

Abstract
Background
Genome‐wide association studies have indicated that most of the currently identified disease and trait‐associated single nucleotide polymorphisms (SNPs) are intronic or intergenic. RegulomeDB is a recently developed database that provides functional annotations for regulatory features of SNPs located in non‐coding regions. We evaluated the potential regulatory SNPs in the EGFR gene region using RegulomeDB and their associations with prognosis after surgery in non‐small cell lung cancer (NSCLC) patients.
Methods
A total of 698 patients with surgically resected NSCLC were enrolled and seven SNPs were selected based on the RegulomeDB database. All SNPs were genotyped using SEQUENOM MassARRAY iPLEX assay.
Results
Among the seven SNPs evaluated, rs9642391 (EGFR ivs19+2851C>G) was significantly associated with survival outcome (adjusted hazard ratio [HR] for overall survival = 0.70, 95% confidence interval [CI] 0.56–0.87, P = 0.001; adjusted HR for disease‐free survival = 0.82, 95% CI 0.70–0.97, P = 0.02; under a codominant model). According to RegulomeDB, rs9642391C>G, which is located in intron 19 of EGFR, was predicted to influence the expression of GBAS but not EGFR. As predicted, rs9642391C>G was associated with GBAS (P = 0.024) but not EGFR messenger RNA expression in tumor tissues.
Conclusion
In conclusion, our study provides evidence that rs9642391C>G in the intron of EGFR is associated with GBAS expression and survival outcomes of patients with surgically resected early‐stage NSCLC.
Keywords: EGFR, lung cancer, RegulomeDB, survival
Introduction
EGFR is a cell surface protein with cytoplasmic kinase activity that transduces important growth factor signals from extracellular regions. EGFR is overexpressed in > 60% of non‐small cell lung cancers (NSCLCs) and plays a crucial role in regulating cell proliferation, survival, motility, and differentiation.1 In addition, EGFR mutations, including mutations in the tyrosine kinase domain (exons 18–21), and increased gene copy numbers, are frequently detected in NSCLC patients.2 Furthermore, specific types of activating mutations are associated with enhanced sensitivity to EGFR‐tyrosine kinase inhibitors (TKIs).3
Several studies have investigated the associations between EGFR polymorphisms and lung cancer.4, 5 However, previous studies have been performed to identify functional polymorphisms that are located in the coding, promoter, and untranslated regions of EGFR.6 Although a few studies have investigated polymorphisms in the EGFR intron or non‐coding regions, these studies mainly focused on single nucleotide polymorphisms (SNPs) in the intron 1 region, which have been shown to potentially influence promoter activity.7
Genome‐wide association studies (GWAS) have reported that most of the currently identified disease and trait‐associated SNPs are intronic or intergenic.8 Post‐GWAS efforts are now focused on performing functional characterization of these associations. Some newly discovered GWAS variants have been annotated as cell‐type‐specific gene enhancer elements by integrating knowledge of regulatory sequences (e.g. histone modification and DNase sensitivity).9, 10, 11 Pomerantz et al. reported that the 8q24 colorectal cancer risk variant, rs6983267, participates in long‐range physical interactions with the MYC proto‐oncogene.11
Data from the Encyclopedia of DNA elements (ENCODE) and the Roadmap Epigenome Project can provide better interpretation of the non‐coding sequences of the genome.12 As a result, the RegulomeDB database, which integrates data from ENCODE and other major databases, was developed to enable regulatory and epigenomic annotation of any set of variants derived from GWAS or genomic sequencing.13 These systematically annotated data have recently been used to elucidate the mechanisms by which GWAS variants that are located in non‐coding regions can influence clinically relevant phenotypes. These studies have identified the potential variants that demonstrate regulatory functions and have provided insights on the mechanisms that underlie the functions of these variants. To further verify the impact of regulatory SNPs in the EGFR gene on lung cancer prognosis, we evaluated the association of the potentially functional SNPs predicted by RegulomeDB and the survival outcomes of surgically resected NSCLC patients.
Methods
Patient characteristics
This study included NSCLC patients who underwent curative surgical resection at the Kyungpook National University Hospital between September 1998 and December 2007 (n = 316) and at the Seoul National University Bundang Hospital between September 2005 and March 2012 (n = 382). The clinicopathologic characteristics of the patients and associations with overall survival (OS) and disease‐free survival (DFS) are shown in Table S1. All patients were of Korean ethnicity. The institutional review boards of the two hospitals approved this study.
Single nucleotide polymorphism (SNP) selection and genotyping
RegulomeDB is a database that functionally annotates the regulatory features of SNPs in the human genome based on experimental datasets derived from ENCODE and other sources, as well as computational predictions and manual annotations.13 RegulomeDB employs a six‐category system to interpret functional variants. Categories 1–3 comprise SNPs with strong evidence of binding based on ChIP‐seq and DNase footprints. However, categories 4–6 still lack experimental evidence to demonstrate that the variant actually disrupts the binding site. We obtained a total of 942 SNPs within the EGFR gene region, NC_000007.13 (55086678.0.55279262, complement) by Genome Reference Consortium Human Build 37 patch release 13 (GRCh37.p13) assembly, using RegulomeDB (http://regulome.stanford.edu). We prioritized 124 SNPs that were classified under categories 1–3 because a lower score suggests stronger evidence of binding and indicates that a variant is located in a functional region. Among the 124 polymorphisms, seven SNPs (rs9642391C>G, rs1554718T>C, rs7792797A>C, rs11534100C>T, rs12718945G>T, rs11977660C>T, and rs2302535C>A) were selected after excluding 111 polymorphisms with minor allele frequency < 0.1 in HapMap JPT and six SNPs in strong linkage disequilibrium (r 2 > 0.8). All SNPs were genotyped using MassARRAY iPLEX assay (Sequenom Inc., San Diego, CA, USA) according to the manufacturer's instructions. For genotype validation, approximately 5% of the cohort samples were randomly selected for repeated genotyping via a restriction fragment length polymorphism assay by a different investigator; the results were 100% concordant.
Quantitative reverse transcription‐PCR
Quantitative reverse transcription (qRT)‐PCR was performed to determine the expression of human EGFR and GBAS. Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from fresh tumors and paired non‐malignant lung tissues of 144 NSCLC patients who underwent surgery at Kyungpook National University Medical Center. Real‐time PCR was performed in triplicate using QuantiFast SYBR Green PCR Master Mix (Qiagen, Hilden, Germany) in a LightCycler 480 (Roche Applied Science, Mannheim, Germany). For each target transcript, PCR was performed with a final reaction volume of 10 μL containing 1 μL of complimentary DNA, 5 μL of mix, and 1 μL of each primer. All complementary DNA samples were prepared in a 1:5 dilution to obtain results within the range of the standard. Relative messenger RNA (mRNA) expression levels were normalized against β‐actin expression and calculated via the 2−ΔΔCt method.
Statistical analysis
Hardy–Weinberg equilibrium was tested by comparing the observed and expected genotype frequencies using a goodness‐of‐fit χ2 test. The Kaplan–Meier method and log‐rank test were used to estimate OS and DFS. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated for multivariate statistical models, with adjustments for age, gender, smoking status, pathologic stage, and adjuvant therapy. A paired t‐test was used to compare EGFR and GBAS mRNA expression between tumor and normal tissues. All analyses were performed using SAS version 9.2 (SAS Institute Inc., Cary, NC, USA).
Results
The clinical characteristics of the 698 patients enrolled in this study are shown in Table S1. There were 209 deaths (29.9%), and the estimated five‐year OS and DFS rates for all patients were 60% (95% CI 55–65%) and 43% (95% CI 38–47%), respectively. Pathological stage was found to be significantly associated with OS and DFS (both log‐rank P [P L‐R] < 0.0001). Gender and smoking status were also associated with OS (P L‐R < 0.0001) and DFS (P L‐R = 0.03).
Statistical analysis of SNPs showed that all genotype frequencies were in Hardy–Weinberg equilibrium. Among the seven SNPs examined, only rs9642391 (EGFR ivs19+2851C>G) was significantly associated with OS and DFS (Table 1). The rs9642391 C>G variant was found to be significantly associated with increased survival (adjusted HR [aHR] for OS = 0.70, 95% CI 0.56–0.87, P = 0.001; aHR for DFS = 0.82, 95% CI 0.70–0.97, P = 0.02; under a codominant model) (Table 2, Fig 1).
Table 1.
Seven SNPs of log‐rank P in overall and disease‐free survival
| Overall survival | Disease‐free survival | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log‐rank P | Log‐rank P | |||||||||||
| SNPs† | Position† | Score‡ | Alleles | CR (%) | MAF | HWE‐P | Global | Dominant | Recessive | Global | Dominant | Recessive |
| rs9642391 | ivs19+2851 | 1d | CG | 99 | 0.36 | 0.78 | 0.01 | 0.01 | 0.03 | 0.05 | 0.06 | 0.03 |
| rs11534100 | ivs2−12589 | 2b | CT | 97 | 0.29 | 0.39 | 0.61 | 0.32 | 0.90 | 0.41 | 0.26 | 0.72 |
| rs12718945 | ivs2−17016 | 3a | GT | 96 | 0.30 | 0.94 | 0.78 | 0.80 | 0.59 | 0.38 | 0.18 | 0.96 |
| rs11977660 | ivs2−47673 | 2a | CT | 98 | 0.33 | 0.77 | 0.09 | 0.03 | 0.22 | 0.49 | 0.23 | 0.69 |
| rs2302535 | ivs2−55291 | 2b | CA | 99 | 0.12 | 0.53 | 0.21 | 0.10 | 0.27 | 0.59 | 0.40 | 0.76 |
| rs1554718 | ivs21−3449 | 2b | TC | 98 | 0.14 | 0.55 | 0.63 | 0.84 | 0.34 | 0.43 | 0.23 | 0.40 |
| rs7792797 | *5601 | 2b | AC | 98 | 0.34 | 0.70 | 0.61 | 0.96 | 0.34 | 0.42 | 0.50 | 0.20 |
Information about single nucleotide polymorphisms (SNPs) and SNP ID were obtained from the National Center for Biotechnology Information database (http://ncbi.nih.gov). The transcription start site was counted as +1 in reference sequences.
Score provided from the RegulomeDB database (http://regulome.stanford.edu).
CR, call rate; HWE, Hardy–Weinberg equilibrium; MAF, minor allele frequency.
Table 2.
Overall and disease‐free survival according to genotypes in patients with non‐small cell lung cancer
| Overall survival | Disease‐free survival | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Position | Genotype | No. of cases | No. of deaths (%) | 5Y‐OSR (%)† | HR (95% CI)‡ | P ‡ | No. of events (%) | 5Y‐DFSR (%)† | HR (95% CI)‡ | P ‡ |
| rs9642391 | ivs19+2851 | CC | 285 | 96 (33.7) | 56 | 1.00 | 140(49.1) | 41 | 1.00 | ||
| CG | 313 | 83 (26.5) | 63 | 0.70 (0.52–0.94) | 0.02 | 145(46.3) | 44 | 0.83 (0.66–1.05) | 0.13 | ||
| GG | 90 | 18 (20.0) | 67 | 0.49 (0.30–0.82) | 0.01 | 35(38.9) | 48 | 0.67 (0.46–0.98) | 0.04 | ||
| Dominant | 0.65 (0.49–0.86) | 0.003 | 0.79 (0.63–0.99) | 0.04 | |||||||
| Recessive | 0.59 (0.36–0.96) | 0.04 | 0.74 (0.52–1.06) | 0.10 | |||||||
| Codominant | 0.70 (0.56–0.87) | 0.001 | 0.82 (0.70–0.97) | 0.02 | |||||||
Proportion of survival derived from Kaplan–Meier analysis.
Calculated using multivariate Cox proportional hazard models adjusted for age, gender, smoking status, tumor histology, stage, and adjuvant therapy.
5Y‐DFSR, 5‐year disease‐free survival rate; 5Y‐OSR, 5‐year overall survival rate; CI, confidence interval; HR, hazard ratio.
Figure 1.
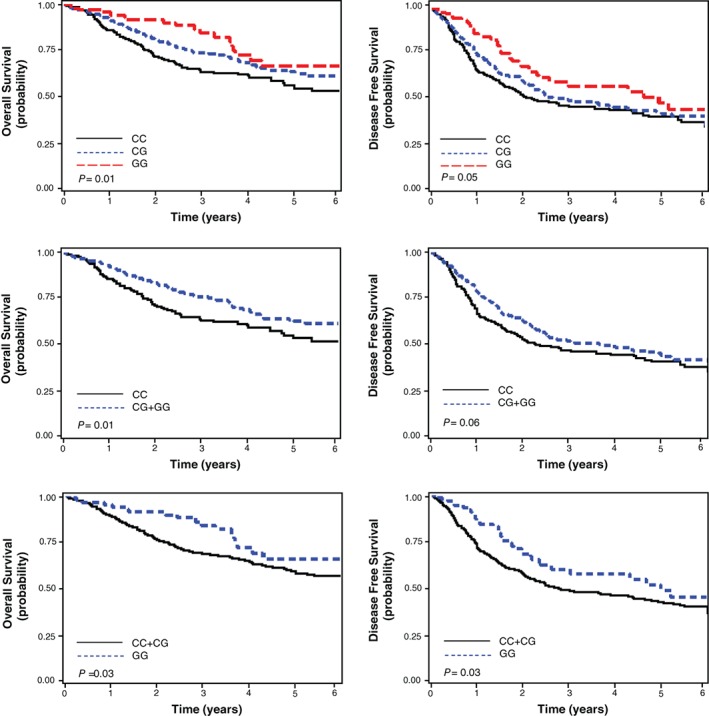
Kaplan–Meier plot of overall and disease‐free survival curves according to EGFR rs9642391C>G genotype. P values, log‐rank test.
The associations of the rs9642391C>G variant with survival outcomes were further analyzed after classifying the patients by age, gender, smoking status, histological type, and pathologic stage. The rs9642391C>G genotypes were significantly associated with survival outcomes in men, smokers, and squamous cell carcinoma patients (Table 3). In adenocarcinoma patients, a correlation was found between the rs9642391C>G genotypes and survival of smoking adenocarcinoma patients (aHR for OS = 0.67, 95% CI 0.44–1.02; P = 0.06). These findings suggested that the rs9642391C>G variant influences the prognosis of smoking lung cancer patients.
Table 3.
Stratified analysis of the effects of the rs9642391C>G genotypes under a codominant model for the minor allele at each polymorphism on survival outcomes by selected variables
| Overall survival | Disease‐free survival | |||
|---|---|---|---|---|
| SNP/Variables | HR (95% CI)† | P † | HR (95% CI)† | P † |
| Age (years) | ||||
| < 64 | 0.62 (0.43–0.88) | 0.01 | 0.80 (0.62–1.04) | 0.09 |
| ≥ 64 | 0.76 (0.58–1.00) | 0.05 | 0.84 (0.67–1.04) | 0.11 |
| Gender | ||||
| Male | 0.69 (0.54–0.87) | 0.002 | 0.80 (0.66–0.96) | 0.02 |
| Female | 0.83 (0.48–1.45) | 0.52 | 1.03 (0.72–1.48) | 0.86 |
| Smoking status | ||||
| Never | 0.94 (0.58–1.52) | 0.79 | 1.12 (0.81–1.54) | 0.51 |
| Ever | 0.66 (0.52–0.84) | 0.001 | 0.75 (0.62–0.91) | 0.004 |
| Histological type | ||||
| SCC | 0.66 (0.49–0.88) | 0.005 | 0.74 (0.59–0.94) | 0.02 |
| AC | 0.78 (0.55–1.08) | 0.14 | 0.90 (0.70–1.15) | 0.38 |
| Smoker AC | 0.67 (0.44–1.02) | 0.06 | 0.69 (0.49–0.96) | 0.03 |
| Never smoker AC | 1.16 (0.66–2.04) | 0.62 | 1.34 (0.94–1.93) | 0.11 |
| Pathologic stage | ||||
| I | 0.76 (0.51–1.15) | 0.20 | 0.85 (0.64–1.14) | 0.28 |
| II | 0.57 (0.39–0.82) | 0.003 | 0.71 (0.52–0.96) | 0.02 |
| IIIA | 0.77 (0.53–1.12) | 0.17 | 0.91 (0.67–1.23) | 0.54 |
Hazard ratios (HRs), 95% confidence intervals (CIs), and corresponding P values were calculated using multivariate Cox proportional hazard models, adjusted for other variables.
AC, adenocarcinoma; P H, P value test for homogeneity; SCC, squamous cell carcinoma; SNP, single nucleotide polymorphism.
The rs9642391C>G variant, which is located in intron 19 of EGFR, was predicted to influence the expression of GBAS based on RegulomeDB data (http://regulome.stanford.edu). Therefore, we evaluated EGFR and GBAS mRNA levels in 144 tumor and paired non‐malignant lung tissues to confirm these predicted results. The expression levels of EGFR and GBAS were significantly higher in tumor than in non‐malignant lung tissues (P = 2 × 10−6 and P = 9 × 10−8, respectively) (Fig 2a). GBAS expression levels were correlated with EGFR expression levels in tumor tissues (r = 0.61; P = 6.5 × 10−14 by Pearson's method) (Fig 2b). GBAS expression levels were significantly higher in GG than in CC and CG genotypes (P = 0.024, under a recessive model) (Fig 2c). However, no significant differences in EGFR expression levels were observed among patients with different rs9642391C>G genotypes.
Figure 2.
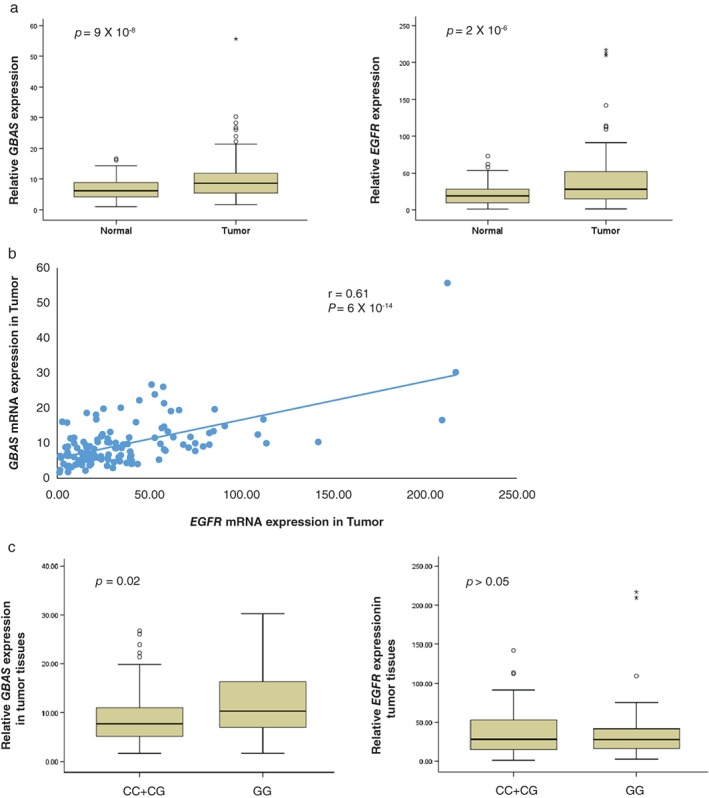
(a) GBAS and EGFR messenger RNA (mRNA) expression levels in tumor and non‐malignant lung tissues. *P < 0.001 by paired t‐test. (b) Relationship between GBAS and EGFR mRNA levels in lung tumor tissue (correlation coefficient = 0.61, P < 0.001 by Pearson's method). (c) GBAS and EGFR mRNA expression levels according to rs9642391C>G genotype in lung tumor tissues.
Discussion
The first step in identifying the function of non‐coding SNPs is to select the related SNPs that lie within regulatory regions, including enhancer and promoter regions.14, 15 In recent years, multiple consortia, such as the ENCODE and the Roadmap Epigenomics Mapping Consortium, have employed a variety of genome‐wide methods to study the chromatin states of non‐coding regions.16 The RegulomeDB database annotates SNPs with known and predicted regulatory elements of the Homo sapiens genome using these public datasets.
In the present study, we investigated the association between potential regulatory SNPs in the EGFR gene region and survival of surgically resected early‐stage NSCLC patients. RegulomeDB employs a six‐category system to interpret functional variants.13 To identify regulatory SNPs, we prioritized SNPs classified under categories 1–3 that were located in the EGFR region. Results revealed a significant association between EGFR rs9642391C>G and prognosis of early‐stage NSCLC patients. A previous study indicated that this polymorphism was associated with breast cancer mortality.17 Consistent with this finding, our results showed that patients harboring the rs9642391 GG or GC genotypes had significantly better OS and DFS compared with those carrying the rs9642391 CC genotype. Our result implies that rs9642391C>G may play an important role in the pathogenesis of lung cancer.
Although the selected SNPs were located in EGFR gene region, RegulomeDB predicted that rs9642391C>G influences the expression of GBAS but not EGFR. Our mRNA expression data was consistent with RegulomeDB. EGFR rs9642391 resides more than 700kb upstream of the GBAS gene, although they are both located in chromosome 7. rs9642391 in intron of EGFR belongs to category 1 variants that are known to express quantitative trait loci that have been experimentally shown to be associated with target gene expression. Using the database, we found that rs9642391 lies in a location that overlaps the binding site for DNA binding proteins, such as POLR2A and NFIC, and regulates GBAS expression (http://regulome.stanford.edu/snp/chr7/55245363). Gene expression is controlled by regulatory elements that can be located further away on the same chromosome or even on other chromosomes.18 Advances in technologies such as chromosome conformation capture (3C)‐based methods could detect physical interactions between chromosomes, providing convincing evidence for the widespread formation of long range interactions between genes and regulatory elements.19, 20 However, further studies are required to establish the functional relevance of these long‐range associations.
A previous study showed that GBAS is coamplified with EGFR in many cancer cell lines, including lung cancer.21 In the present study, EGFR and GBAS mRNA expression consistently showed a significant positive correlation. GBAS has an identifiable signal peptide and transmembrane motifs, as well as two tyrosine phosphorylation sites, suggesting that the encoded protein acts as a substrate for tyrosine kinases.21 Some studies have reported that the GBAS protein is localized to mitochondria and plays a role in oxidative phosphorylation.22, 23 However, very little is known about the function of the GBAS gene in cancer. According to The Cancer Genome Atlas database and other sources, GBAS expression is upregulated compared to normal tissues in many types of cancer, including lung cancer. Although this may suggest that GBAS could be a potential oncogene, more direct evidence is needed to confirm its oncogenic function. A study reported that GBAS overexpression increases the centrosome amplification rate and facilitates migration and invasion in bladder cancer cell lines, and that high GBAS expression correlates with poor survival in bladder cancer patients.24 However, when survival curves were generated using the Kaplan–Meier Plotter online tool (http://kmplot.com/analysis/), which utilizes public databases, higher GBAS expression was significantly associated with better survival in lung and gastric cancers and poor survival in breast and ovarian cancers.25 Therefore, GBAS may have tissue and context specific functions in the pathogenesis of various types of cancers. Future studies are warranted to elucidate the role of GBAS in lung carcinogenesis and to validate the mechanism of association between the regulatory variant and GBAS.
In conclusion, our results show that the rs9642391C>G variant, which is located in the intron region of EGFR, is associated with GBAS expression and survival outcomes in surgically resected early‐stage NSCLC patients.
Disclosure
No authors report any conflict of interest.
Supporting information
Table S1 Univariate analysis of overall and disease‐free survival by age, gender, smoking status, histological type, pathologic stage, and adjuvant therapy.
Acknowledgments
This research was supported in part by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C0402) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF‐2015R1D1A1A01058280).
Contributor Information
Shin Yup Lee, Email: shinyup@knu.ac.kr.
Jae Yong Park, Email: jaeyong@knu.ac.kr.
References
- 1. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–81. [DOI] [PubMed] [Google Scholar]
- 2. Gazdar AF. Activating and resistance mutations of EGFR in non‐small‐cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009; 28 (Suppl 1): S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu W, Wu X, Zhang W et al Relationship of EGFR mutations, expression, amplification, and polymorphisms to epidermal growth factor receptor inhibitors in the NCI60 cell lines. Clin Cancer Res 2007; 13: 6788–95. [DOI] [PubMed] [Google Scholar]
- 5. Brandt B, Meyer‐Staeckling S, Schmidt H, Agelopoulos K, Buerger H. Mechanisms of EGFR gene transcription modulation: Relationship to cancer risk and therapy response. (Published erratum appears in Clin Cancer Res 2007; 13:3759) Clin Cancer Res 2006; 12:7252–60. [DOI] [PubMed] [Google Scholar]
- 6. Buckland PR. The importance and identification of regulatory polymorphisms and their mechanisms of action. Biochim Biophys Acta 2006; 1762: 17–28. [DOI] [PubMed] [Google Scholar]
- 7. Jou YS, Lo YL, Hsiao CF et al Association of an EGFR intron 1 SNP with never‐smoking female lung adenocarcinoma patients. Lung Cancer 2009; 64: 251–6. [DOI] [PubMed] [Google Scholar]
- 8. Hindorff LA, Sethupathy P, Junkins HA et al Potential etiologic and functional implications of genome‐wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009; 106: 9362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cowper‐Sal lari R, Zhang X, Wright JB et al Breast cancer risk‐associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet 2012; 44: 1191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schödel J, Bardella C, Sciesielski LK et al Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat Genet 2012; 44: 420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pomerantz MM, Ahmadiyeh N, Jia L et al The 8q24 cancer risk variant rs6983267 demonstrates long‐range interaction with MYC in colorectal cancer. Nat Genet 2009; 41: 882–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyle AP, Hong EL, Hariharan M et al Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012; 22: 1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol 2012; 30: 1095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tak YG, Farnham PJ. Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non‐coding regions of the human genome. Epigenetics Chromatin 2015; 30: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roadmap Epigenomics Consortium . Integrative analysis of 111 reference human epigenomes. Nature 2015; 518: 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Connor AE, Baumgartner RN, Baumgartner KB et al Epidermal growth factor receptor (EGFR) polymorphisms and breast cancer among Hispanic and non‐Hispanic white women: The Breast Cancer Health Disparities Study. Int J Mol Epidemiol Genet 2013; 4: 235–49. [PMC free article] [PubMed] [Google Scholar]
- 18. Miele A, Dekker J. Long‐range chromosomal interactions and gene regulation. Mol Biosyst 2008; 4: 1046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bartkuhn M, Renkawitz R. Long range chromatin interactions involved in gene regulation. Biochim Biophys Acta 2008; 1783: 2161–6. [DOI] [PubMed] [Google Scholar]
- 20. Wei Z, Huang D, Gao F et al Biological implications and regulatory mechanisms of long‐range chromosomal interactions. J Biol Chem 2013; 288: 22369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang XY, Smith DI, Liu W, James CD. GBAS, a novel gene encoding a protein with tyrosine phosphorylation sites and a transmembrane domain, is co‐amplified with EGFR . Genomics 1998; 49: 448–51. [DOI] [PubMed] [Google Scholar]
- 22. Martherus RS, Sluiter W, Timmer ED, VanHerle SJ, Smeets HJ, Ayoubi TA. Functional annotation of heart enriched mitochondrial genes GBAS and CHCHD10 through guilt by association. Biochem Biophys Res Commun 2010; 402: 203–8. [DOI] [PubMed] [Google Scholar]
- 23. Smits P, Rodenburg RJ, Smeitink JA, van der Heuvel LP. Sequence variants in four candidate genes (NIPSNAP1, GBAS, CHCHD1 and METT11D1) in patients with combined oxidative phosphorylation system deficiencies. J Inherit Metab Dis 2010; 33(Suppl 3): S13–9. [DOI] [PubMed] [Google Scholar]
- 24. Matsumoto H, Fujii N, Kobayashi K et al GBAS as a novel regulator for targeting centrosome amplification and malignant behavior in bladder cancer. J Clin Oncol 2016; 34 (2 Suppl): Abstract 429. [Google Scholar]
- 25. Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. (Published erratum appears in PLoS One 2014;9:e111842) PLoS One 2013; 8: e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Univariate analysis of overall and disease‐free survival by age, gender, smoking status, histological type, pathologic stage, and adjuvant therapy.