

Abstract
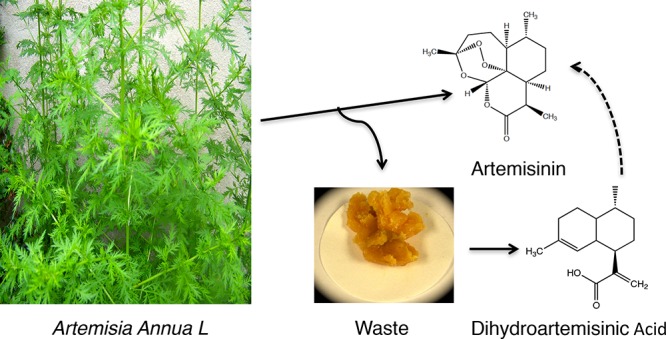
High-performance liquid chromatography, liquid chromatography–mass spectrometry, and gas chromatography–mass spectrometry methods were developed to analyze the process waste streams of Artemisia Annua extraction. Results from these methods suggested that the final waste from the extraction process could serve as a source of dihydroartemisinic acid (DHAA) that could be converted to additional artemisinin. Two additional impurities were isolated and identified in the waste material as well as in A. annua leaf samples. That these impurities also appear as side-products in chemical transformations of DHAA to artemisinin supports the conclusion that the in vivo transformation proceeds as nonspecific oxidations. These impurities do not appear in isolated artemisinin. A simple, high-yielding procedure for recovery of DHAA from the primary waste stream was developed.
Introduction
Artemisinin is a natural product that serves as the starting material for antimalarial active pharmaceutical ingredients in artemisinin combination therapy treatments. As an agricultural product, its supply and price may be subject to disruptive price fluctuations. In response to these concerns about the impact of price fluctuations on efforts to treat and even eradicate malaria, the Bill and Melinda Gates Foundation funded a cooperative effort to develop an alternate source of artemisinin. The results of the research toward semisynthetic artemisinin (SSA) represent a technical tour de force encompassing development of fermentation processes1 to produce artemisinic acid (AA, 1) and a chemical conversion to artemisinin developed by Sanofi2 (3). In the semisynthetic approach (Scheme 1), AA produced by fermentation is reduced selectively to dihydroartemisinic acid (2) (DHAA), which is then converted to artemisinin via a chemical conversion which mimics the synthetic pathway used by the plant.3
Scheme 1.

Despite the technical merit of SSA, it has not been widely adopted, as the market for plant-derived artemisinin has stabilized at prices lower than those attainable by this approach. There are several reports4−8 of alternative approaches to the chemical conversion of AA to artemisinin that may provide savings relative to the process described by Sanofi. Current estimates put fermentation-derived AA cost at $50–75/kg. Conversion to DHAA results in costs for this intermediate in the $100–150/kg range. For the SSA approach to be competitive with plant-based artemisinin, the cost of these inputs needs to be much lower. Current prices for natural artemisinin average around $150/kg. With current conversion methods, this means that DHAA needs to be at most $75/kg. Because this does not seem to be possible from current fermentation routes, alternative sources should be explored.
There are numerous claims of high levels of AA and DHAA in Artemisia annua leaves giving rise to suggestions that the plant could be a viable source for these starting materials.5 These claims largely trace back to unpublished observations by Haynes & Vonwiller,9 who claimed that AA was present at levels as high as 2.6%, 10-fold higher than artemisinin. They did not provide data to support this assertion. This claim of high levels of AA has often been cited but never substantiated. In more recent reports, Ferreira10,11 showed levels of ∼0.2% DHAA and ∼0.02% AA in leaves containing 1–1.2% artemisinin. These results are consistent with most unpublished observations, including our own. The discussion was reignited by a report by Suberu et al.12 suggesting that DHAA is present at 5% by weight in leaves. Larson et al.13 countered this with their survey of a large number of A. annua varieties and found levels of artemisinin, DHAA, and AA consistent with those reported by Ferreira. Although there may be points in the plant life cycle where AA and DHAA concentrations exceed those of artemisinin, these points occur before maturation when the total mass of leaves is low. The DHAA and AA levels never exceed the final concentration of artemisinin at maturity. Given the low levels of artemisinin precursors in leaves, it would be counter-productive to modify current extraction procedures to recover these compounds at the expense of artemisinin.
It may however be useful to consider recovering DHAA and AA from waste streams from the existing extraction processes. Given that DHAA and AA are not observed in isolated artemisinin, it is likely that they will appear in the waste from the extraction process. The objective of this work was to ascertain if sufficient levels of DHAA and AA are available for recovery from the extraction process of A. annua. If this were found to be true, an economically feasible recovery process would be sought.
Results and Discussion
Analysis of Leaves and Waste Process Streams
Details of artemisinin extraction processes are often proprietary but most follow a general approach of extraction with a nonpolar solvent such as hexane or petroleum distillate, use of an inert solid such as silica to remove waxy materials, rudimentary chromatography to remove baseline materials, and isolation by crystallization. The order of these operations may vary along with the solvents and solid supports used. These variations might affect the amounts of DHAA and AA present in the process streams and impact their isolation.
Botanical Extracts-EPZ Ltd (Athi River, Kenya) kindly provided samples of leaves and their artemisinin extraction process stream for analysis. Using higher performance liquid chromatography (HPLC), it was found that leaves from three different batches contained artemisinin between 1.0 and 1.4 wt %, together with 0.24 to 0.36 wt % of DHAA and 0.04 to 0.09 wt % of AA. Similar results were obtained for these samples using analysis conditions described by Suberu et al.12 The levels of DHAA, AA, and artemisinin are in general agreement with those reported by Ferreira et al.10,11
Analysis of the supplied spent leaf demonstrated that the extraction protocol used by Botanical Extracts-EPZ Ltd effectively extracts >95% of the available acids and artemisinin. Samples from various points in the extraction process were analyzed with the goal of determining if there is an optimal point in the process for the isolation of DHAA and AA. Although DHAA and AA were observed at a few points in the extraction process, it was found that mother liquors from the final artemisinin crystallization steps contained the highest levels of both DHAA and AA (5.0 and 0.9%, respectively). This represents an opportunity to recover these acids without altering the current artemisinin extraction process. Mother liquor concentrates are typically discarded after solvent recovery.
The corresponding materials obtained from Guilin Pharmaceutical Company represent a different geographic source of A. annua, as well as a different extraction process. It should be expected to contain different impurities as well. Characterized only as “final waste,” its precise stage in the extraction process is less clear. It was denser than the Botanical Extracts-EPZ material, possibly because of a more thorough solvent removal prior to shipment. The process waste from Guilin contained 8.0 wt % DHAA and 0.88 wt % AA.
Bionexx in Madagascar provided two distinct samples of “extraction waste”. The extraction protocol used by this firm reportedly differs from most extractors because it is described in general terms as using a supercritical fluid. The two samples, identified as polar and nonpolar wastes, were indeed thin oils rather than the gummy solids presented by other extractions. The Bionexx nonpolar waste was found to contain 6.4 wt % DHAA and 1.4 wt % AA.
Differences in DHAA and AA content may derive from differences in extraction processes or from differences in Artemisia strains extracted. The levels are however high enough to justify efforts to develop a straightforward procedure to recover these acids from waste material.
Figure 1 shows an HPLC chromatogram of a methanol extraction of waste material from Guilin. Three major peaks along with numerous smaller peaks are observed. Peaks eluting at 6.4 and 7.2 min were verified to be DHAA and AA, respectively, by coinjection of the reference material and liquid chromatography–mass spectrometry (LC–MS). Note that the response factor for AA is 2.5× that of DHAA; therefore, its content is lower than it appears. A significant unknown peak was observed at 5.4 min. A similar profile was observed in other waste samples.
Figure 1.

HPLC chromatogram of methanol extraction of Guilin process waste.
Identification of Components in Waste Material
The observation of the unknown component eluting at RT 5.4 min by HPLC led to speculation regarding its identity. Several reports14−16 indicate that arteannuin B is a common impurity, and there are further reports16,17 that this compound can be converted into artemisinin. The impurity is fairly cleanly extracted into heptane and might provide an additional source of starting material for artemisinin synthesis if these reports are correct. A sample of a heptane extraction was concentrated to an oil for further identification work. Gas chromatography–mass spectrometry (GC–MS) analysis of this oil (Figure 2) revealed two major components eluting at 4.09 and 5.46 min. Thin-layer chromatography showed these major components as well as several lower level compounds. Using a silica gel column and 10% ethyl acetate in heptane eluent, we were able to cleanly isolate the two major components. The fraction collected with Rf 0.34 and the fraction with Rf 0.16 correspond to GC–MS peaks at 4.09 and 5.46 min, respectively.
Figure 2.

GC–MS of initial heptane phase.
The major unknown component observed in GC–MS eluting at 4.09 min was identified as dihydro-epi-deoxyarteannuin B (4) rather than arteannuin B. The mass spectrum of this peak showed molecular ion peak at m/z 234 (5%) with a major fragment at m/z 190 (80%), which can be attributed to a decarboxylation of the lactone moiety of compound (4). 13C NMR spectra were obtained and were in agreement with those reported by Brown & Sy17 (see the Supporting Information for comparison of 13C NMR data of this isolated component with literature data18 of dihydro-epi-deoxyarteannuin B as well as its stereoisomer, dihydro-deoxyarteannuin B). This compound was identified in their seminal studies as the major side-product in the biosynthetic conversion of DHAA to artemisinin in the plant. It was also identified by Levesque & Seeberger5 as a major side-product in the chemical transformation of DHAA to artemisinin. Depending on the solvent used, its level could vary between 10 and 50% relative to artemisinin.19
The minor unknown component observed only in the GC–MS at RT = 5.45 min was identified as deoxyartemisinin (5). The mass spectrum of this peak showed a weak molecular ion peak at m/z 266 (3%), consistent with the molecule mass of deoxyartemisinin. Further, 13C NMR spectra were obtained and were in agreement with those reported by Brown & Sy.18 These authors also found this to be the second largest side-product in their studies. Scheme 2 shows the proposed conversion of DHAA to artemisinin by both chemical and biosynthetic pathways.
Scheme 2. Common Route of Conversion of DHAA to Artemisinin.

GC–MS indicated that all process waste samples contained both dihydro-epi-deoxyartennuin B and deoxyartemisinin, as did all leaf samples tested. Samples of crystallized artemisinin did not show any of these impurities, indicating that isolation procedures effectively reject these impurities.
Recovery of Artemisinin Precursors from Process Waste
Our work toward recovering DHAA and AA from the extraction process waste material initially focused on solubilizing all material in water-immiscible solvents, with the intent to take advantage of the ionizable nature of the target compounds through acid/base partitioning. In attempts to completely dissolve the process waste, heptane and toluene gave emulsions or incomplete dissolution, whereas tetrahydrofuran (THF), 2-methyl-THF, and methyl t-butyl ether gave clear solutions at 0.5 g in 10 mL concentration.
Efforts to apply acid/base partitioning from solvents able to effect complete dissolution were not successful, as the acids would not partition cleanly into aqueous base (0.1 N NaOH). There were problems with getting clean-phase splits, and considerable rag layers were observed. Heptane partition experiments with pure DHAA and AA dissolved in aqueous acid or base also showed appreciable losses to an interfacial layer. Losses because of rag layers were even greater with the provided extraction samples. We subsequently discovered that adding methanol to the aqueous layer (1:1 by volume) eliminated the rag layer without sacrificing extraction efficiency.
We had previously observed that methanol extracted most of the available DHAA and AA while leaving behind large amounts of waxy material. In hopes that cleaner phase splits would result by not solubilizing this waxy material, extraction into methanol was further pursued. Using 100 mL methanol per 5 g waste it was found that stirring at 55–60 °C enhances extraction efficiency, and further, that subsequent chilling of this extract precipitates remaining waxy solids without loss of solubilized DHAA and AA.
Addition of aqueous sodium hydroxide (1:1 by volume) to the filtered methanol extract gives the desired mixture for partitioning against heptane. This results in clear phase cuts, with DHAA and AA retained in the lower aqueous layer as carboxylates. Subsequent acidification of the aqueous phase with HCl then allows clean extraction of DHAA and AA into heptane.
When this approach was applied to a sample of Guilin process waste, HPLC assay of the methanol filtrate indicated the bulk of the available DHAA, and AA was solubilized from the initial waxy material. Chilling of the methanol extract allowed the filtration of solubilized waxy material to give a clear solution.
The methanolic filtrate was diluted with an equal volume of aqueous 0.1 N sodium hydroxide solution, and the resultant mixture was extracted with an equal volume of heptane to remove nonacidic components. The peak identified as dihydro-epi-deoxyarteannuin B extracts cleanly into this initial heptane phase (see the Supporting Information). The pH of the remaining aqueous phase was then adjusted to ∼2 with 6 N hydrochloric acid, followed by extraction twice with equal volumes of heptane. Both DHAA and AA are effectively extracted into the heptane phase. These heptane phases were combined and concentrated to a clear, light-yellow oil.
The HPLC analysis of process streams samples (see the Supporting Information) showed excellent recovery through this recovery procedure. The initial methanol filtrate contained essentially all of the available DHAA and AA. No acid was lost to the first heptane extraction of the basic aqueous methanol solution. The first heptane extraction of the acidified aqueous layer showed 80–90% of the available acids with the second heptane extraction increasing this to recovery levels seen in the final oil. Recovery from the Guilin process waste was quantitative for DHAA and AA. Similar recoveries were observed from the Botanical Extracts-EPZ waste material. Recovery values for AA were more variable because of the low levels of this acid. Recoveries from the Bionexx extracts material was lower at around 80%.
This recovery procedure provides excellent recovery of DHAA and AA from artemisinin extraction wastes in a straightforward process that could be economical to perform at scale. Crystalline DHAA with >99.0% purity was obtained by concentrating the second heptane phase to an oil followed by crystallization from cold acetonitrile. About 34% of the DHAA was lost to the mother liquors. Crystallization from other solvents gave DHAA with lower purity and similar losses in the mother liquors. Such losses would likely make this approach economically untenable.
Recent studies suggest that high-purity DHAA might not be required for conversion to artemisinin. Kopetzki et al. reported that their flow photochemistry approach is capable of converting DHAA in a similar extract to artemisinin in good yield.19 Salvaging AA may be more problematic, as most reduction processes use metal catalysts that are likely to be poisoned by impurities present in the final oil. Feth et al.20 reported the reduction of AA to DHAA using diimide, thus avoiding the need for a catalyst. When we applied this chemistry to AA in the presence of DHAA, we observed over-reduction of the DHAA resulting in a negative yield. There is a promising recent report21 using this diimide in flow chemistry with controlled addition to prevent over-reduction of formed DHAA. Despite this, it is unclear whether it is worthwhile converting the low levels of AA present in the waste material.
If the DHAA contained in a filtered methanol extract of waste material could be converted to artemisinin without further isolation, this approach might be economically feasible. This assumes that coextracted impurities do not interfere with the transformation and that this could be accomplished on the site of the extraction facility. If additional purification is required, it is unlikely to be worthwhile. It is not unexpected that these impurities would not interfere in the transformation to artemisinin as they are formed in the same transformation in the plant.
It is worth noting that the side-products formed in the conversion of DHAA to artemisinin are similar in both the plant and chemical reactors. This suggests that no enzyme is involved in the final biosynthetic step of artemisinin synthesis. Artemisinin may just be the byproduct of in vivo scavenging of singlet oxygen generated during photosynthesis. Recent work22 demonstrated that chlorophylls extracted from Artemisia leaves function at least as well as other photosensitizers in ex vivo flow photochemical conversion of DHAA to artemisinin. DHAA reacts with singlet oxygen ultimately giving artemisinin and other related compounds. DHAA and other sesquiterpenes act as antioxidants and defensive agents in the plant. It is just fortuitous that artemisinin is useful to humans.
Conclusions
Procedures were developed to recover DHAA from the waste from extraction of artemisinin from plant sources. With the goal of providing DHAA as a starting material for conversion to artemisinin, it is essential to consider the conversion costs making for a maximum target price of $75/kg DHAA. Although waste material may be considered free of cost, the processing costs to isolate DHAA must be considered. This can be carried out by using a rough estimate of facility usage coupled with an estimate of a charge rate for this usage. Under described conditions a kilogram of waste may be extracted into 20 L methanol, chilled and filtered in 3 h. A typical usage rate for a pharmaceutical plant is roughly $0.10 per hour per liter. Applying this rate gives $6 to process 1 kg of waste material to this point. If the waste material contains 8% DHAA processing to this point costs $75/kg DHAA. Clearly, further purification of DHAA beyond this point is not economically viable.
Conversion of DHAA from artemisinin extraction waste material needs to be able to utilize it in the presence of coextracted impurities. Thus far, this has not been demonstrated. The described extraction process was designed with the goal of obtaining purified DHAA, and it is likely that the process can be modified to reduce processing costs if the subsequent conversion of DHAA in the raw extract can be demonstrated.
Experimental Section
A. annua Leaves and Extraction Process Streams
Three batches of A. annua leaves, a batch of spent leaves, and samples taken at various points of the extraction process including final mother liquors were kindly provided by Botanical Extracts-EPZ Ltd (Athi River, Kenya). A sample of final process waste was provided by Guilin Pharmaceutical Co. in China. Waste stream samples were also provided by Bionexx in Madagascar.
HPLC Conditions
HPLC analysis of Artemisia leaves, process streams, and waste extractions were accomplished using an Agilent Technologies 1100 equipped with a diode array detector. An Agilent Eclipse XDB-C18 column (150 mm L × 4.6 mm i.d., 5 μm particle size) was used with a mobile phase consisting of 55:45 acetonitrile: 0.1% phosphoric acid pumped at 1.5 mL/min. The column oven was set at 40 °C. Injection volume was 5 μL and detection was at 210 nm.
HPLC–MS Conditions
LC–MS was used to verify the identity of DHAA and AA in Artemisia leaves and extracts. This was accomplished using an Advion Expression CMS with negative electrospray ionization (ESI) detection. The capillary temperature was set at 250 °C with a voltage of 180. The gas temperature was set at 300 °C with an ESI voltage of 3000. Two different HPLC conditions were used. A mobile phase of 25:75 acetonitrile/0.1% ammonium hydroxide provided typical negative ionization, whereas 80:20 methanol/0.1% phosphoric acid allowed “wrong-way around” ionization. In both cases, an Agilent Eclipse XDB-C18 column (2.1 mm i.d. × 150 mm L, 3.5 μm particle size) was used.
GC–MS Conditions
GC–MS was used to identify impurities present in Artemisia leaves and extraction waste. This was accomplished using an Agilent 6890N GC equipped with an Agilent HP-5MS 5% phenylmethyl siloxane (30 M × 0.25 mm i.d.; 0.25 μm film thickness) column with helium carrier gas and an isothermal oven temperature of 210 °C. Detection was via Agilent 5975C VL MSD.
NMR Spectroscopy
13C NMR spectra were obtained at the field strengths of 15 MHz using a Magritek Spinsolve 60 NMR spectrometer. Samples were dissolved in CDCl3 with tetramethylsilane as standard.
Leaf and Process Stream Assays
Artemisia leaves were extracted with THF (5 mL/g leaf using sonication (30 min). The resulting clear green solution was filtered prior to injection. Process stream samples were diluted with acetonitrile to give approximate 1 mg/mL concentrations of DHAA and AA. Reference materials for these acids were prepared by recrystallization of acid samples.
Acknowledgments
The authors acknowledge the support of the Bill and Melinda Gates Foundation, grant OPP1138478.
Glossary
Abbreviations
- DHAA
dihydroartemisinic acid
- AA
artemisinic acid
- HPLC
high-performance liquid chromatography
- LC–MS
liquid chromatography–mass spectrometry
- GC–MS
gas chromatography–mass spectrometry
- API
active pharmaceutical ingredient
- ACT
artemisinin combination therapy
- SSA
semisynthetic artemisinin
- NMR
nuclear magnetic resonance
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00974.
13C NMR spectroscopic data for identified impurities; HPLC chromatograms of partition samples; and HPLC assay results for partition samples (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ro D.-K.; Paradise E. M.; Ouellet M.; Fisher K. L.; Newman K. L.; Ndungu J. M.; Ho K. A.; Eachus R. A.; Ham T. S.; Kirby J.; Chang M. C. Y.; Withers S. T.; Shiba Y.; Sarpong R.; Keasling J. D. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- Turconi J.; Griolet F.; Guevel R.; Oddon G.; Villa R.; Geatti A.; Hvala M.; Rossen K.; Göller R.; Burgard A. Semisynthetic artemisinin, the chemical path to industrial production. Org. Process Res. Dev. 2014, 18, 417–422. 10.1021/op4003196. [DOI] [Google Scholar]
- Wallaart T. E.; van Uden W.; Lubberink H. G. M.; Woerdenbag H. J.; Pras N.; Quax W. J. Isolation and identification of dihydroartemisinic acid from Artemisia annua and its possible role in the biosynthesis of artemisinin. J. Nat. Prod. 1999, 62, 430–433. 10.1021/np980370p. [DOI] [PubMed] [Google Scholar]
- Yadav J. S.; Thirupathaiah B.; Srihari P. A concise stereoselective total synthesis of (+)-artemisinin. Tetrahedron 2010, 66, 2005–2009. 10.1016/j.tet.2010.01.051. [DOI] [Google Scholar]
- Levesque F.; Seeberger P. H. Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem., Int. Ed. 2012, 51, 1706–1711. 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Cook S. P. A concise synthesis of (+)-artemisinin. J. Am. Chem. Soc. 2012, 134, 13577–13579. 10.1021/ja3061479. [DOI] [PubMed] [Google Scholar]
- Chen H.-J.; Han W.-B.; Hao H.-D.; Wu Y. A facile and scalable synthesis of qinghaosu (artemisinin). Tetrahedron 2013, 69, 1112–1114. 10.1016/j.tet.2012.11.056. [DOI] [Google Scholar]
- Paddon C. J.; Westfall P. J.; Pitera D. J.; Benjamin K.; Fisher K.; McPhee D.; Leavell M. D.; Tai A.; Main A.; Eng D.; Polichuk D. R.; Teoh K. H.; Reed D. W.; Treynor T.; Lenihan J.; Jiang H.; Fleck M.; Bajad S.; Dang G.; Dengrove D.; Diola D.; Dorin G.; Ellens K. W.; Fickes S.; Galazzo J.; Gaucher S. P.; Geistlinger T.; Henry R.; Hepp M.; Horning T.; Iqbal T.; Kizer L.; Lieu B.; Melis D.; Moss N.; Regentin R.; Secrest S.; Tsuruta H.; Vazquez R.; Westblade L. F.; Xu L.; Yu M.; Zhang Y.; Zhao L.; Lievense J.; Covello P. S.; Keasling J. D.; Reiling K. K.; Renninger N. S.; Newman J. D. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- Haynes R. K.; Vonwiller S. C. Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans. R. Soc. Trop. Med. Hyg. 1994, 88, 23–26. 10.1016/0035-9203(94)90466-9. [DOI] [PubMed] [Google Scholar]
- Ferreira J. F. S.; Gonzalez J. M. Analysis of underivatized artemisinin and related sesquiterpene lactones by high-performance liquid chromatography with ultraviolet detection. Phytochem. Anal. 2009, 20, 91–97. 10.1002/pca.1101. [DOI] [PubMed] [Google Scholar]
- Ferreira J. F. S.; Luthria D. L. Drying affects artemisinin, dihydroartemisinic acid, artemisinic acid and the antioxidant capacity of Artemisia annua L. leaves. J. Agric. Food Chem. 2010, 58, 1691–1698. 10.1021/jf903222j. [DOI] [PubMed] [Google Scholar]
- Suberu J.; Song L.; Slade S.; Sullivan N.; Barker G.; Lapkin A. A. A rapid method for the determination of artemisinin and its biosynthetic precursors in Artemisia annua L crude extracts. J. Pharm. Biomed. Anal. 2013, 84, 269–277. 10.1016/j.jpba.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Larson T. R.; Branigan C.; Harvey D.; Penfield T.; Bowles D.; Graham I. A. A survey of artemisinic and dihydroartemisinic acid contents in glasshouse and global field-grown populations of the artemisinin-producing plant Artemisia annua L. Ind. Crops Prod. 2013, 45, 1–6. 10.1016/j.indcrop.2012.12.004. [DOI] [Google Scholar]
- Jeremić D.; Jokić A.; Behbud A.; Stefanović M. A new type of sesqiterpene lactone isolated from Artemisia annua L. Arteannuin B. Tetrahedron Lett. 1973, 32, 3039–3042. 10.1016/s0040-4039(01)96314-2. [DOI] [Google Scholar]
- Acton N.; Klayman D. L.; Rollman I. J.; Novotny J. F. Isolation of artemisinin (qinghaosu) and its separation from artemisitene using the Ito multilayer coil separator-extractor and isolation of arteannuin B. J. Chromatogr. 1986, 355, 448–450. 10.1016/s0021-9673(01)97352-2. [DOI] [Google Scholar]
- Nair M. S. R.; Basile D. V. Bioconversion of arteannuin B to artemisinin. J. Nat. Prod. 1993, 56, 1559–1556. 10.1021/np50099a015. [DOI] [PubMed] [Google Scholar]
- Nowak D. M.; Lansbury P. T. Synthesis of (+)-artemisinin and (+)-deoxoartemisinin from arteannuin B and arteannuic acid. Tetrahedron 1998, 54, 319–336. 10.1016/s0040-4020(97)10286-1. [DOI] [Google Scholar]
- Brown G. D.; Sy L.-K. In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron 2004, 60, 1139–1159. 10.1016/j.tet.2003.11.070. [DOI] [Google Scholar]
- Kopetzki D.; Lévesque F.; Seeberger P. H. A continuous-flow process for the synthesis of artemisinin. Chem.—Eur. J. 2013, 19, 5450–5456. 10.1002/chem.201204558. [DOI] [PubMed] [Google Scholar]
- Feth M. P.; Rossen K.; Burgard A. Pilot plant PAT approach for the diastereoselective diimide reduction of artemisinic acid. Org. Process Res. Dev. 2013, 17, 282–293. 10.1021/op300347w. [DOI] [Google Scholar]
- Pieber B.; Glasnov T.; Kappe C. O. Continuous flow reduction of artemisinic acid utilizing multi-injection strategies-closing the gap towards a fully continuous synthesis of antimalarial drugs. Chem.—Eur. J. 2015, 21, 4368–4376. 10.1002/chem.201406439. [DOI] [PubMed] [Google Scholar]
- Triemer S.; Gilmore K.; Vu G. T.; Seeberger P. H.; Seidel-Morgenstern A. Literally green chemical synthesis of artemisinin from plant extracts. Angew. Chem., Int. Ed. 2018, 57, 5525–5528. 10.1002/anie.201801424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.