

Abstract
Long interspersed nuclear element‐1 (LINE‐1 or L1) reactivation is linked to poor prognosis in non‐small‐cell lung carcinoma (NSCLC), but the molecular bases of this response remain largely unknown. In this report, we show that challenge of human bronchial epithelial cells (HBECs) with the lung carcinogen, benzo(a)pyrene (BaP), shifted the L1 promoter from a heterochromatic to euchromatic state through disassembly of the nucleosomal and remodeling deacetylase (NuRD) complex. Carcinogen challenge was also associated with partial displacement of constituent proteins from the nuclear to the cytoplasmic compartment. Disruption of NuRD corepression by genetic ablation or carcinogen treatment correlated with accumulation of L1 mRNA and proteins. Mi2β bound directly to the L1 promoter to effect retroelement silencing, and this response required the DNA‐ and ATPase‐binding domains of Mi2β. Sustained expression of L1 in HBECs was tumorigenic in a human–SCID mouse xenograft model, giving rise to tumors that regressed over time. Together, these results show that functional modulation of the NuRD constituent proteins is a critical molecular event in the activation of L1 retrotransposon. L1 expression creates a microenvironment in HBECs that is conducive to neoplasia and malignant transformation.
Keywords: benzo(a)pyrene, euchromatin, heterochromatin, long interspersed nuclear element‐1, nucleosomal and remodeling deacetylase complex, retrotransposon
Abbreviations
- BaP
benzo(a)pyrene
- HBECs
human bronchial epithelial cells
- Line‐1/L1
long interspersed nuclear element‐1 (LINE‐1 or L1)
- MBD
methyl‐CpG‐binding domain proteins
- MTA
metastasis‐associated proteins
- NuRD
nucleosomal and remodeling deacetylase
- RNPS
ribonucleoprotein particles (RNPs)
Introduction
Human L1 is ~6 kb and consists of an internal promoter located within the 5′untranslated region (5′UTR), two open reading frames (ORFs) encoding ORF1p and ORF2p, and a 3′UTR with a poly(A) tail and signal (Dombroski et al., 1991; Scott et al., 1987). L1 elements constitute a large family of mammalian retrotransposons that have continuously replicated for over 100 Myr (Boissinot et al., 2000). Approximately 100 retrotransposition‐competent L1s remain in the human genome that function as autonomous elements through a copy‐and‐paste mechanism (Brouha et al., 2003). The human L1 promoter is highly conserved among active L1s, except for a single nucleotide deletion at position 74, and a single nucleotide polymorphism (SNP) at 711 (t/c), both of which differentiate Ta‐1d from the Ta‐Ind, Ta‐0, and Pre‐Ta families (Boissinot et al., 2000; Swergold, 1990). The active human Ta family arose ~4MYA and subsequently differentiated into two major subfamilies, Ta‐0 and Ta‐1, each of which contains additional members. Ta‐1 is younger than Ta‐0 and now accounts for at least 50% of the Ta family (Boissinot et al., 2000).
A cycle of retrotransposition involves transcription of both ORFs from a common 5′UTR by RNA PolII, followed by translation in the cytoplasm (Swergold, 1990). The L1‐encoded proteins exhibit either cis‐ (major) or trans‐ (minor) preference to bind L1 mRNA or SINEs and other cellular mRNAs, to form ribonucleoprotein particles (RNPs) (Hohjoh and Singer, 1996; Wei et al., 2001). The RNP‐containing L1 mRNA, or other mRNAs, mainly localizes to the cytoplasm, but can translocate into the nucleus where the endonuclease domain of ORF2p nicks a single strand of genomic DNA to expose a 3′‐OH group that can then be used by the reverse transcriptase domain of ORF2p to prime and reverse synthesize L1 cDNA from the 3′ end (Feng et al., 1996; Hattori et al., 1986). This process can lead to full‐length or truncated insertions of L1 sequences throughout the genome (Moran et al., 1996).
Because uncontrolled retrotransposition can be genotoxic and detrimental to the host, active L1 elements are silenced in most somatic cells via DNA methylation and histone covalent modifications (Montoya‐Durango et al., 2009; Teneng et al., 2011). In addition, mammalian cells evolved several mechanisms against retrotransposition, including the deamination by APOBEC proteins, L1 mRNA degradation, and preferential targeting of L1 sequences to gene‐poor regions (Bojang et al., 2014; Esnault et al., 2006). L1 is reactivated by tobacco carcinogens such as benzo(a)pyrene (BaP) through a process that is orchestrated by the aryl hydrocarbon receptor (AHR) and retinoblastoma (RB) family of proteins to influence the epigenetic machinery (Lu and Ramos, 2003; Montoya‐Durango et al., 2009; Stribinskis and Ramos, 2006; Teneng et al., 2007). L1 reactivation can reshuffle the genome to afford a survival advantage or mediate acquisition of stem cell‐like properties and epithelial‐to‐mesenchymal transition (EMT), as seen in non‐small‐cell lung cancers (NSCLCs) (Bojang et al., 2013; Ikeda et al., 2013; McClintock, 1984; Saito et al., 2010). We recently showed that L1 couples EMT with oncogenesis in human bronchial epithelial cells (HBECs) through a signaling cascade initiated by TGF‐β1 (Reyes‐Reyes et al., 2017).
Given the possible roles of L1 in lung pathogenesis, elucidation of the molecular mechanisms of L1 regulation is of interest. In this context, we have shown that the nucleosomal and remodeling deacetylase (NuRD) multiprotein complex is enriched in the 5′untranslated DNA sequence of L1 in human and murine cells. Further, genetic ablation of RB proteins destabilizes interactions within the NuRD complex, and mediates nuclear rearrangement of Mi2β, an ATP‐dependent helicase subunit with nucleosome remodeling activity (Montoya‐Durango et al., 2016). The NuRD multiprotein complex consists of six core components, including the chromodomain‐helicase‐DNA‐binding proteins 3 or 4 (also known as Mi2α and Mi2β, respectively); methyl‐CpG‐binding domain proteins 1/2/3 (MBD1/2/3); metastasis‐associated proteins 1/2/3 (MTA1/2/3); histone‐binding proteins (Rbbp4/7, RbAp46/48), histone deacetylases 1/2 (HDAC1/2), and nuclear zinc finger proteins (Gata2a/2b(p66α/β)) (Xue et al., 1998; Zhang et al., 1999). HDAC1/2 and Rbbp4/7 are not exclusive to the NuRD complex and are also present in the SIN3 and CoREST multiprotein complexes, while Mi2α/β, MBDs, and MTAs are relatively specific to the NuRD complex, and their interactions with core components and other accessory proteins mediate recruitment and functionally of the NuRD complex (Humphrey et al., 2001).
The present studies were conducted to elucidate structural and functional determinants of NuRD corepression within the L1 promoter in HBECs following reactivation by the tobacco carcinogen, BaP. Evidence is presented here that L1 reactivation in HBECs by BaP shifts the L1 promoter from a heterochromatic to euchromatic state through disassembly and delocalization of the NuRD multiprotein complex. This response leads to cellular accumulation of ORF1 and ORF2 mRNA and proteins. The corepressor function of NuRD is dependent upon the DNA and ATPase domains of Mi2β, working together with MBD2/3 to regulate the state of L1 chromatin. Expression of a constitutively active L1 in HBECs was tumorigenic in a SCID mouse xenograft model, a finding that directly implicates L1 reactivation in neoplasia and malignant transformation of lung epithelial cells.
Materials and methods
Plasmids and cloning
Mi2β wild‐type (WT) and a biologically inactive W508A chromodomain mutant were kind gifts from J. Hagman at the University of Colorado (Ramírez et al., 2012). DNA (E874A)‐ and ATPase (W879A)‐binding domain mutants were created as described previously using PCR primers as specified in Table 1 (Ramírez et al., 2012). Mutant fragments were joined by nested PCR and cloned into a Age I and pSHA I linearized MSCV vector by infusion cloning (Clontech). L1‐5′UTR promoter was cloned into the BglII site of pGL4.15 (luc2P/Hygro) vector using infusion cloning and primers listed in Table 1. Cloned fragments were sequenced in reverse and forward directions to confirm identity and orientation. HBECs were transfected with pB001CTR or pB015WT using Lipofectamine, as previously described. The pB015WT and pB001CTR (i.e., pGL4.15 backbone) vectors were used to create stably transfected HBECs constitutively expressing L1‐ORF1 and ORF2 proteins, as confirmed by indirect immunofluorescence.
Table 1.
Cloning of Mi2β and L1‐5′UTR
| Sequence name | Sequence |
|---|---|
| DNA (E874A)‐IF | 5′‐GAAATTCCATGTGCTGCTGACAGCCTATGAATTGATC‐3′ |
| DNA (E874A)‐IR | 5′‐CATGTCAATG GTGATCAATTCATAGGCTGTCAGCAG‐3′ |
| ATPase (W879A)‐1F | 5′‐CTGAGCCCTATGCAGAAGAA ATACTACGCGTACATCCTC‐3′ |
| ATPase (W879A)‐1R | 5′‐CTTCAAAATTTCGAGTGAGGATGTACGCGTA GTATTTCTT C‐3′ |
| 5′‐GTACAGACAGCAGTCTTCCTGTATTCCCTT TACAAGG‐3′ | |
| 5′‐GGTCACAAACCGG TAGATCATTACCTTTTTATTTTGC‐3′ | |
| pGL4.15Luc‐1F | 5′‐GAGGATATCAAGATCTGGGGGGAGGAGCCAAGATGGCCG‐3′ |
| pGL4.15Luc‐1R | 5′‐GCCGAGGCCAGATCTCTTTGTGTTTTATCTACTTTT GGTC‐3′ |
Real‐Time (RT) PCR, siRNA treatment, western analysis, and indirect immunofluorescence
BEAS‐2B cells (CRL#: 9609, ATCC) is a nontransformed HBEC line immortalized with a replication‐defective SV‐40/adenovirus 12 hybrid (Cat#: 15338100, Thermo Scientific). Despite the presence of T‐antigen in replication‐defective lines (Butel and Rapp, 1966), BEAS‐2B cells remain RB/p53 competent (Pan et al., 2011), and are representative of squamous epithelial cells within the proximal airway. The integrity of RB and p53 signaling was confirmed in studies showing intact RB phosphorylation and p53 acetylation in BEAS‐2B cells (data not shown). Cells were grown to 70% confluence and treated with scrambled siRNA or siRNAs directed against Mi2β, MBD2/3, and MTA1/2/3 for 48 h. Total RNA was extracted and 0.5–1 μg used for cDNA synthesis (Invitrogen Superscript II) with OligoDT primers (Tables 2 and 3). The homogeneity of PCR products was confirmed using an RT‐PCR melting curve for SYBR probes. For TaqMan probes, ΔΔCT was used to calculate the relative quantity of L1. For SYBR probes, a standard curve method was used to calculate relative quantity. For western blot analysis, HBECs were lysed with RIPA buffer and 15–30 μg total protein loaded onto 4–12% gradient gels (Invitrogen). A polyclonal antibody against L1‐ORF1 generated by New England Peptide and validated in previous studies was used in all experiments (Ramos et al., 2011; Reyes‐Reyes et al., 2016). The optimization of L1‐ORF1 detection by Western analysis in HBECs is presented in Fig. S1. The overexpression of L1 proteins in cells transfected with pB015WT was confirmed by Western and documented previously (Reyes‐Reyes et al., 2017). Mi2β (Cat#: SAB5300324), MBD2/3 (Cat #: SAB2101140/SAB1300141), and MTA2/3 (Cat#: M7569, WH0057504MI) antibodies were obtained from Sigma or Cell Signaling. Primary antibodies against NuRD proteins were diluted 1/1000, while the ORF1 antibody was diluted 1/500 and incubated overnight. The antibody against GAPDH was obtained from Santa‐Cruz (Cat#: sc‐47724). Indirect immunofluorescence was conducted as described in detail previously (Bojang et al., 2013).
Table 2.
L1‐ORF1/2 TaqMan primers
| Sequence name | Sequence |
|---|---|
| L1‐ORF1 Taqm ‐1F | AAT GTT AAG GGC AGC CAG AG |
| L1‐ORF1 Taqm ‐1R | CTC TTC TGG CTT GTA GGG TTT C |
| L1‐ORF1 Taqm PRB‐1 | /56‐FAM/AC CCT CAA A/ZEN/G GAA AGC CCA TCA GA/3IABkFQ/ |
| L1‐ORF1 Taqm ‐2F | CCT CAC CAG CAA CAG AAC AA |
| L1‐ORF1 Taqm ‐2R | CCT CCC GTA GCT CAG AGT AAT |
| L1‐ORF1 Taqm PRB‐2 | /56‐FAM/TT GAC GAG C/ZEN/T GAG AGA AGA AGG CT/3IABkFQ/ |
| L1‐ORF2 Taqm ‐1F | CTC AGA CCA CAG TGC AAT CA |
| L1‐ORF2 Taqm ‐1R | GTC ATT CAG GAG CAG GTT GT |
| L1‐ORF2 Taqm PRB‐1 | /56‐FAM/AG CCG CTC A/ZEN/A CTA CAT GGA AAC TGA/3IABkFQ/ |
Table 3.
L1‐ORF1/2 SYBR Green Primers
| Sequence name | Sequence |
|---|---|
| L1‐ORF1‐1F | CCA AGTTGGAAAACACTCTGC |
| L1‐ORF1‐1R | TGTGGCGTTCTCTGTATTTCC |
| L1‐ORF2‐1F | TCGACACATACACTCTCCCAAG |
| L1‐ORF2‐1R | TGGTCCTGGACTCTTTTTGG |
Chromatin immunoprecipitation assay followed by RT‐PCR
Approximately 1 × 106 HBECs were grown to 70% confluence and treated with 1% formaldehyde for 10 min at 37 °C and washed 2x with ice‐cold PBS supplemented with protease and phosphatase inhibitor cocktails (Cat#: 539‐134‐1SET). Chromatin immunoprecipitation assay (ChIP) was conducted as described before (Montoya‐Durango et al., 2009; Teneng et al., 2011). After incubation with respective antibodies for NuRD components, complexes were pelleted, the supernatant removed, and samples kept on ice. Immune complexes were eluted by addition of 250 μL elution buffer (1% SDS 0.1 M NaHCO3), vortexed briefly, and incubated at room temperature for 15 min with gentle rotation. The eluate was combined with 20 μL of 5M NaCl and incubated at 65 °C for 4 h to reverse formaldehyde cross‐linking of DNA/protein complexes. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. DNA was resuspended in nuclease‐free H2O and quantified by Cytation 3 machine. Serial dilutions were completed to confirm a linear relationship while accounting for proper amplification of template to ensure equal amounts of starting template for all samples. RT‐PCR was used to determine enrichment for L1‐5′UTR sequences using tiling primers (P1‐P4) that span the 5′UTR promoter region of L1. The specificity of NuRD target recognition was examined in previous studies (Montoya‐Durango et al., 2009) and validated by DNA sequencing (Table 4 and Fig. S3). ChIP measurements in BaP‐ and DMSO‐treated samples used the P3 primer to focus on the RB/E2F recognition site within the promoter.
Table 4.
PRIMERS FOR NuRD component used in RT‐PCR
| Sequence name | Sequence |
|---|---|
| HCHD4‐1F | CTC GGG CCA GTG TAG AGG TC |
| HCHD4‐1R | TCT TCA TTT TCT GGG TGG GGT |
| HMTA2‐1F | GGC ATT ATG GTG AAA CGG GC |
| HMTA2‐1R | GGG CAT CAG CTG GGT TTA GT |
| HMTA3‐1F | AGT TGA GGC TGA CTT GAC CG |
| HMTA3‐1R | TCC CCT GAT ATG TGT TGC GG |
| HMBD2‐1F | CAA CCG GTA ACC AAA GTC ACA |
| HMBD2‐1R | GCT GAC GTG GCT GTT CAT TC |
| HMBD3‐1F | AAG ATG AAC AAG AGC CGC CA |
| HMBD3‐1R | TGG TAA TCT TGG TCA CCG GC |
| ACTIN‐1F | CCGGACGCTGAGTATGTTCA |
| ACTIN‐1R | TCTCCTGTCCTTCAGCCACTCT |
| 18S‐1F | AGTCCCTGCCCTTTGTACACA |
| 18S‐1R | GATCCGAGGGCCTCACTAAAC |
Formaldehyde‐assisted isolation of regulatory elements
Formaldehyde‐assisted isolation of regulatory elements (FAIRE) was used to study L1 sequences associated with regulatory activity based on preferential formaldehyde cross‐linking of nucleosome‐bound DNA relative to nucleosome‐depleted regions of the genome (Giresi et al., 2007). HBECs were grown to 70% confluence and treated with 0.1 μm BaP or equivalent volume of DMSO for 12 h. The medium was removed and the cells cross‐linked with 1% formaldehyde for 10 min at 37 °C. Cells were washed 2X with ice‐cold DPBS (Dulbecco's phosphate‐buffered saline pH 7.4) supplemented with protease and phosphatase inhibitor cocktails (Cat#: 539‐134‐1SET) before being scraped and pelleted for 5 min at 800 g at 4 °C and resuspended in DPBS. Two 500‐μL aliquots of lysate were set aside for RNA analysis and input control. A total of 500 μL of lysate was resuspended in FAIRE lysis buffer (1% SDS, 100 mm NaCL, 50 mm Tris pH 8, 2 mm EDTA, and 2 mm EGTA supplemented with protease and phosphatase inhibitor cocktails) and sonicated as described for 30 sec and cooled in ice for ~5 min. Unbound DNA and DNA cross‐linked to proteins were separated by phenol/chloroform extraction. Unbound DNA in the aqueous phase was ethanol precipitated with 1/10 of 3M sodium acetate (NaAc) and 2.5X of 95% ethanol (ETOH) overnight followed by centrifugation at 14000 g for 20 min. DNA was washed with 70% ETOH once, air‐dried, and resuspended in 50 μL of sterile nuclease‐free H2O. Total DNA was quantified and serial dilutions of 2.5, 5, and 10 ng used for RT‐PCR analysis using a primer set (P4) that spans the CpG shore region of the L1 promoter (Table 5). For L1 transcript analysis, RNA was isolated as previously described using RNeasy Plus Mini Kit (Cat#: 74134). Total gDNA for use as input control was isolated using Purelink genomic DNA isolation kit (Cat#: K1820‐01).
Table 5.
Mi2β ChIP RT‐PCR primers
| Sequence name | Sequence |
|---|---|
| HL1UTR‐1f | GGAACAGCTCCGGTCTACAG |
| HL1UTR‐1R | TCACCCCTTTCTTTGACTCG |
| HL1UTR‐2f | GAGTTCCCTTTCCGAGTCAA |
| HL1UTR‐2R | TGTGCTAGCAATCAGCGAGA |
| HL1UTR‐3f | CACGGAATCTCGCTGATTG |
| HL1UTR‐3R | CCAGAGGTGGAGCCTACAGA |
| HL1UTR‐4f | CAGGGAAGCTCGAACTGG |
| HL1UTR‐4R | AGCTGCAGGTCTGTTGGAAT |
Viral transfection and luciferase analysis
HEK293T‐17 cells were used to generate virus titers for subsequent transduction of the virus containing Mi2β and mutant plasmids into BEAS‐2B cells. HEK293T‐17 cells were grown to 90% confluence and transfected with Mi2βWT/mutants retroviral vectors (6 μg), combined with pSPAX‐2 packaging vector (3 μg) and pVSV‐G envelope vector (1.5 μg) diluted in 500 μL of dilution media (10 mm HEPES/150 mm NaCl, pH 7.05). Retroviral packaging vectors (pSPAX‐2 and pVSV‐G) were a kind gift from Schenten at the University of Arizona (Addgene plasmid # 12260 and Addgene plasmid # 36399, respectively). An aliquot of 42 μL of PEI (1.0 μg/μL) was added to each transfection and incubated at room temperature for 15 min. DNA/PEI complexes were added to the cells and incubated for 18 h. Transfection media were removed and new media containing 10% FBS added. The viral supernatant was collected the next day and filtered through 0.45‐μm PES filter tubes. Viral titer was incubated with one volume of X‐concentrator (Clontech) overnight and centrifuged at 1500 g for 45 min. The supernatant was removed and virus resuspended in 100 μL PBS to make 10X viral concentrate. A total of 10 μL of viral titer was used to transiently transduce transfected cells. HBECs were grown to 70% confluence and transiently transfected with 0.75 μg of luciferase and 0.25 μg of Renilla vectors. Transfection media were removed 18 h later. Cells were allowed to recover for 24 h and then transduced with Mi2βWT or mutant retroviral vectors or cotreated with BaP and retroviral vectors for 24 h. Cells were lysed passively for 15 min at room temperature and luciferase and Renilla luminescence measured using a Cytation 3 machine. Relative luminescence units (RLU) were calculated as a ratio of firefly luciferase to Renilla.
Mouse xenografts and immunohistochemistry
HBECs (6 × 106) cells stably expressing control (pB001ctr) or wild‐type (WT) L1 plasmids (pB015wt) were mixed with Matrigel and injected subcutaneously into anesthetized SCID mice at the University of Arizona Cancer Center Core Facility. Mouse weights and tumor burden were monitored twice weekly for up to 90 days, with tumor volumes estimated using the following formula: [(width)2 × length]/2. A 1 : 50 dilution of ORF1p antibody was used to monitor L1‐ORF1 protein in tumors derived from grafted cells or human tissues by immunohistochemistry (IHC). Images were taken using an Axiovision inverted microscope at 40 × magnification.
Statistics
The differences between groups were evaluated using the Mann–Whitney U‐test to compare samples drawn from the same distribution. Mann–Whitney U‐test is a nonparametric alternative test often used to evaluate normalized data because it does not require the assumption of normal distribution when trying to determine differences between two independent samples. The number of replicates and independent measures for each of experiment are included in the figure legends.
Results
L1 reactivation by BaP disrupts NuRD promoter occupancy and partially shifts constituent proteins from the nuclear to cytoplasmic compartment
We have previously shown that the silenced L1‐Ta promoter is enriched in repressive heterochromatin marks, namely histone‐3 lysine‐9 trimethylation (H3K9Me3) and histone‐4 lysine‐20 trimethylation (H4K20Me3) (Teneng et al., 2011). We also showed that RB proteins stabilize the NuRD multiprotein complex to effect L1 silencing and that BaP interferes with RB functions via AHR‐dependent mechanisms (Montoya‐Durango et al., 2009, 2016; Teneng et al., 2007). AHR is present in the repressor complex assembled by RB on the L1 promoter in human and murine cells (Montoya‐Durango et al., 2009). However, the structural and functional determinants of NuRD corepression during L1 reactivation by tobacco carcinogens in HBECs have not been elucidated. To fill this gap, experiments were initiated to study L1 reactivation in HBECs challenged with BaP, a carcinogenic aromatic hydrocarbon present in tobacco smoke.
The retention of NuRD constituent proteins on the L1‐5′UTR promoter in HBECs challenged with 0.1 μm BaP is presented in Fig. 1A. This concentration was chosen based on dose–response and time–response experiments in BEAS 2B cells showing significant induction of L1 in the range of 0.1 to 3 μm (data not shown). Marked reductions in enrichment for Mi2β, MBD2/3, and MTA2 were seen 12 h after carcinogen challenge, indicating that BaP displaces NuRD macromolecular complex constituents from the L1 promoter. Next, FAIRE was employed to separate unbound and bound DNA sequences based on their relative affinities for the aqueous and organic phases, respectively (Giresi et al., 2007). BaP treatment reduced protein trapping at genomic regions containing L1‐5′UTR DNA sequence (Fig. 1B), indicating a shift of L1 chromatin from a heterochromatic to euchromatic state. These changes were associated with transcriptional activation and accumulation of L1 ORF1 and ORF2 mRNA (Fig. 1C). The lower levels of ORF2 mRNA compared to ORF1 mRNA are consistent with the presence of cryptic polyadenylation sites in ORF2 that cause early termination of transcription (Kazazian et al., 1988).
Figure 1.
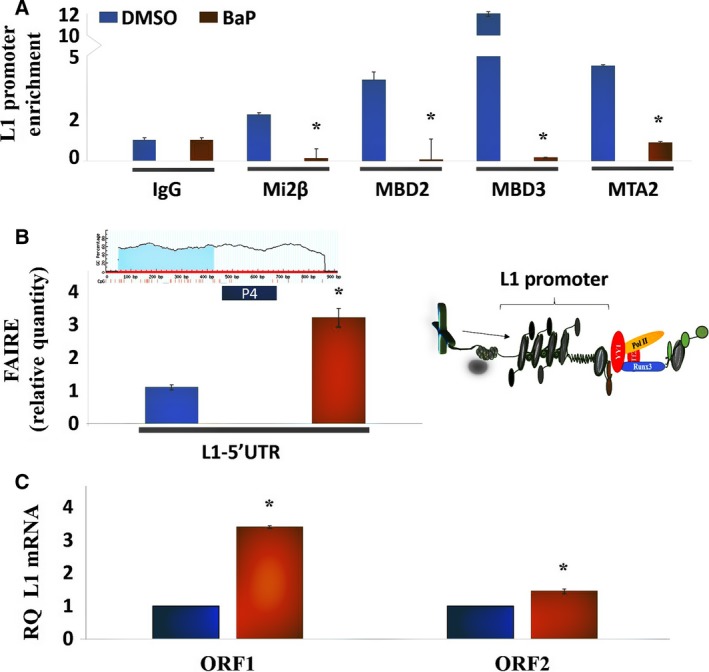
Displacement of NuRD macromolecular complex constituents from the L1 promoter by BaP shifts L1 sequences from heterochromatin to euchromatin and leads to transcriptional activation. (A) ChIP experiments of L1 promoter using primer 3 in HBECs treated with DMSO control or 0.1 μm BaP for 12 h. Fold enrichment was calculated relative to IgG control. Each assay was performed at least three times in triplicate, and the values shown are representative of combined experiments. Primer sequences are provided in Table 4. (B) FAIRE analysis of HBECs under DMSO control or BaP‐treated conditions using primer 4 (P4) to examine protein‐bound versus unbound L1 DNA sequences. Similar results were seen in three independent experiments using triplicates in each of these experiments. (C) RT‐PCR analysis of ORF1 and ORF2 in DMSO control or BaP‐treated cells. Similar results were seen in three independent experiments using triplicates in each experiment. Primer sequences are provided in Tables 2 and 3. RQ, relative quantity. Differences between groups were evaluated by the Mann–Whitney U‐test to compare samples from the same distribution. Statistical significance is denoted by (*).
L1 reactivation in HBECs following carcinogen challenge showed time‐dependent profiles (Fig. S2), with induction of L1 mRNA peaking at 12 h and returning to baseline by 48 h. Western analysis at 12 h showed accumulation of L1 ORF1 and ORF2 proteins in cells challenged with BaP (Fig. S2), confirming effective translation of mRNA into proteins. CYP1A1 protein was detected by immunofluorescence in BEAS 2B cells (Fig. S4), suggesting that BaP metabolites may contribute to the L1 induction response. We have previously shown that CYP‐derived metabolites of BaP readily activate L1 and are significantly more potent than the parent compound in vascular cells (Lu and Ramos, 2003). To further evaluate the disruption of NuRD corepressor complex interactions by BaP, the localization of NuRD constituent proteins was examined by indirect immunofluorescence. In these studies, HP1γ staining was measured as an index of the structural integrity of heterochromatin domains (Lechner et al., 2000; Mansfield et al., 2011). HP1γ staining in vehicle‐treated HBECs showed distinct focal accumulations, while staining in BaP‐treated cells was diffuse and considerably less pronounced (Fig. 2A,D, column 1). Mi2β and MTA2 staining strongly colocalized with DAPI staining, and was visibly nuclear in DMSO‐treated cells, indicating that under basal conditions these proteins preferentially reside within the nuclear compartment (Fig. 2A,C, columns ii and iv). In keeping with previous reports (Takeshita et al., 2016), the nuclei of BaP‐treated cells appeared swollen at times. Staining for Mi2β and MTA2 in HBECs treated with BaP was distributed between the nuclear and cytoplasmic compartments (Fig. 2B,D, columns ii and iv), suggesting a partial shift to the cytoplasmic compartment. To confirm this interpretation, nuclear fluorescence intensity was quantified and subtracted from total fluorescence intensity using imagej software (imagej.nih.gov). Increased fluorescence intensity for cytoplasmic Mi2β and MTA2 was observed in cells treated with BaP compared to DMSO controls (Fig. 2E,F, respectively). Thus, challenge of HBECs with BaP is associated with disintegration of heterochromatin leading to disassembly and detachment of NuRD components from the L1 promoter. These changes in turn were associated with a partial shift of constituent proteins from the nuclear to cytoplasmic compartment. The exact mechanism responsible for translocation of NuRD constituent proteins is not known, but may involve nuclear export signals and exportins, as shown previously for MBD2‐interacting proteins (Lechner et al., 2000).
Figure 2.
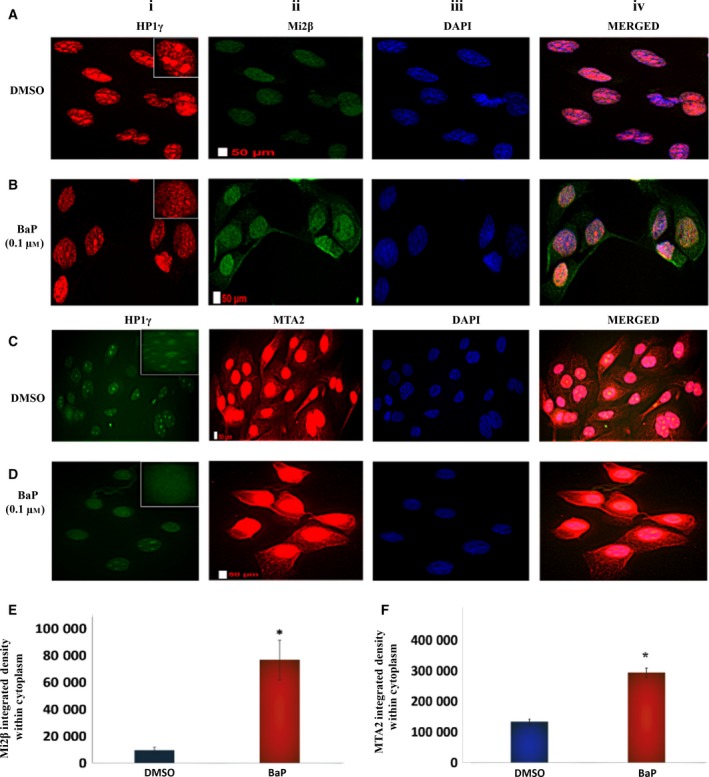
Benzo(a)pyrene disassembles the NuRD complex and shifts constituent proteins from the nuclear to cytoplasmic compartment. (A, B) Mi2β and (C, D) MTA2 immunofluorescence staining in HBECs treated with DMSO or 0.1 μm BaP for 12 h. (E, F) Densitometric measurement of fluorescence intensity for Mi2β and MTA2 in the cytoplasmic compartment of control and BaP‐treated HBECs, respectively. HP1γ was used as an index of structural integrity of heterochromatin domains. DAPI was used to stain the nuclei, with merged signals denoting colocalization of proteins. The staining for HP1γ was switched from red to green and vice versa to control for nonspecific staining by the secondary antibody. Similar results were seen in three independent experiments. The differences between groups were evaluated by the Mann–Whitney U‐test to compare samples drawn from the same distribution (n = 32 cells/experiment). Scale bars are 50 μm. Statistical significance is denoted by (*).
Mi2β binding to the L1 promoter in HBECs effects L1 retroelement silencing
Mi2β is an ATP‐dependent helicase subunit with nucleosome remodeling activity that is specific to the NuRD complex. Therefore, the next set of experiments was completed to characterize the influence of Mi2β binding to the L1 promoter in HBECs by ChIP using tiling primers (P1–P4) that spanned the complete L1‐Ta 5′UTR promoter sequence coupled with antibodies specific to Mi2β (Fig. 3A). L1‐Ta 5′UTR sequences were highly enriched in samples treated with Mi2β antibody compared to IgG control for all primer sets used to amplify promoter sequences (Fig. 3B). DNA electrophoresis and sequencing were completed to confirm the identity and size (P1: 224 bp, P2: 175 bp, P3: 188 bp, and P4: 315 bp) of amplified sequences (Fig. S3). Thus, chromatin‐bound Mi2β is present across the entire length of the L1 promoter.
Figure 3.
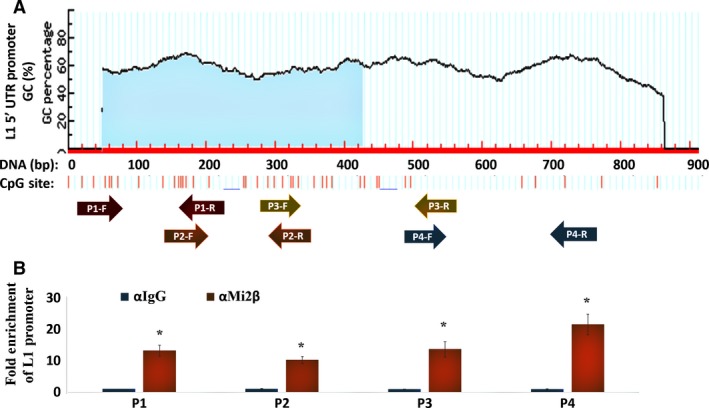
Mi2β Interacts with L1 Promoter Sequences. (A) Schematic representation of the location of tiling primers used to characterize Mi2β binding along the full‐length L1‐5′UTR (Table 4). CpG islands and shores within the promoter are also shown. (B) Solubilized chromatin was immunoprecipitated with antibodies against Mi2β or mouse IgG. After isolation and normalization of genomic DNA, RT‐PCR was performed using tiling primers specific to L1‐5′UTR promoter. Similar results were seen in three independent experiments. Statistical significance is denoted by (*).
Genetic ablation of Mi2α/β mRNA and protein using target‐specific siRNA (Fig. 4A, panels i and iii, respectively) increased the levels of L1 ORF1 mRNA and protein (Fig. 4A, panels ii and iv, respectively). L1 ORF1 mRNA and protein were detectable under unstimulated conditions (See Fig. S1) and significantly upregulated by siRNA treatment. The functional role of MBD2/3 and MTA2/3 was examined next using gene‐specific siRNA directed against these proteins (Fig. 5A,B). L1‐ORF1 mRNA and protein were markedly upregulated by MBD2/3 knockdown (Fig. 5A, panel ii), while ablation of MTAs was without effect (Fig. 5B, panel ii). These findings identify Mi2β and MBDs as the key architectural elements that effect L1 silencing. The negative regulatory function of Mi2β likely reflects its activity within the NuRD macromolecular complex, while MBDs may exert regulatory control through binding to methyl groups in DNA. Collectively, these findings show that functional modulation of Mi2β and MBDs is a fundamental molecular event in the activation of L1 in HBECs.
Figure 4.
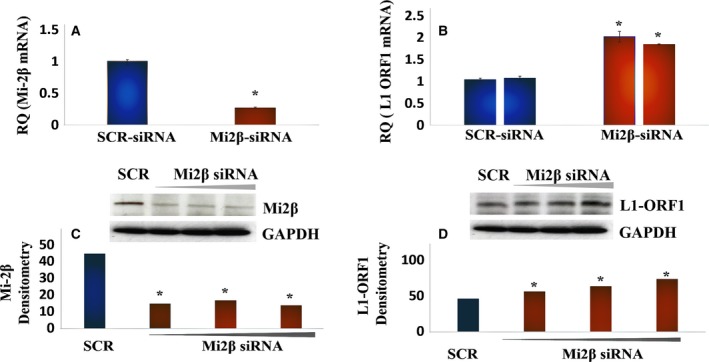
Mi2β Binding to L1 Promoter Sequences Effects L1 Retroelement Silencing. (A) RT‐PCR analysis of cells treated with scrambled (SCR) siRNA or siRNA against M12β. Similar results were seen in three independent experiments using triplicates in each of these experiments. (B) RT‐PCR analysis of L1 mRNA levels in cells treated with scrambled siRNA or siRNA against M12β. Similar results were seen in three independent experiments using triplicates in each of these experiments. (C) Western blot analysis of Mi2β levels following siRNA silencing. Similar results were seen in three independent experiments. (D) Western blot analysis of L1 ORF1p following Mi2β silencing. The image showed the same membrane probed for Mi2β, L1 ORF1p, and GAPDH. Similar results were seen in three independent experiments. Differences between groups were evaluated by the Mann–Whitney U‐test to compare samples from the same distribution. Statistical significance is denoted by (*).
Figure 5.
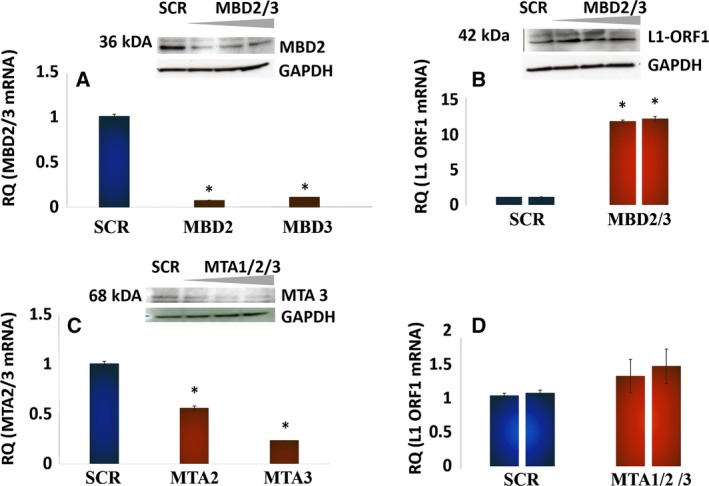
MBD2/3, but not MTA1/2/3, also participates in L1 Retroelement Silencing. (A) Western blot for MBD2 and RT‐PCR analysis of MBD2/3 in HBECs treated with scrambled siRNA or siRNA against MBD2/3. Similar results were seen in three independent experiments using triplicates in each of these experiments. (B) L1 ORF1p mRNA and protein in HBECs treated with scrambled siRNA or siRNA against MBD2/3. Similar results were seen in three independent experiments. (C) Western blot for MTA3 and RT‐PCR analysis of MTA2/3 in HBECs treated with scrambled siRNA or siRNA against MTA1/2/3. Similar results were seen in three independent experiments using triplicates in each of these experiments. (D) L1 ORF1p mRNA in HBECs treated with scrambled siRNA or siRNA against MTA1/2/3. Similar results were seen in three independent experiments. The primer sequences used are presented as supplementary data in tabular form. Differences between groups were evaluated by the Mann–Whitney U‐test to compare samples from the same distribution. Statistical significance is denoted by (*).
Mutations in the DNA‐ and ATPase‐binding domains, but not the chromodomain of Mi2β negatively regulate NuRD corepressor activity
To further confirm the role Mi2β in L1 suppression, the suppressor activity of Mi2βWT was compared to that of mutants within the DNA‐binding domain, ATPase‐binding domain, and chromodomain (Ramírez et al., 2012). L1‐Ta 5′UTR promoter‐driven luciferase and Renilla vector (internal control) were transiently transfected into HBECs. Cells recovered from transfection for 24 h before treatment with retroviral vectors encoding Mi2βWT or mutants for an additional 24 h. Transduction efficiency was optimal 24 h after exposure (data not shown). A significant, but modest, reduction in L1 reporter activity was seen in Mi2β WT‐transfected cells (Fig. 6A). The modest response likely implicates significant contributions by other accessory proteins to basal activation of the L1 promoter. Importantly, mutants in the DNA‐ and ATPase‐binding domains of Mi2β lost their ability to suppress luciferase reporter gene expression, while the chromodomain mutant was without effect (Fig. 6A). Cotreatment of cells transfected with Mi2βWT retroviral vectors significantly reduced both basal‐ and BaP‐driven luciferase expression (Fig. 6B), confirming that disruption of Mi2β activity by BaP plays an important role in regulating the repressive functions of the NuRD complex on L1. The partial reversal seen upon reconstitution with Mi2β confirms that combinatorial functions with other members of the repressor complex are essential for L1 repression.
Figure 6.
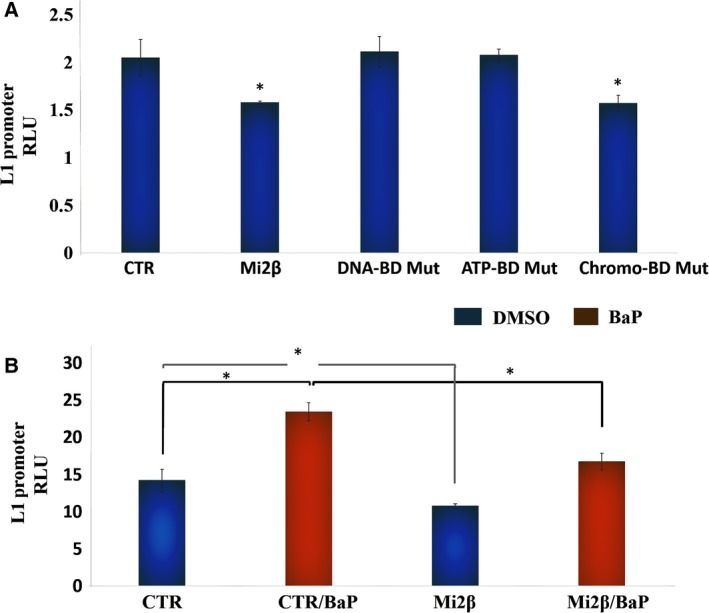
The DNA and ATPase domains, but not the chromodomain, are necessary for L1 promoter silencing. (A) Mi2β and mutant retroviral vectors (i.e., W508A chromodomain mutant, E874A DNA‐binding mutant and W879A ATPase domain mutant) were transduced into HBECs transiently transfected with L1‐5′UTR promoter‐driven luciferase and Renilla vector as internal control. RLU values were calculated as a ratio of luciferase luminescence over Renilla luminescence. Similar results were seen in three independent experiments using triplicates in each of these experiments. (B) Influence of Mi2β on L1 Reactivation by BaP. Cotreatment of HBECs with BaP or DMSO control with Mi2βWT retroviral vectors in cells transiently transfected with L1‐5′UTR promoter‐driven luciferase gene. Control (CTR) consisted of pSPAX‐2 packaging vector and pVSV‐G envelope vector. Transfection efficiency was controlled by Renilla vector. Similar results were seen in three independent experiments using triplicates for each of these experiments. Differences between groups were evaluated by the Mann–Whitney U‐test to compare samples from the same distribution. Statistical significance is denoted by (*).
Expression of a constitutively active synthetic L1 is oncogenic in a SCID xenograft mouse model
The final set of experiments examined the oncogenic potential of L1 in a SCID mouse xenograft model. Given that L1 activation in HBECs by BaP peaks at 12 h and returns to baseline by 48 h (Fig. S2), an in vitro model of constitutive L1 activation that replicates the smoking behavior of habitual users who smoke ≥12 cigarettes per day was examined. Our estimation is consistent with previous reports showing regular patterns of smoking frequency of smokers (Chandra et al., 2007). SCID mice were injected subcutaneously with 6 × 106 HBECs stably expressing empty pB001ctr vector (n = 6) or a constitutively active wild‐type L1 construct (pB015WT) (n = 15) (Bojang et al., 2013). pB015WT consists of L1 RP sequence cloned into a pGL4.15 vector backbone with L1 ORF1 and ORF2 proteins tagged with streptavidin–hemagglutinin (Strep–HA) and flag‐myc double tags, respectively (Fig. 7A). A cytomegalovirus (CMV) promoter was placed upstream of the L1‐5′UTR for constitutive expression. No differences in animal weights were seen between the two groups (data not shown). L1 was tumorigenic in SCID mice, with tumor burden first detected one week after inoculation and sustained for up to eight weeks (Fig. 7B). No tumors were seen in control mice at any time during the experiment, confirming the nontumorigenic phenotype of HBECs (i.e., BEAS‐2B cells), and the innocuous nature of the control vector. Peak tumor incidence occurred four weeks after inoculation (12/15 mice), with significant tumor growths ranging in size from 1.47 to 42.0 mm2 in 6 of 15 mice during week five. Variable degrees of tumor regression and recurrence were seen over the course of the eight‐week experiment (Fig. 7B). The expression of L1‐ORF1 protein was examined in the grafted tumors by IHC to confirm molecular phenotype (Fig. 7C). L1‐ORF1 protein expression was positive in all mouse tumors examined (n = 6). Representative samples are shown in Fig. 7C, panels i and ii. Normal human lung tissue and p53 null lung tissue were used as negative and positive controls, respectively (Fig. 7C, panels iii and iv). High levels of L1‐ORF1 protein were detected in p53 null tissue, but not in normal lung. These findings demonstrate that activation of L1 in HBECs creates a microenvironment that supports oncogenic transformation and implicate molecular interactions between L1 and p53 as critical players in lung cancer.
Figure 7.
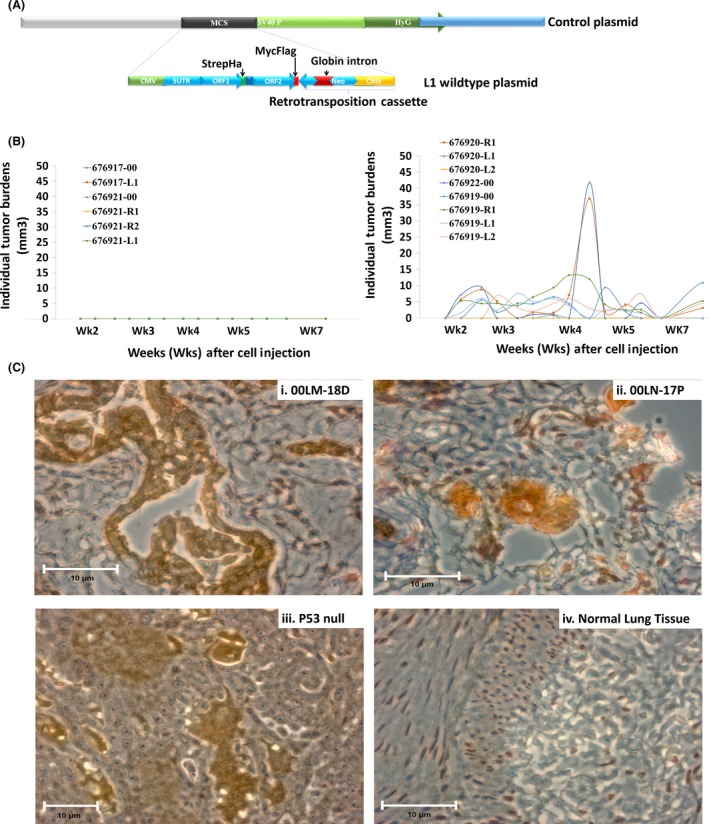
Oncogenicity of Constitutively Active L1 in Human Bronchial Epithelial Cells. (A) Schematic representation of the construct utilized for stable expression of wild‐type L1 or empty vector. (B) Tumor burden in SCID mice injected once subcutaneously with control (n = 6) or wild‐type L1 (n = 15) vectors. (C) Immunohistochemistry of L1‐ORF1p staining in (i) tumors isolated from 00LM‐18 and (ii) 00LN‐17P, SCID mice (iii) p53‐null lung tissue, (iv) normal lung tissue. Scaled bars represent 10 μm.
Discussion
Evidence is presented here establishing a functional linkage between NuRD macromolecular complex disassembly, L1‐Ta reactivation, and malignant transformation in HBECs challenged with the lung carcinogen BaP. These findings build upon earlier reports by our group showing that L1 reactivation involves the following: 1) enrichment of activating H3K4Me3 and H3K9Ac marks; 2) increased histone H3 acetylation of the 5′UTR; 3) reduced DNMT1 recruitment; and 4) CpG hypomethylation in both transformed and nontransformed epithelial cells (Montoya‐Durango et al., 2009; Teneng et al., 2011). Thus, the association of NuRD complexes with the L1 promoter may serve as a key regulatory signal for the formation of regional domains of heterochromatin that can be disrupted by the lung carcinogen BaP during the course of malignant progression. NuRD subunits are known to associate with oncogenic transcription factors, and the loss of NuRD function has been linked to various types of malignancies (Lai and Wade, 2011). Furthermore, Mi2β binds to H3K9Me3, a modification characteristic of the silenced L1 promoter in transformed cells (Mansfield et al., 2011; Teneng et al., 2011), and activating histone marks delocalize Mi2β and MDBs and disrupt architectural anchoring of the NuRD complex to DNA (Allen et al., 2013; Baubec et al., 2013).
The ability of BaP to interfere with the assembly of NuRD subunits and their function likely involves AHR, a ligand‐activated transcription factor regulated by BaP during the course of L1 reactivation (Teneng et al., 2007). AHR was first identified as the nuclear receptor responsible for transcriptional regulation of cytochromes P450 by dioxin and related aromatic hydrocarbons, and later shown to bind a variety of endogenous and exogenous ligands to effect molecular control of proliferation, apoptosis, differentiation, adhesion, and pluripotency. We have previously shown that AHR is present in the repressor complex assembled by RB on the L1 promoter in human and murine cells (Montoya‐Durango et al., 2009). Others have shown that AHR interacts with NuRD complex subunits to regulate differentiation‐specific genes that interfere with early embryonic programming (Gialitakis et al., 2017). Thus, AHR may be the critical effector used by BaP to interfere with NuRD functions in HBECs. This suggestion is supported by our own studies showing that genetic ablation of AHR under basal conditions in BEAS‐2B cells dramatically increases L1 mRNA levels (Bojang and Ramos, unpublished data). As such, constitutive AHR may support critical protein–protein interactions that are disrupted in the presence of BaP to facilitate disassembly of the NuRD repressor complex. Furthermore, AHR interacts with TGF‐β1, an upstream regulator of L1 and inducer of EMT in HBECs (Chang et al., 2007; Ramos et al., 2011; Reyes‐Reyes et al., 2017). Thus, structural and functional interactions between AHR and NuRD complex subunits may cooperate with TGF‐β1 in a highly contextual manner to regulate L1. Wang et al. (2009) reported that NuRD complexes regulate TGF‐β1 signaling by enlisting LSD1, a lysine‐specific demethylase that affords histone demethylase activity to the chromatin remodeling capacity of the NuRD complex (Wang et al., 2009). AHR may also participate in the regulation of CYP genes involved in BaP metabolism, a suggestion consistent with the expression of CYP1A1 protein in BEAS‐2B cells and our previous report that oxidative BaP metabolites activate L1 expression. Although the relative contributions of unmetabolized versus metabolized BaP to the tumorigenic response in BEAS‐2B cells were not examined, the 12‐h lag time for induction of L1 in BEAS‐2B cells suggests that metabolism of the parent hydrocarbon may contribute to the L1 reactivation response.
Based on our collective findings, we conclude that combinatorial interactions between AHR, RB, p53, and NuRD subunits effect epigenetic silencing of L1 and are direct targets of the lung carcinogen BaP during the course of L1 reactivation and malignant transformation (Lai and Wade, 2011; Montoya‐Durango et al., 2009; Teneng et al., 2007). The immortalization of BEAS‐2B cells with SV40 large T‐antigen likely sensitizes RB and p53 to create a permissive environment for L1 reactivation by the carcinogen. This is consistent with the appearance of L1 protein in p53‐null lung cancer, but not normal tissue, and the exquisite sensitivity of BEAS‐2B cells to BaP. However, despite SV40 immortalization, the BEAS‐2B cell line is not tumorigenic in the absence of L1 or carcinogen, remains RB and p53 competent during serial culture, and retains intact Akt signaling (Pan et al., 2011). We previously showed that forced expression of L1 in embryonic kidney cells disrupts differentiation programming and alters growth factor responsiveness (Ramos et al., 2011), suggesting that L1 alone is sufficient to drive the appearance of oncogenic phenotypes. NuRD complex disassembly has been linked to malignant progression through disruption of cellular differentiation and transformation (Kaur et al., 2007), while L1 reactivation has been linked with genomewide reprogramming, EMT, malignant transformation, and poor prognosis in NSCLC patients (Bojang et al., 2013; Ikeda et al., 2013; Saito et al., 2010). Further, disassembly of the NuRD complex from promoters is known to recruit SWI/SNF complexes that regulate EMT (Mohd‐Sarip et al., 2017). The reactivation and mobilization of L1 can predispose lung cancer genomes to aberrant epigenetic control by creating sloppy DNA methylation shores that influence the methylation status and expression of surrounding loci (Grandi et al., 2015). In accord with this view, de novo somatic L1 retrotransposition occurs at higher frequencies in NSCLCs than in other tumors, and site‐specific hypomethylation of L1s is associated with pathological features of NSCLC patients (Ikeda et al., 2013; Saito et al., 2010). Thus, the carcinogenicity of BaP and its impact on NSCLC evolution are likely tied to the disruption of NuRD complex functions and L1 reactivation.
While the repressive role of the NuRD complex has been extensively studied, it is not yet known how the loss of one or more of its components affects overall function. Some of the components have been shown to be mutually exclusive, while others exert opposing functions (Denslow and Wade, 2007). The results of our siRNA analyses showed that loss of Mi2β and MBD2/3 increases L1 mRNA, while loss of MTAs was without effect. These results are in line with the organization of the NuRD complex in a repressive state, where proteins are anchored to the DNA through Mi2β and MBD2/3, and MTAs do not directly contact DNA (Lai and Wade 2011). MTAs have been shown to interact with RBBP4/7 and GATAD2α/β, which in turn interact with MBD2/3 to anchor MTAs to the complex (Lai and Wade 2011; Allen et al., 2013). Therefore, promoter‐specific analysis would be required to determine the functional impact of NuRD‐related functions for other genetic targets, as well as its functionality in different cellular contexts.
Transcriptional regulation of L1 is highly contextual, cell type specific, and dependent upon the activation stimulus (Teneng et al., 2007). Given the functional linkage between NuRD core proteins and L1 retroelement reactivation, regulatory control of L1 is likely coupled to the expression of accessory proteins involved in the regulation of NuRD core protein functions (Lai and Wade 2011; Allen et al., 2013). For instance, the interaction of HIF1α with NuRD activates transcription, while interactions between NuRD and p53 are repressive (Lai and Wade 2011; Allen et al., 2013). This is in keeping with our finding that functional interactions between RB and AHR proteins repress L1 through interactions with the NuRD complex under basal conditions (Montoya‐Durango et al., 2009, 2016). Mi2β plays a prominent role in the regulation of L1 expression upon carcinogen activation, with the DNA‐binding and ATPase enzymatic activities, but not the chromo protein–protein interaction domain, serving to interfere with ectopic L1 expression. The minimal role of the chromodomain is consistent with the function and architecture of the NuRD complex, where NuRD core proteins mediate not only protein–protein interactions, but also remodeling of nucleosomes, histone deacetylation and demethylation, and interactions with methylated DNA (Baubec et al., 2013; Xue et al., 1998). Our data suggest that DNA methylation is a prerequisite for L1 suppression, especially in the context of MBD functions (Jiang et al., 2004; Teneng et al., 2011). This assertion is consistent with the finding that cotreatment of HBECs with Mi2β and BaP did not completely abolish BaP‐driven luciferase activity.
Elevated expression of L1 retrotransposon has been associated with oncogenic transformation and L1 hypomethylation is known to induce stem cell‐like properties and EMT in NSCLCs (Ikeda et al., 2013; Saito et al., 2010). However, the contextual determinants that define L1 functions as a driver or passenger event in tumorigenesis are not fully understood. Our in vivo studies examined these relationships using a constitutive model of L1 activation in HBECs that mimics the repeated cycles of exposure seen in smokers who consume 12 or more cigarettes per day (Chandra et al., 2007; Nanez et al., 2011). Evidence was obtained that expression of constitutively active L1 in HBECs is oncogenic in a SCID mouse xenograft model, confirming that L1 can function as an initiating event during malignant transformation of lung epithelial cells. The oncogenic activity of L1 may be exerted via retrotransposition‐dependent and retrotransposition‐independent mechanisms, with the former involving insertions, deletions, and structural damage, and the latter involving oncogenic signaling pathways that couple with EMT (Reyes‐Reyes et al., 2017).
Interestingly, tumor growth in mice grafted with cells expressing L1 was restricted over time, suggesting that the oncogenic response initiated by L1 cooperates with other pathways that sustain the malignant response. In this regard, the ability of BaP to damage DNA and to mutate both p53 and KRAS in HBECs may amplify the DNA damage response, propagate waves of genomic instability, and disrupt genetic regulatory networks of growth and differentiation. In fact, p53 is known to restrict retrotransposon activity through interaction with components of the piRNA (PIWI Interacting RNA) pathway (Wylie et al., 2016). Our own evidence supports this as L1‐encoded ORF‐1p was increased in human lung tumors null for p53, a finding consistent with previous reports (Wylie et al., 2016). We recently showed that a reverse transcriptase mutant of L1 can induce tumors in athymic mice that outgrow those induced by wild‐type L1 (Reyes‐Reyes et al., 2017), suggesting that retrotransposition and genotoxicity from the unregulated endonuclease activity of wild‐type L1 pose significant restrictions on the tumorigenic process. This is an area worthy of exploration as novel chemotherapeutic agents can be designed to exploit this property in the clinical management of lung tumors.
In conclusion, evidence is presented here showing that challenge of HBECs with the lung carcinogen BaP shifts the L1‐Ta promoter from a heterochromatic to euchromatic state through disassembly of the NuRD complex and partial displacement of constituent proteins from the nucleus to the cytoplasm. Disruption of NuRD corepression correlates with accumulation of L1 mRNA and proteins and tumorigenesis.
Author contributions
KSR conceived the study, KSR and PBJr designed the study, PBJr acquired the data, KSR and PBJr analyzed and interpreted the data, and KSR and PBJr wrote the manuscript.
Supporting information
Fig. S1. Optimization of L1‐ORF1 in HBECs by Western analysis.
Fig. S2. Time‐dependent analysis of L1 mRNA accumulation in HBECs challenged with 0.1 μm BaP.
Fig. S3. DNA and Sequence analysis of L1‐5′UTR sequences.
Fig. S4. Analysis of CYP1A1 expression after 12 h of BaP treatment by indirect immunofluorescence.
Acknowledgements
We are grateful to Drs. Schenten and Hagman at the University of Arizona and the University of Colorado for viral packaging vectors and Mi2βWT and chromodomain vectors, respectively. This work was supported in part by funding from University of Arizona Health Sciences to KSR and P30 CA023074.
References
- Allen HF, Wade PA and Kutateladze TG (2013) The NuRD architecture. Cell Mol Life Sci 70, 3513–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Ivanek R, Lienert F and Schubeler D (2013) Methylation‐dependent and ‐independent genomic targeting principles of the MBD protein family. Cell 153, 480–492. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Chevret P and Furano AV (2000) L1 (LINE‐1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 6, 915–928. [DOI] [PubMed] [Google Scholar]
- Bojang P Jr, Anderton MJ, Roberts RA and Ramos KS (2014) De novo LINE‐1 retrotransposition in HepG2 cells preferentially targets gene poor regions of chromosome 13. Genomics 104, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojang P Jr, Roberts RA, Anderton MJ and Ramos KS (2013) Reprogramming of the HepG2 genome by long interspersed nuclear element‐1. Mol Oncol 7, 812–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz‐Prigge S, Farley AH, Moran JV and Kazazian HH Jr (2003) Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA 100, 5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butel JS and Rapp F (1966) Replication in simian cells of defective viruses in an SV40‐adenovirus “Hybrid” population. J Bacteriol 91, 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Shiffman S, Scharf DM, Dang Q and Shadel WG (2007) Daily smoking patterns, their determinants, and implications for quitting. Smok Stat‐ Exp Clin Psychopharmacol 15, 67–80. [DOI] [PubMed] [Google Scholar]
- Chang X, Fan Y, Karyala S, Schwemberger S, Tomlinson CR, Sartor MA and Puga A (2007) Ligand‐independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol Cell Biol 27, 6127–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denslow SA and Wade PA (2007) The human Mi‐2/NuRD complex and gene regulation. Oncogene 26, 543–5438. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Mathias SL, Nanthakumar E, Scott AF and Kazazian HH Jr (1991) Isolation of an active human transposable element. Science 254, 1805–1808. [DOI] [PubMed] [Google Scholar]
- Esnault C, Millet J, Schwartz O and Heidmann T (2006) Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res 34, 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH Jr and Boeke JD (1996) Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87, 905–916. [DOI] [PubMed] [Google Scholar]
- Gialitakis M, Tolaini M, Li Y, Pardo M, Yu L, Toribio A, Choudhary JS, Niakan K, Papayannopoulos V and Stockinger B (2017) Activation of the aryl hydrocarbon receptor interferes with early embryonic development. Stem Cell Reports 9, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR and Lieb JD (2007) FAIRE (Formaldehyde‐Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi FC, Rosser JM, Newkirk SJ, Yin J, Jiang X, Xing Z, Whitmore L, Bashir S, Ivics Z, Izsvak Z et al (2015) Retrotransposition creates sloping shores: a graded influence of hypomethylated CpG islands on flanking CpG sites. Genome Res 25, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Kuhara S, Takenaka O and Sakaki Y (1986) L1 family of repetitive DNA sequences in primates may be derived from a sequence encoding a reverse transcriptase‐related protein. Nature 321, 625–628. [DOI] [PubMed] [Google Scholar]
- Hohjoh H and Singer MF (1996) Cytoplasmic ribonucleoprotein complexes containing human LINE‐1 protein and RNA. EMBO 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y and Howard BH (2001) Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta‐L1. J Biol Chem 276, 6817–6824. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Shiraishi K, Eguchi A, Shibata H, Yoshimoto K, Mori T, Baba Y, Baba H and Suzuki M (2013) Long interspersed nucleotide element 1 hypomethylation is associated with poor prognosis of lung adenocarcinoma. Annals Thoracic Surg 96, 1790–1794. [DOI] [PubMed] [Google Scholar]
- Jiang CL, Jin SG and Pfeifer GP (2004) MBD3L1 is a transcriptional repressor that interacts with methyl‐CpG‐binding protein 2 (MBD2) and components of the NuRD complex. J Biol Chem 279, 52456–52464. [DOI] [PubMed] [Google Scholar]
- Kaur E, Gupta S and Dutt S (2007) Clinical implications of MTA proteins in human cancer. Cancer Metastasis Rev 33, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Kazazian HH Jr, Wong C, Youssoufian H, Scott AF, Phillips DG and Antonarakis SE (1988) Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332, 164–166. [DOI] [PubMed] [Google Scholar]
- Lai AY and Wade PA (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner MS, Begg GE, Speicher DW and Rauscher FJ 3rd (2000) Molecular determinants for targeting heterochromatin protein 1‐mediated gene silencing: direct chromoshadow domain‐KAP‐1 corepressor interaction is essential. Mol Cell Biol 20, 6449–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP and Ramos KS (2003) Redox regulation of a novel L1Md‐A2 retrotransposon in vascular smooth muscle cells. J Biol Chem 278, 28201–28209. [DOI] [PubMed] [Google Scholar]
- Mansfield RE, Musselman CA, Kwan AH, Oliver SS, Garske AL, Davrazou F, Denu JM, Kutateladze TG, Mackay JP (2011) Plant homeodomain (PHD) fingers of CHD4 are histone H3‐binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem 286, 11779–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B (1984) Significance of responses of the genome to challenge. Science 226, 792–801. [DOI] [PubMed] [Google Scholar]
- Mohd‐Sarip A, Teeuwssen M, Bot AG, De Herdt MJ, Willems SM, Baatenburg de Jong RJ, Looijenga LHJ, Zatrenu D, Bexstarosti K, Van Riet J et al (2017) DOC1‐dependent recruitment of NURD reveals antagonism with SWI/SNF during epithelial‐mesenchymal transition in oral cancer cells. Cell Rep 20, 61–75. [DOI] [PubMed] [Google Scholar]
- Montoya‐Durango DE, Liu Y, Teneng I, Kalbfleisch T, Lacy ME, Steffen MC and Ramos KS (2009) Epigenetic control of mammalian LINE‐1 retrotransposon by retinoblastoma proteins. Mutat Res 665, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya‐Durango DE, Ramos KA, Bojang P Jr, Ruiz L, Ramos IN and Ramos KS (2016) LINE‐1 silencing by retinoblastoma proteins is effected through the nucleosomal and remodeling deacetylase multiprotein complex. BMC Cancer 16, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD and Kazazian HH Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell 87, 917–927. [DOI] [PubMed] [Google Scholar]
- Nanez A, Ramos IN and Ramos KS (2011) A mutant Ahr allele protects the embryonic kidney from hydrocarbon‐induced deficits in fetal programming. Environ Health Perspect 119, 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JJ, Chang QS, Wang X, Son YO, Liu J, Zhang Z, Bi YY and Shi X (2011) Activation of Akt/GSK3β and Akt/Bcl‐2 signaling pathways in nickel‐transformed BEAS‐2B cells. Int J Oncol 39, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Ramírez J, Dege C, Kutateladze TG and Hagman J (2012) MBD2 and multiple domains of CHD4 are required for transcriptional repression by Mi‐2/NuRD complexes. Mol Cell Biol 32, 5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos KS, Montoya‐Durango DE, Teneng I, Nanez A, Stribinskis V (2011) Epigenetic control of embryonic renal cell differentiation by L1 retrotransposon. Birth Defects Res A Clin Mol Teratol 91:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Reyes E, Aispuro I, Tavera‐Garcia M, Field M, Moore S, Ramos I and Ramos KS (2017) LINE‐1 couples EMT programming with acquisition of oncogenic phenotypes in human bronchial epithelial cells. Oncotarget 8, 103828–103842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Reyes EM, Ramos IN, Tavera‐Garcia MA and Ramos KS (2016) The aryl hydrocarbon receptor agonist benzo(a)pyrene reactivates LINE‐1 in HepG2 cells through canonical TGF‐β1 signaling: implications in hepatocellular carcinogenesis. Am J Cancer Res 6, 1066–1077. [PMC free article] [PubMed] [Google Scholar]
- Saito K, Kawakami K, Matsumoto I, Oda M, Watanabe G and Minamoto T (2010) Long interspersed nuclear element 1 hypomethylation is a marker of poor prognosis in stage IA non‐small cell lung cancer. Cancer Res 16, 2418–2426. [DOI] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, Cooley T, Heath P, Smith KD and Margolet L (1987) Origin of the human L1 elements: proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stribinskis V and Ramos KS (2006) Activation of human long interspersed nuclear element 1 retrotransposition by benzo(a)pyrene, an ubiquitous environmental carcinogen. Can Res 66, 2616–2620. [DOI] [PubMed] [Google Scholar]
- Swergold GD (1990) Identification, characterization, and cell specificity of a human LINE‐1 promoter. Mol Cell Biol 10, 6718–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita K, Ogawa HI and Maeda T (2016) Structural chromosome aberrations cause swelling of the nucleus. Genes Environ 38, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneng I, Montoya‐Durango DE, Quertermous JL, Lacy ME and Ramos KS (2011) Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 6, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneng I, Stribinskis V and Ramos KS (2007) Context‐specific regulation of LINE‐1. Genes Cells 12, 1101–1110. [DOI] [PubMed] [Google Scholar]
- Wang Y1, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L et al (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672. [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD and Moran JV (2001) Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A, Jones AE, D'Brot A, Lu WJ, Kurtz P, Moran JV, Rakheja D, Chen KS, Hammer RE, Comerford SA, et al (2016) p53 genes function to restrain mobile elements. Genes Dev 30, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Cote J and Wang W (1998) NURD, a novel complex with both ATP‐dependent chromatin‐remodeling and histone deacetylase activities. Mol Cell 2, 851–861. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument‐Bromage H, Tempst P, Bird A and Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Optimization of L1‐ORF1 in HBECs by Western analysis.
Fig. S2. Time‐dependent analysis of L1 mRNA accumulation in HBECs challenged with 0.1 μm BaP.
Fig. S3. DNA and Sequence analysis of L1‐5′UTR sequences.
Fig. S4. Analysis of CYP1A1 expression after 12 h of BaP treatment by indirect immunofluorescence.