, R1140–R1148. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Thaning P, Nyberg M, Saltin B & Hellsten Y (2011). Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol 589, 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL & Sarelius IH (2002). Multiple dilator pathways in skeletal muscle contraction‐induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol 282, R969–R978. [DOI] [PubMed] [Google Scholar]
- Nelson MT & Quayle JM (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268, C799–C822. [DOI] [PubMed] [Google Scholar]
- Ngo AT, Jensen LJ, Riemann M, Holstein‐Rathlou N‐H & Torp‐Pedersen C (2010). Oxygen sensing and conducted vasomotor responses in mouse cremaster arterioles in situ. Pflugers Arch 460, 41–53. [DOI] [PubMed] [Google Scholar]
- Ozkor MA, Murrow JR, Rahman AM, Kavtaradze N, Lin J, Manatunga A & Quyyumi AA (2011). Endothelium‐derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation 123, 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkor MA & Quyyumi AA (2011). Endothelium‐derived hyperpolarizing factor and vascular function. Cardiol Res Pract 2011, 156146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W, Han J, Kim N, Ko J, Kim S & Earm Y (2005). Activation of inward rectifier K+ channels by hypoxia in rabbit coronary arterial smooth muscle cells. Am J Physiol Hear Circ Physiol 289, H2461–H2467. [DOI] [PubMed] [Google Scholar]
- Rådegran G & Hellsten Y (2000). Adenosine and nitric oxide in exercise‐induced human skeletal muscle vasodilation. Acta Physiol Scand 168, 575–591. [DOI] [PubMed] [Google Scholar]
- Ray CJ, Abbas MR, Coney AM & Marshall JM (2002). Interactions of adenosine, prostaglandins and nitric oxide in hypoxia‐induced vasodilatation: in vivo and in vitro studies. J Physiol 544, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JC, Crecelius AR, Larson DG, Luckasen GJ & Dinenno FA (2017). Impaired peripheral vasodilation during graded systemic hypoxia in healthy older adults: role of the sympathoadrenal system. Am J Physiol ‐ Hear Circ Physiol 312, H832–H841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JC, Luckasen GJ, Larson DG & Dinenno FA (2014). Role of α‐adrenergic vasoconstriction in regulating skeletal muscle blood flow and vascular conductance during forearm exercise in ageing humans. J Physiol 21, 4775–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JAL & Saltin B (1999). Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol 276, H438–H445. [DOI] [PubMed] [Google Scholar]
- Rongen GA, Smits P & Thien T (1994). Characterization of ATP‐induced vasodilation in the human forearm vascular bed. Circulation 90, 1891–1898. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Saltin B, Kiens B & Christensen NJ (1986). Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am J Physiol Heart Circ Physiol 251, H1038–H1044. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Dietz NM & Joyner MJ (2006). Effects of combined inhibition of ATP‐sensitive potassium channels, nitric oxide, and prostaglandins on hyperemia during moderate exercise. J Appl Physiol 100, 1506–1512. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner N Jr & Costin J (1971). Interactions between oxygen, potassium, and osmolality in regulation of skeletal muscle blood flow. Circ Res 28, 73–85. [PubMed] [Google Scholar]
- Smani T, Hernandez A, Urena J, Castellano AJ, Franco‐Obregon A, Ordonez A & Lopez‐Barneo J (2002). Reduction of Ca2+ channel activity by hypoxia in human and porcine coronary monocytes. Cardiovasc Res 53, 97–104. [DOI] [PubMed] [Google Scholar]
- Spector AA (2009). Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res 50(Suppl), S52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilk S, Herr MD, Sinoway LI & Leuenberger UA (2013). Endothelium‐derived hyperpolarizing factor contributes to hypoxia‐induced skeletal muscle vasodilation in humans. Am J Physiol Heart Circ Physiol 301, H1639–H1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K & Piantadosi CA (1997). Blood flow regulation by S‐nitrosohemoglobin in the physiological oxygen gradient. Science 276, 2034–2037. [DOI] [PubMed] [Google Scholar]
- Straub AC, Zeigler AC & Isakson BE (2014). The myoendothelial junction: connections that deliver the message. Physiology 29, 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P, Collier J & Moncada S (1989). Effects of endothelium‐derived nitric oxide on peripheral arteriolar tone in man. Lancet 334, 997–1000. [DOI] [PubMed] [Google Scholar]
- Victor RG & Seals DR (1989). Reflex stimulation of sympathetic outflow during rhythmic exercise in humans. Am J Physiol Heart Circ Physiol 257, H2017–H2024. [DOI] [PubMed] [Google Scholar]
- Weston AH, Félétou M, Vanhoutte PM, Falck JR, Campbell WB & Edwards G (2005). Bradykinin‐induced, endothelium‐dependent responses in porcine coronary arteries: Involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol 145, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Pike TL, Martin EA, Curry TB, Ceridon ML & Joyner MJ (2008). Exercise intensity‐dependent contribution of beta‐adrenergic receptor‐mediated vasodilatation in hypoxic humans. J Physiol 586, 1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Schrage WG, Liu Z, Hancock KC & Joyner MJ (2006). Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol 101, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter P & Dora KA (2007). Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol 582, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KC, Cortese‐Krott MM, Kovacic JC, Noguchi A, Liu VB, Wang X, Raghavachari N, Boehm M, Kato GJ, Kelm M & Gladwin MT (2013). Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb Vasc Biol 33, 1861–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR & Roman RJ (1996). Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K(+)‐channel activity. Am J Physiol Renal Physiol 270, F822–F832. [DOI] [PubMed] [Google Scholar]