

Abstract
Background
Increasing evidence has demonstrated that circular RNAs (circRNAs) may play an important role in oncogenesis and tumor development; however, their role in lung adenocarcinoma (LUAD) remains unclear. We identified the differentially expressed circRNAs in LUAD and investigated the potential mechanisms for cancer progression.
Methods
We examined differentially expressed circRNAs in LUAD and paired normal tissues using downloaded circRNA microarrays from the Gene Expression Omnibus. We constructed gene co‐expression networks based on the degree of Pearson correlation to predict the critical circRNA in LUAD. Gene Ontology analysis was performed on the genes in the network. We observed one novel circRNA upregulated in LUAD, hsa_circ_0000792, as well as its potential sponged microRNA, miR‐375. Subsequent real‐time quantitative PCR was used to verify the bioinformatics analysis.
Results
Several circRNAs showed significantly different expression levels in LUAD tissues. Real‐time quantitative PCR and further co‐expression network analysis of 42 matched tissue samples showed a significant difference in expression between LUAD and normal tissues in hsa_circ_0000792 (P < 0.001). We built a network of hsa_circ_0000792‐targeted miRNA gene interactions, including miR‐375 and the corresponding messenger RNAs. Gene Ontology analysis revealed that hsa_circ_0000792 could participate in signal transduction and cell communication during LUAD development. Larger area under the curve by receiver operating characteristic curve analysis of hsa_circ_0000792 and miR‐375 (0.815 and 0.772, respectively) in LUAD indicated greater potential as biomarkers.
Conclusions
We identified hsa_circ_0000792 as a potential LUAD biomarker; however, further studies are required to determine the mechanism of this circRNA in LUAD development.
Keywords: Circular RNA, lung adenocarcinoma, microarray expression, prognostic biomarker
Introduction
Lung cancer is one of the most common and fatal cancers in the world. In 2012, up to 1.8 million new cases were diagnosed, causing more than 1.6 million deaths globally, representing a sharp increase from 2008.1 Non‐small cell lung cancer, including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), remains the most common pathological subtype of lung cancer. Different from other histological types, LUAD is the most common subtype among never‐smokers.2 Despite vast research into cancer therapy, LUAD patient survival is still far from satisfactory.3 Patients with LUAD lacking specific clinical symptoms are usually diagnosed at a very late stage, leaving little opportunity for effective treatment. Thus, identifying a novel cancer‐specific biomarker for LUAD patients could help make timely diagnoses and guide clinical treatment.
Circular RNAs (circRNAs) are a novel type of endogenous non‐coding RNA molecule.4 CircRNAs were located in 1976 in an RNA virus as the by‐products of abnormal RNA splicing and splicing errors as a result of dysregulation.5 With newly developed technologies of high throughput sequencing and computational approaches, particularly RNA‐sequencing, up to 30 000 circRNAs have been identified.6 As a result of recent discovery of their abundance, various functions, and tissue‐specific expression, circRNAs have drawn much interest. Compared with linear RNA expression, circRNA expression is extraordinary high in isoforms of the same gene.7 A recent study proved that some circRNAs regulate gene expression by sponging specific microRNAs (miRNAs) or relieving their repression of messenger RNA (mRNA) targets.8 Some circRNAs can even encode peptides.9 It is reasonable to hypothesize that circRNA dysregulation influences the progress of various diseases, including cancer.10 Importantly, the unique construction of circRNAs makes them insensitive to ribonucleases; therefore circRNAs can exist in tissues and serum, and as a result could serve as biomarkers for human cancer.11 Increasing evidence demonstrates that the circRNA expression profile is different in tumor tissues compared to their adjacent non‐tumor tissues in many cancers,1 including LUAD.12 Recent studies have demonstrated that dysregulation of circRNAs has a potential correlation with development and survival in multiple cancers, including LUAD.13
In this study, we investigated circRNA expression profiles in LUAD and paired noncancerous tissues from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). We identified several circRNAs dysregulated in LUAD tissues compared to adjacent normal lung tissues. These circRNAs may play an important role in the development of LUAD malignancy and progression. We identified and validated a potential sponged microRNA, miR‐375, and a new circRNA, hsa_circ_ 0000729, involved in the process of LUAD development and propose that these new circRNAs could be used as potential targets in LUAD therapy.
Methods
Identification of differentially expressed RNAs in lung adenocarcinoma (LUAD) samples
RNA sequencing data from five LUAD and adjacent matched samples were obtained from the GEO. GSE101586 from the platform GPL19978 was analyzed using Agilent‐069978 Arraystar Human CircRNA microarray V1 (Agilent Technologies, Santa Clara, CA, USA). Five LUAD patients were included in this study and provided five LUAD and five non‐tumorous adjacent lung tissue samples.
R version 3.3.3 (R Foundation for Statistical Computing, Vienna, Austria) was used to perform statistical analysis. The limma package (https://git.bioconductor.org/packages/limma) was used to process data. Differences in expression of circRNAs between LUAD and adjacent normal tissues were determined by fold changes and associated P values. Dysregulated RNA was assigned fold changes > 2 or < 0.5, respectively, with P < 0.05.
Patient samples
Forty‐two LUAD and paired adjacent normal tissue samples were obtained from the Department of Thoracic Surgery, Qingdao University Hospital. The clinical information of LUAD patients is summarized in Table 1. Samples were immediately snap‐frozen in liquid nitrogen after resection and then stored at −80°C until RNA extraction. The diagnosis of LUAD was confirmed pathologically. Thirty‐four pairs of samples were used for quantitative real‐time (qRT) PCR validation. All patients provided informed consent prior to sample collection and the ethics committee of Qingdao University approved the study.
Table 1.
Correlation between hsa_circ_0000792 expression and clinical parameters in LUAD
| hsa_circ_0000792 expression | ||||
|---|---|---|---|---|
| Clinicopathological features | Number of patients | Low, n(%) | High, n(%) | P |
| Age | ||||
| < 60 | 20 | 11 (55) | 9 (45) | 0.9241 |
| > 60 | 22 | 10 (45.5) | 12 (54.5) | |
| Gender | ||||
| Male | 18 | 9 (50) | 9 (50) | 0.6551 |
| Female | 24 | 10 (41.7) | 14 (58.3) | |
| T stage | ||||
| T1 | 30 | 13 (43.4) | 17 (56.6) | 0.03407 |
| ≥ T2 | 12 | 7 (58.3) | 5 (41.7) | |
| Distant metastasis | ||||
| Absent | 4 | 2 (50) | 2 (50) | 0.0000196 |
| Present | 38 | 16 (42.1) | 22 (57.9) | |
| Smoking status | ||||
| Non‐smoker | 23 | 8 (34.8) | 15 (65.2) | 0.4298 |
| Smoker | 19 | 9 (47.4) | 10 (52.6) | |
LUAD, lung adenocarcinoma.
Construction of the ceRNA network and functional assessment
The ceRNA network was constructed based on the relationship among circRNAs, messenger RNAs (mRNAs), and microRNAs (miRNAs). Targetscan (http://www.targetscan.org/) and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/) were used to predict the mRNAs targeted by miRNAs, while circRNA‐miRNA interaction was predicted using CircNet tools (http://circnet.mbc.nctu.edu.tw/). Furthermore, miRNAs that regulated expression of both circRNAs and mRNAs were chosen to construct the network. Cytoscape version 3.4 (National Institute of General Medical Sciences, Bethesda, MD, USA) was used to construct and visualize the ceRNA network.14 To understand the underlying mechanism of the ceRNA network, the Database for Annotation, Visualization and Integrated Discovery (http://david.abcc.ncifcrf.gov/) was used for functional enrichment analysis, in which Gene Ontology analysis was defined at a significance level of P < 0.01 and an enrichment score > 2.
Quantitative real‐time reverse transcription PCR
Total RNAs were extracted from tissue samples with Trizol reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcription was performed using a QuantiTect Reverse Transcription Kit (Invitrogen, USA). CircRNA expression was measured by qRT‐PCR using the SYBR Green PCR kit on an ABI StepOnePlus thermocycler (ABI, Oyster Bay, NY, USA). The PCR primers used in this study were as follows: circRNA_0000792, 5′‐TCT GAT GGT GGC TCC TAT TCG GGA‐3′ (forward) and 5′‐TCG TCC GTA GTC CCA GAC TGG TGC‐3′ (reverse); miR‐375, 5′‐AGC CGT TTG TTC GTT CGG CT‐3′ (forward) and 5′‐GTG CAG GGT CCG AGG T‐3′ (reverse); and U6 RNA, 5′‐CGC TTC GGC AGC ACA TAT AC‐3′ (forward) and 5′‐TTC ACG AAT TTG CGT GTC AT‐3′ (reverse). Samples were run in triplicate for analysis. Data was calculated using the comparative 2−ΔΔCT method.
Statistical analysis
Results were all expressed as means ± standard deviation. Differences between groups were assessed using a paired t‐test. Receiver operating characteristic (ROC) curves and logistic regression analysis were utilized to analyze specificity, sensitivity, and area under the curve (AUC) with 95% confidence intervals (CIs). A P value < 0.05 was considered statistically significant. All data were analyzed using R software version 3.3.3 (R Foundation, Austria), Excel 2013 (Microsoft, Washington, DC, USA) and GraphPad Prism 6 (GraphPad, La Jolla, CA, USA).
Results
Identification of differentially expressed circular RNAs (circRNAs)
We initially performed differential expression analysis of the patient samples by comparing circRNA expression in LUAD and adjacent normal lung tissues in the GEO. The criteria to identify differentially expressed circRNAs were fold changes > 2 and P values < 0.05. We obtained 98 differentially expressed circRNAs (25 upregulated and 73 downregulated) between LUAD and adjacent normal lung tissues (Fig 1). These differentially expressed circRNAs were selected to construct the ceRNA network.
Figure 1.
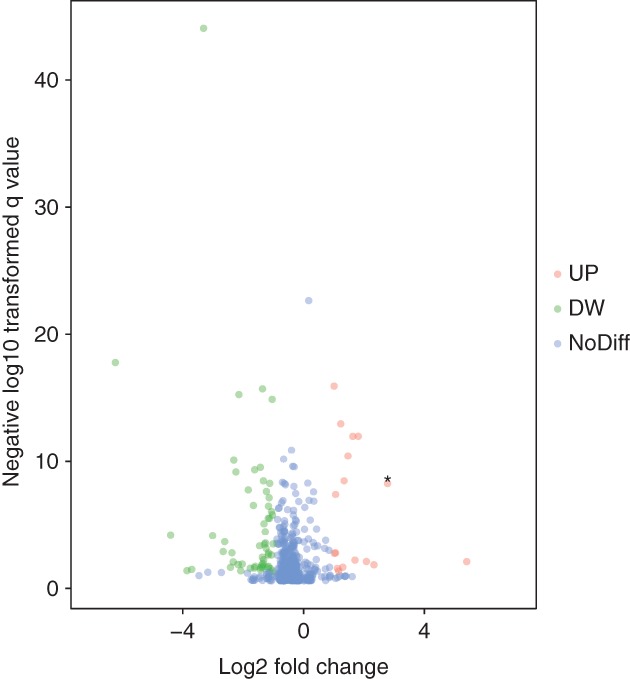
Differentially expressed circular RNAs (circRNAs) in five lung cancer and adjacent normal tissue samples. Volcano plots of aberrantly expressed circRNAs. Ninety‐eight circRNAs showed significantly aberrant expression (≥ two‐fold change); P < 0.05 and a false discovery rate < 0.05. The red and green nodes correspond to upregulated (UP) and downregulated (DW) circRNAs, respectively. The labeled nodes in the plots represent the significantly differentially expressed circRNA, hsa_circ_0000792.
Co‐expression network to predict critical circRNAs in LUAD tissues
To date only a few of the functions of circRNAs have been identified. Thus, we built a gene co‐expression network on the basis of the degree of correlation to predict the critical circRNAs in LUAD tissues. The core of the co‐expression networks was constructed in accordance with the normalized intensities of circRNAs and mRNAs. In the co‐expression network, each node represents one gene in the network, with a string connecting every two related genes, meaning a tight correlation between these genes and a possible regulatory relationship (Fig 2a). We performed Gene Otology analysis with the genes in the network and identified that hsa_circ_0000792 had a high possibility of involvement in the biological function process of signal transduction and cell communication (Fig 2b). The co‐expression network showed that hsa_circ_0000729 was closely correlated with a novel microRNA, miR375, suggesting an interaction between critical circRNAs and miRNAs in LUAD.
Figure 2.
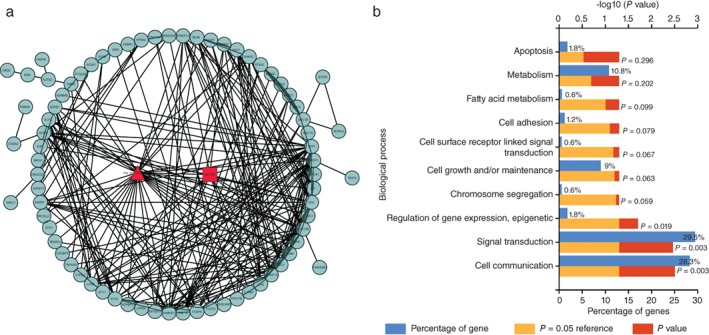
(a) The predicted hsa_circ_0000792 targeted circular RNA‐microRNA‐messenger RNA (circRNA‐miRNA‐mRNA) gene network based on sequence‐pairing prediction. A round node represents a protein‐coding gene, a triangular node represents a circRNA, and a squad node represents an miRNA. Lines between two nodes indicate correlated interactions. (b) Gene Ontology analysis based on the hsa_circ_0000792‐miRNA‐mRNA network. The top 10 significantly enriched biological processes and their scores (logarithm of P value) are listed on the x and y axes, respectively. The horizontal axis represents the significant level of Gene Ontology.
Validation of the differential expression levels of circRNAs
In order to verify the accuracy of our predictions, we measured the expression level of the critical circRNA, hsa_circ_0000729, and its potential sponged microRNA, miR‐375, in 42 clinical samples of LUAD and paired adjacent normal liver tissues by qRT‐PCR. The expression levels of hsa_circ_0000729 in LUAD tissues were significantly higher than those in the corresponding normal tissues; the opposite result occurred in the microRNA group (Fig 3a). The level of hsa_circ_0000792 expression was associated with LUAD clinical stage and distant metastasis (Table 1). Upregulated hsa_circ_0000729 expression means a lower expression of its potential sponge miR‐375 (Fig 3b). All findings were consistent with those of microarray analysis.
Figure 3.
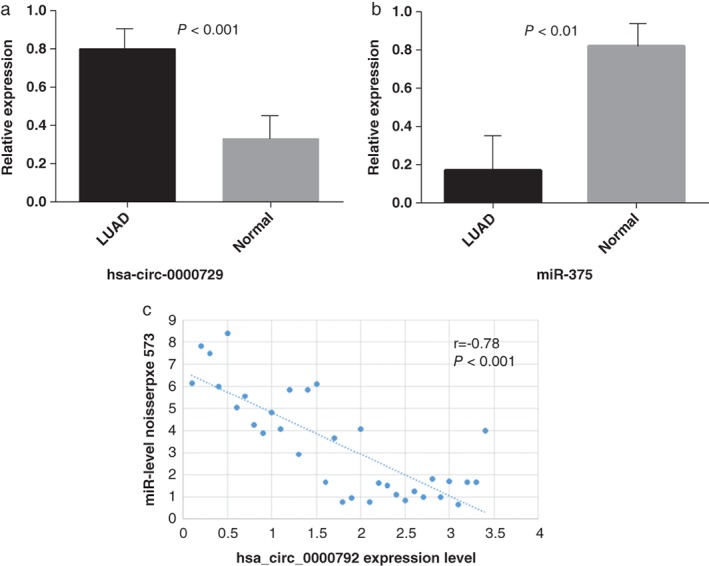
Relative expression of hsa_circ_0000792 and miR‐375 by quantitative real‐time PCR. (a) The hsa_circ_0000792 expression levels are significantly higher (P < 0.001) and (b) the miR‐375 expression levels are significantly lower than in corresponding normal tissues (P < 0.01). (c) Correlation between hsa_circ_0000792 and miR‐275 expression in tissue specimens. LUAD, lung adenocarcinoma.
Diagnostic value of hsa_circ_0000729 and miR‐375
We further assessed the diagnostic value of the novel circRNA and its potential sponged microRNA for LUAD by plotting an ROC curve (Fig 4). We found that hsa_circ_0000729 and miR‐375 had significant diagnostic accuracy, with area under the curve (AUC) values of 0.815 (95% CI 0.805–0.862) and 0.772 (95% CI 0.707–0.806), respectively.
Figure 4.
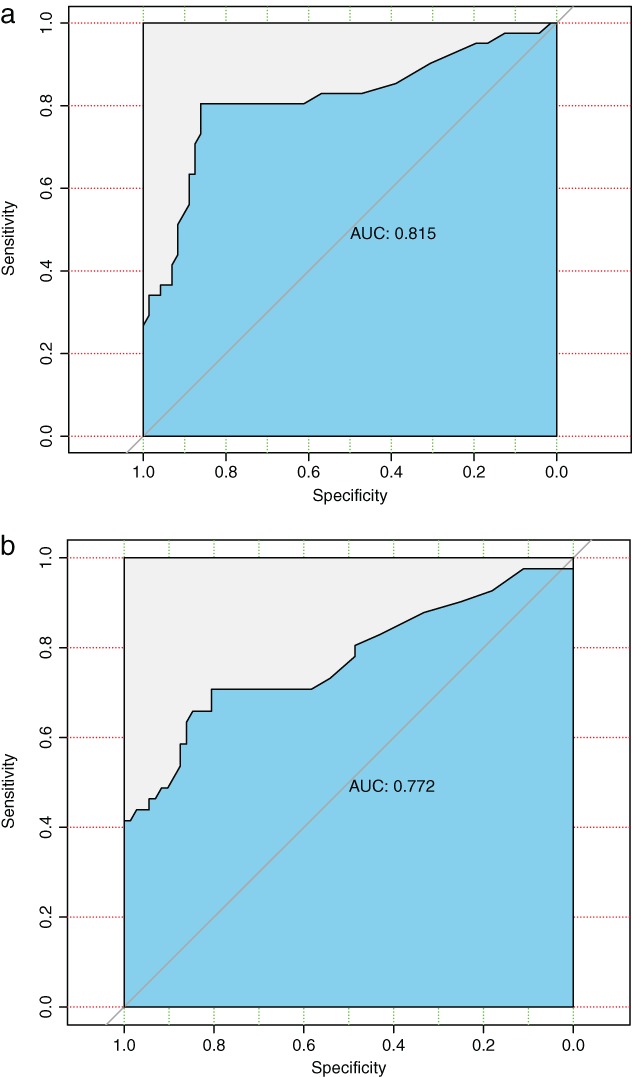
Receiver operating characteristic curve analysis of hsa_circ_0000792 (a) and miR‐375 (b) for discriminative ability between lung cancer cases (hsa_circ_0000792: area under the curve [AUC] = 0.815, 95% confidence interval [CI] 0.805–0.862; P < 0.001) and normal controls (miR‐;375: AUC = 0.772, 95% CI 0.707–0.806; P < 0.001).
Discussion
As a result of the discovery of high‐throughput RNA sequencing and various bioinformatics methods, increasing attention has been focused on circRNAs, the result of splicing errors, in cancer‐related research.15 Recent studies have shown that circRNAs exhibit tissue and stage specific expression; therefore, dysregulation of circRNAs in various cancers could play an important role in cancer initiation and development.16 Further, inaccurate expression of circRNAs is related to malignant cancer phenotypes. Different from the traditional biomarkers, circRNAs can compensate for low organ specificity as a result of diversity and tissue‐specific expression. Studies have proven that some circRNAs play vital roles in the process of tumor development acting as miRNA sponges, such as CDR1as or ciRS‐7 and circ‐ITCH.17, 18 For example, the first study of circRNAs found that circRNAs acted as an miRNA sponge in CDR1, with at least 70 conserved mir‐7 binding sites.19 When miRNA expression significantly exceeds that of the ceRNA, most targeted mRNAs are totally suppressed. A great deal of evidence suggests that dysregulation of miRNA expression plays an essential role in the development of various human cancers. A recent study found that circ‐ITCH is upregulated in lung cancer tissue and can act as a sponge of miR‐214 and miR‐7, competing with the two miRNAs for binding sites of ITCH, thus inhibiting activation of the Wnt/β‐catenin signaling pathway.20
In this study, we explored a downloaded microarray expression profile of circRNAs in five LUAD and paired noncancerous tissue samples to investigate the underlying molecular mechanisms of LUAD. Ninety‐eight DE‐circRNAs (25 upregulated and 73 downregulated) were significantly differentially expressed, revealing an important role of circRNAs in LUAD. Bioinformatics methods identified a novel circRNA, hsa_circ_0000792, which was significantly upregulated and may play an important role in LUAD. In particular, after further RT‐PCR experimentation, we identified that hsa_circ_0000792 is indeed significantly upregulated in LUAD tissue. We constructed a network of circRNA‐miRNA interactions and found that miR‐375 interacted with most nodes. Based on our assumption that hsa_circ_0000792 functions as an miRNA sponge for its predicted targeted mRNAs, we examined the target genes using GO analysis. We found that there is a high possibility that hsa_circ_0000792 is involved in the biological function process of signal transduction and cell communication in LUAD development. Similarly, we performed RT‐PCR and determined that miR‐375 is significantly downregulated in LUAD tissues, which has a negative correlation with the circRNA. Therefore, we speculate that hsa_circ_0000792 might competitively bind with miR‐375 and abrogate the inhibiting effect on their associated target genes.
The circRNAs profiled in the GEO and examined in this study have not previously been investigated in relation to their affect on LUAD. Researchers have found that microRNA 375 can regulate proliferation and migration in colon cancer by suppressing the CTGF‐EGFR signaling pathway.21 Meanwhile, a recent study reported that epigenetic repression of miR‐375 may be the dominant mechanism for sustaining activation of the PDPK1/RPS6KA3 signaling axis in multiple myeloma.22 Accordingly, we present the theory that the mRNA‐microRNA‐circRNA axis may be the potential molecular regulatory mechanism of LUAD. Further research on the novel circRNA mechanisms and other associated functions is required.
In summary, the results of our study reveal the hsa_circ_0000792 level in LUAD patients and suggest its potential value as a biomarker for LUAD diagnosis and prognosis. Further, our results indicate that hsa_circ_0000792 may be utilized as a novel biomarker for LUAD with a high degree of accuracy, specificity, and sensitivity. More prospective studies are required to validate these results.
Disclosure
No authors report any conflict of interest.
Acknowledgment
The Department of Thoracic Surgery, Affiliated Hospital of Qingdao University supported this study.
Contributor Information
Jia Liu, Email: dadaliujia@qdu.edu.cn.
Wenjie Jiao, Email: xwkjiao@126.com.
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Wu C, Xu B, Zhou Y et al Correlation between serum IL‐1beta and miR‐144‐3p as well as their prognostic values in LUAD and LUSC patients. Oncotarget 2016; 7: 85876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30. [DOI] [PubMed] [Google Scholar]
- 4. Yang F, Zhu P, Guo J et al Circular RNAs in thoracic diseases. J Thorac Dis 2017; 9: 5382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patop IL, Kadener S. circRNAs in cancer. Curr Opin Genet Dev 2018; 48: 121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang M, Yu F, Wu W et al Circular RNAs: A novel type of non‐coding RNA and their potential implications in antiviral immunity. Int J Biol Sci 2017; 13: 1497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang G, Li S, Yang N, Zou Y, Zheng D, Xiao T. Recent progress in circular RNAs in human cancers. Cancer Lett 2017; 404: 8–18. [DOI] [PubMed] [Google Scholar]
- 8. Hansen TB, Jensen TI, Clausen BH et al Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495: 384–8. [DOI] [PubMed] [Google Scholar]
- 9. Granados‐Riveron JT, Aquino‐Jarquin G. The complexity of the translation ability of circRNAs. Biochim Biophys Acta 2016; 1859: 1245–51. [DOI] [PubMed] [Google Scholar]
- 10. Szabo L, Salzman J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat Rev Genet 2016; 17: 679–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, Li C, Tan C, Liu X. Circular RNAs: A new frontier in the study of human diseases. J Med Genet 2016; 53: 359–65. [DOI] [PubMed] [Google Scholar]
- 12. Yao JT, Zhao SH, Liu QP et al Over‐expression of circRNA_100876 in non‐small cell lung cancer and its prognostic value. Pathol Res Pract 2017; 213: 453–6. [DOI] [PubMed] [Google Scholar]
- 13. Jin X, Guan Y, Sheng H, Liu Y. Crosstalk in competing endogenous RNA network reveals the complex molecular mechanism underlying lung cancer. Oncotarget 2017; 8: 91270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franz M, Lopes CT, Huck G, Dong Y, Sumer O, Bader GD. Cytoscape.js: A graph theory library for visualisation and analysis. Bioinformatics 2016; 32: 309–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barrett SP, Salzman J. Circular RNAs: Analysis, expression and potential functions. Development 2016; 143: 1838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell‐type specific features of circular RNA expression (Published erratum appears in PLoS Genet 2013; 9: 10.1371/annotation/f782282b-eefa-4c8d-985c-b1484e845855). PLoS Genet 2013; 9: e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang X, Xiong Q, Wu Y, Li S, Ge F. Quantitative proteomics reveals the regulatory networks of circular RNA CDR1as in hepatocellular carcinoma cells. J Proteome Res 2017; 16: 3891–902. [DOI] [PubMed] [Google Scholar]
- 18. Guo W, Zhang J, Zhang D et al Polymorphisms and expression pattern of circular RNA circ‐ITCH contributes to the carcinogenesis of hepatocellular carcinoma. Oncotarget 2017; 8: 48169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang W, Ji M, He G et al Silencing CDR1as inhibits colorectal cancer progression through regulating microRNA‐7. Onco Targets Ther 2017; 10: 2045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wan L, Zhang L, Fan K et al Circular RNA‐ITCH suppresses lung cancer proliferation via inhibiting the Wnt/beta‐catenin pathway. Biomed Res Int 2016; 2016: 1579490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alam KJ, Mo JS, Han SH et al MicroRNA 375 regulates proliferation and migration of colon cancer cells by suppressing the CTGF‐EGFR signaling pathway. Int J Cancer 2017; 141: 1614–29. [DOI] [PubMed] [Google Scholar]
- 22. Tatekawa S, Chinen Y, Ri M et al Epigenetic repression of miR‐375 is the dominant mechanism for constitutive activation of the PDPK1/RPS6KA3 signalling axis in multiple myeloma. Br J Haematol 2017; 178: 534–46. [DOI] [PubMed] [Google Scholar]