

Abstract

Boronic acids (BAs) are a prominent functionality extensively used to design biologically active compounds and functional biomaterials. Boronic acids open shell can lead to unspecific reactivity of BAs with endogenous nucleophiles and to undesired off-target effects. Here, diazaborines are presented as a new class of boron-based warheads for serine proteases inhibition, in which the boron function is stabilized in the form of an aromatic boron-based heterocycle. In this study, diazaborines were readily synthesized in a single step in yields up to 96%, without any chromatographic operation and were shown to reversibly inhibit human neutrophil elastase (HNE) serine protease with IC50 values in the low μM range. Diazaborines were also selective toward HNE, as no inhibition was observed against a panel of five closely related serine proteases. Synthetic and theoretical studies performed on this system suggest that, similar to BAs, the reaction mechanism involves the formation of a reversible covalent bond between the diazaborine boron center and the catalytic serine oxygen. Finally, different from BAs, diazaborines were shown to be very stable in pH 7.4 phosphate buffer and human plasma.
Introduction
Serine proteases are one of the largest and more important groups of enzymes found in eukaryotes and prokaryotes that selectively catalyze the hydrolysis of peptide bonds.1 Proteolysis plays a key role in pathologies such as rheumatoid arthritis, bacterial and viral infections, cancer, and Alzheimer’s disease.1 Therefore, the discovery of innovative protease inhibitors stands as an invaluable tool for advanced therapeutic intervention.2 In the past, boronic acids (BAs) emerged as a very promising class of serine protease inhibitors that readily form a negatively charged tetravalent borate upon reaction with nucleophiles at the enzyme active site (Scheme 1A).3 Rather surprisingly and despite being extensively used, with the notable exception of benzoxaboroles,4 this chemical warhead periphery remained basically unaltered for more than 40 years.3 This lack of diversity reflects the intrinsic chemical properties required for the interaction with the protein active site. In detail, the carbon substituent is essential to improve the recognition by the biologic target, while the boron open shell confers Lewis acidity to BAs and enables the reversible covalent ligation with the active site nucleophiles.3 However, also because of this, BAs react unspecifically with other endogenous molecules such as vicinal diols or proteins and this leads to unfavorable pharmacokinetics. This reactive profile constitutes a major obstacle in the development of boron-based therapeutics.5 Therefore, the discovery of a new BA scaffold that is generally unreactive but retains the ability to form a reversible covalent bond with oxygen-based nucleophiles at the protein active site is of fundamental importance to discover the next generation of serine protease inhibitors. On the basis of this, we anticipated that a strategy to overcome the unspecific reactivity of BAs would be to embed this function in an aromatic heterocycle where the boron vacant orbital may be stabilized by the lone pair of electrons of an adjacent nitrogen atom.6 Careful tuning of this boron-based (B–N) heterocycle stereo-electronic properties would not only contribute to an improved stability but, more importantly, would offer a mechanism to expose the BA at the enzyme active site, exploring the selective disruption of the B=N bond catalyzed by the serine hydroxyl group (Scheme 1B).6
Scheme 1. (A) Formation of a Negatively-Charged Tetrahedral Boron Center upon Reaction with a Serine at Enzyme Active Site; (B,C) Development of a Stable B–N Heterocycle Capable of Selective Enzyme Recognition.
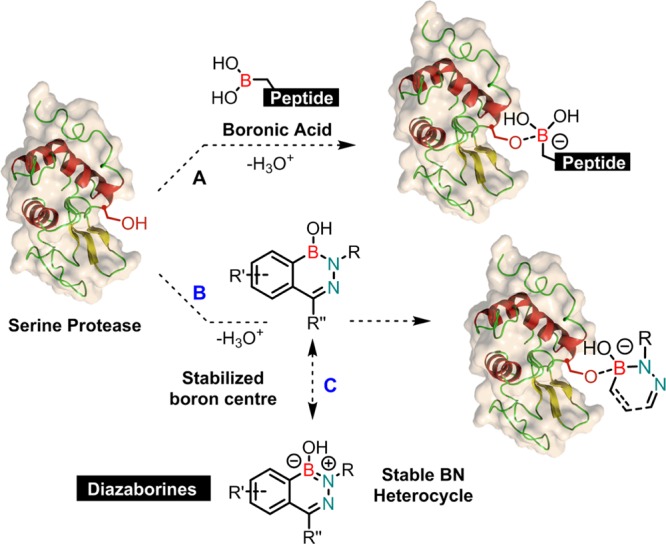
In the early 80s, diazaborines (Scheme 1C) emerged as a promising class of antibacterial agents that target enoyl reductase via formation of a covalent bond between the nicotinamide ribose hydroxyl group and the boron atom.7 Encouraged by this mode of action, we initiated a study to determine if this family B–N heterocycles could also be used as a replacement of aromatic BAs in the design of serine protease inhibitors.
Results and Discussion
To test this hypothesis, we selected human neutrophil elastase (HNE) which is an important serine protease identified as a potential therapeutic target in several inflammatory diseases, and for which, there is still no efficient inhibitor marketed.8 As shown in Scheme 2, following reported methodologies, diazaborines 1 and 2 were synthesized in excellent yields, simply by combining in aqueous media, 2-carbonyl benzene BAs with hydrazine.9 Once prepared, we evaluated these heterocycles as well as phenyl BA and benzoxoborole against HNE. Although phenyl BA and benzoxoborole were inactive against HNE up to a concentration of 100 μM, diazaborines 1 and 2 effectively inhibited the enzyme with IC50 values of 24 and 48 μM, respectively.
Scheme 2. Phenyl BA, Benzoxoborole, and Diazaborines 1 and 2 Evaluation against HNE.

These results were further corroborated by in silico molecular docking studies that posed the phenyl BA and the benzoxoborole at the interface of the entrance for the primary recognition S1 pocket but at the opposite side to the catalytic serine. This positioning suggests that these compounds can hardly establish a ligation with the serine oxygen and support their inactivity. Differently, diazaborines 1 and 2 adopt a pose inside the HNE binding site which allows a closer contact of the boron atom with the oxygen of Ser195 (∼3.5 Å). As shown in Figure 1, both the distance and orientation of these compounds support the formation of a B–OSer195 covalent bond. This mechanism of action was further supported by covalent docking calculations which showed that upon coordination with the oxygen of Ser195, the boron adopts a tetrahedral geometry that promotes the formation of important new interactions with Phe192, and Gly193, and the blockage of the access to the catalytic serine.
Figure 1.

Best noncovalent docking poses of phenyl BA (A), benzoxoborole (B), diazaborines 1 (C), and 2 (D), and best covalent docking poses of diazaborines 1 (E) and 2 (F) compounds inside the HNE binding site.
Considering these promising results and taking advantage of the well-established and straightforward assemblage of diazaborines,9 we set out to explore the electronic and chemical space of the boron center. With this objective, 2-carbonyl benzene BAs were reacted with different hydrazines as shown in Scheme 3. The expected heterocycles were all obtained in good yields in one step without any chromatographic purification. In terms of HNE inhibition, the heterocycles prepared with 2-acetyl-benzene BA were generally more active than diazaborines 3–5, assembled from 2-formyl-benzene BA. The use of hydrazines featuring electron-withdrawing N-substituents also improved the heterocycle activity up to an IC50 value of 11.7 μM (compound 8, Scheme 3).
Scheme 3. Modular Assembly of Diazaborines 3–8 (Benzene-BA Series) and Inhibitory Activity against HNE.

Docking studies performed on this series revealed that the most active molecules, 5 and 8, consistently fit the heterocycle in the S1 subsite, while the N-substituents are either flipped to the S2 or the S1′ subpockets in the noncovalent and covalent models, respectively (Figure 2). On the basis of this, we anticipated that a smaller heterocycle would better fit the S1 pocket and improve the overall affinity of the inhibitor. On the basis of this rational, a series of thienyl diazaborines (9–16)7 were studied against the HNE active site. Similarly, to the benzene-BA series (3–8), apart from compounds 10 and 14 that fit the phenyl group inside the S1 subsite, all other diazaborines pose the thienyl moiety inside the S1 cavity, the N-substituents in the S2 subpocket and the boron atom at a distance between 3.4 and 4 Å of the serine oxygen. Upon the formation of the covalent B–OSer195, the heterocycle pose is maintained, while the N-substituent moves to the S1′ subpocket and additional interactions are established with Cys42 and Phe192. According to this model, the thienyl diazaborines exhibit slightly higher affinities for the HNE active site than the benzene-BA series (scores between 44 and 52), probably because of an extra interaction that the thienyl moiety (S atom) is able to establish with Ser195 nitrogen.
Figure 2.

Best noncovalent docking poses of diazaborines 8 (A) and 12 (B) and best covalent docking poses of diazaborines 8 (C) and 12 (D) compounds inside the HNE binding site.
Taking these results in consideration, we performed the synthesis of diazaborines 9–16 and evaluated their inhibitory potential against HNE. As shown in Scheme 4, these compounds elicited higher potency than the previous diazaborines (3–8), and heterocycle 12 featuring the N-p-toluenesulfonyl substituent became the most active molecule with an IC50 of 3.4 μM. The inhibitory kinetic profile of 12 was studied using the progress curve method under pseudo-first order conditions (Supporting Information, Figure S4). Under these conditions, diazaborine 12 displayed a time-dependent inhibition of HNE and a second-order rate constant, kon, of 2.68 M–1 s–1. This value, albeit modest when compared to large peptidomimetic-based inhibitors,10 combined with a ligand efficiency (LE) value of 0.39 and a ligand-lipophilicity efficiency (LLE) value of 3.4, suggests that diazaborines are a good starting point for further medicinal chemistry optimization.11,12
Scheme 4. Modular Assembly of Diazaborines 9–16 (Thienyl-BA Series) and Activity against HNE.

Diazaborines are often regarded as stable heterocycles, though the B–N bond is potentially reversible, namely, when the nitrogen is substituted with electron-withdrawing groups.13 Therefore, we questioned if the observed activity of compound 12 could be due to the in situ hydrolysis of the B–N bond. To study this, the thienyl BA hydrazone 17 was prepared and tested against HNE. As shown in Scheme 4, the BA hydrazone 17 (IC50 = 12.0 μM) was able to inhibit the enzyme, with only slightly less efficiency than the corresponding diazaborine 12 (IC50 = 3.4 μM) but with a significantly higher second-order rate constant (216 vs 2.86 M–1 s–1). To rationalize this difference, we docked both molecules against the enzyme active site. As shown in Figure 3, compound 17 shows a completely different binding pose inside HNE active site. While compound 12 fits the thienyl core deep inside the S1 cavity and accommodates the tosyl group in the S1′, compound 17 rotates the tosyl group to S2 cavity, losing the interactions with Cys42, but establishing a π–π stacking interaction with His57.
Figure 3.
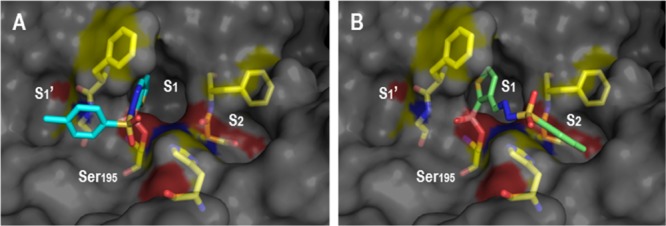
Best covalent docking poses of diazaborines 12 (A) and 17 (B) compounds inside the HNE binding site.
Once we established diazaborines as a valuable scaffold to design HNE inhibitors, we access their potency, selectivity, and chemical stability against open shell BAs. With this objective, diazaborine 18 and the oxime BA 19 that is unable to form a six-member heterocycle, were prepared and tested against HNE. Very gratifyingly, both compounds exhibited similar potencies (18 IC50 = 2.7 μM vs 19 IC50 = 0.7 μM) but significantly different second-order rate constants (18kon = 12.8 M–1 s–1 vs 19kon = 97.0 M–1 s–1). The docking calculations performed on these systems revealed that when covalently bounded to HNE, these compounds place the thienyl moiety inside the S1 cavity and place the benzyl moiety either in the S1′ (18) or in the S2 cavities (19). The affinity of compound 19 for HNE is slightly better than the affinity of diazaborine 18 (scores 53.9 vs 51.3), and this is probably a result of an extra interaction with Phe215 in the S2 subpocket. Overall, both compounds (18 and 19) show a good spatial fitting of the binding pocket, however, occupying perpendicular subpockets (S1′ and S2) (Figure 4).
Figure 4.

HNE inhibition data and cytotoxicity for diazaborines 18 and 19 and best covalent docking poses of diazaborines 18 (A) and 19 (B) compounds inside the HNE binding site.
Regarding the selectivity, 18 and 19 were tested against a panel of closely related serine proteases (Supporting Information, Table S1). In this assay, at a concentration of 100 μM, both the diazaborine and oxime BA were proven to be inactive toward urokinase, trypsin, thrombin, kallikrein, and chymotrypsin. Similarly, in a standard cytotoxicity assay using a human cell line HEK 293T (Supporting Information, Table S2), these compounds did not induce any appreciable toxicity even after a 48 h exposure period and up to a concentration of 100 μM.
Finally, we addressed the chemical stability of both compounds in biocompatible conditions. BAs are well-known for binding carbohydrates; therefore, compounds 18 and 19 were incubated with 1 equiv of fructose in phosphate-buffered saline (PBS) at pH 7.4. As shown in Figure 5, under these conditions, the open-shell BA 19 exhibits a half-life of 2 h, while the diazaborine remained mostly intact over a 24 h period, which is in agreement with earlier reports that thienyl groups display a good chemical stability.14 Encouraged by these results and aiming to establish the stability of the diazaborine in a more complex system, compound 18 was incubated in human plasma at 37 °C (400 μM) for 10 h. The concentration was monitored by high-performance liquid chromatography (HPLC), and over this time, no degradation of the diazaborine was detected, which supports the initial hypothesis that the formation of the B–N heterocycle improves the BA stability, while maintaining a mechanism for the interaction with enzyme nucleophiles.
Figure 5.

(A,B) UV–vis spectra of compounds 18 and 19 (80 μM) in PBS buffer pH 7.4 at 37 °C in the presence of fructose over 24 and 5.5 h, respectively; (C) stability of compound 18 (400 μM) in human plasma (diluted in PBS buffer pH 7.4) at 37 °C analyzed by HPLC over 9 h; (D) calculation of compound 19’s half-life by plotting graphic B at 366 nm.
Conclusions
In summary, here, we disclosed the use of diazaborines as serine protease inhibitors and demonstrate that this class of compounds can be effectively used as BA replacements, eliciting similar potencies and selectivities but improved stabilities in complex mediums. In detail, a series of diazaborines were prepared in a simple step without chromatographic purifications (yields 49–97%) and were shown to inhibit HNE reversibly in the low micromolar range. Alcoholysis of diazaborines, used here as a model for the reaction with the catalytic serine residue, led to the rapid and quantitative formation of the corresponding boronate ester, thus strongly suggesting that diazaborines are covalent inhibitors of HNE (Figure S1). Furthermore, docking studies performed on this system support the interaction of diazaborines with the HNE binding site via the occupancy of the S1 subsite and the formation of a covalent B–OSer195 bond. A direct comparison between diazaborine 18 and its open-shell BA counterpart, 19, revealed that both compounds inhibited the enzyme with similar potencies, though both exhibit very distinct stability profiles in favor of the diazaborine that was stable in human plasma over a 10 h period. Additional evidence for the unique features displayed by diazaborines as enzyme inhibitors comes from the observation that BAs 17 and 19 are equipotent but significantly less reactive toward HNE than their cyclic counterparts 12 and 18. This is consistent with their lower dissociation rate constants, koff, and thus they have longer residence times than their BA counterparts.15−17 These results, combined with appropriate LE and LLE values, highlight the potential of diazaborines as excellent starting points to develop selective HNE inhibitors.
Acknowledgments
We acknowledge the financial support of Fundação para a Ciência e a Tecnologia (FCT) Portugal (grants: PTDC/QEQ-MED/5512/2014, PTDC/QEQ-QOR/1434/2014, PTDC/BBB-BEP/2463/2014; iMed.ULisboa grant UID/DTP/04138/2013; MedChemTrain grant PD/BD/128239/2016). We acknowledge the useful discussions with Dr. Francesco Montalbano and Dr. Susana D. Lucas.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00702.
Procedures and compound characterization; stability studies; biochemical studies; and docking calculations (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Powers J. C.; Asgian J. L.; Ekici O. D.; James K. E. Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev. 2002, 102, 4639–4750. 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- Smoum R.; Rubinstein A.; Dembitsky V. M.; Srebnik M. Boron Containing Compounds as Protease Inhibitors. Chem. Rev. 2012, 112, 4156–4220. 10.1021/cr608202m. [DOI] [PubMed] [Google Scholar]
- a Baker S. J.; Tomsho J. W.; Benkovic S. J. Boron-containing inhibitors of synthetases. Chem. Soc. Rev. 2011, 40, 4279–4285. 10.1039/c0cs00131g. [DOI] [PubMed] [Google Scholar]; b Touchet S.; Carreaux F.; Carboni B.; Bouillon A.; Boucher J.-L. Aminoboronic acids and esters: from synthetic challenges to the discovery of unique classes of enzyme inhibitors. Chem. Soc. Rev. 2011, 40, 3895–3914. 10.1039/c0cs00154f. [DOI] [PubMed] [Google Scholar]; c Matteson D. S. α-Amido boronic acids: A synthetic challenge and their properties as serine protease inhibitors. Med. Res. Rev. 2008, 28, 233–246. 10.1002/med.20105. [DOI] [PubMed] [Google Scholar]; d Leung I. K. H.; Brown T. Jr.; Schofield C. J.; Claridge T. D. W. An approach to enzyme inhibition employing reversible boronate ester formation. Med. Chem. Commun. 2011, 2, 390–395. 10.1039/c1md00011j. [DOI] [Google Scholar]
- a Peters J. A. Interactions between boric acid derivatives and saccharides in aqueous media: Structures and stabilities of resulting esters. Coord. Chem. Rev. 2014, 268, 1–22. 10.1016/j.ccr.2014.01.016. [DOI] [Google Scholar]; b Adamczyk-Woźniak A.; Borys K. M.; Sporzyński A. Recent Developments in the Chemistry and Biological Applications of Benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]; c Tomsho J. W.; Pal A.; Hall D. G.; Benkovic S. J. Ring Structure and Aromatic Substituent Effects on the pKa of the Benzoxaborole Pharmacophore. ACS Med. Chem. Lett. 2012, 3, 48–52. 10.1021/ml200215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang W.; Gao X.; Wang B. Boronic acid compounds as potential pharmaceutical agents. Med. Res. Rev. 2003, 23, 346–368. 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]; b Poplawski S. E.; Lai J. H.; Sanford D. G.; Sudmeier J. L.; Wu W.; Bachovchin W. W. Pro-Soft Val-boroPro: A Strategy for Enhancing in Vivo Performance of Boronic Acid Inhibitors of Serine Proteases. J. Med. Chem. 2011, 54, 2022–2028. 10.1021/jm100972f. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Groziak M. P.; Chen L.; Yi L.; Robinson P. D. Planar Boron Heterocycles with Nucleic Acid-Like Hydrogen-Bonding Motifs. J. Am. Chem. Soc. 1997, 119, 7817–7826. 10.1021/ja963784i. [DOI] [Google Scholar]; b Kanichar D.; Roppiyakuda L.; Kosmowska E.; Faust M. A.; Tran K. P.; Chow F.; Buglo E.; Groziak M. P.; Sarina E. A.; Olmstead M. M.; Silva I.; Xu H. H. Synthesis, Characterization, and Antibacterial Activity of Structurally Complex 2-Acylated 2,3,1-Benzodiazaborines and Related Compounds. Chem. Biodiversity 2014, 11, 1381–1397. 10.1002/cbdv.201400007. [DOI] [PubMed] [Google Scholar]; c Rombouts F. J. R.; Tovar F.; Austin N.; Tresadern G.; Trabanco A. A. Benzazaborinines as Novel Bioisosteric Replacements of Naphthalene: Propranolol as an Example. J. Med. Chem. 2015, 58, 9287–9295. 10.1021/acs.jmedchem.5b01088. [DOI] [PubMed] [Google Scholar]; d Stojanović M.; Baranac-Stojanović M. B. Aromaticity of Diazaborines and Their Protonated Forms. J. Org. Chem. 2016, 81, 197–205. 10.1021/acs.joc.5b02499. [DOI] [PubMed] [Google Scholar]
- a Baldock C.; Rafferty J. B.; Sedelnikova S. E.; Baker P. J.; Stuitje A. R.; Slabas A. R.; Hawkes T. R.; Rice D. W. A mechanism of drug action revealed by structural studies of enoyl reductase. Science 1996, 274, 2107–2110. 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]; b Baldock C.; de Boer G.-J.; Rafferty J. B.; Stuitje A. R.; Rice D. W. The mechanism of action of diazaborines. Biochem. Pharmacol. 1998, 55, 1541–1550. [DOI] [PubMed] [Google Scholar]
- Lucas S. D.; Costa E.; Guedes R. C.; Moreira R. Targeting COPD: advances on low-molecular-weight inhibitors of human neutrophil elastase. Med. Res. Rev. 2013, 33, E73–E101. 10.1002/med.20247. [DOI] [PubMed] [Google Scholar]
- a Stress C. J.; Schmidt P. J.; Gillingham D. G. Comparison of boron-assisted oxime and hydrazone formations leads to the discovery of a fluorogenic variant. Org. Biomol. Chem. 2016, 14, 5529–5533. 10.1039/c6ob00168h. [DOI] [PubMed] [Google Scholar]; b Dilek O.; Lei Z.; Mukherjee K.; Bane S. Rapid formation of a stable boron–nitrogen heterocycle in dilute, neutral aqueous solution for bioorthogonal coupling reactions. Chem. Commun. 2015, 51, 16992–16995. 10.1039/c5cc07453c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Fincke A.-C.; Blaut M.; Braune A.; Gütschow M. A BODIPY-Tagged Phosphono Peptide as Activity-Based Probe for Human Leukocyte Elastase. ACS Med. Chem. Lett. 2018, 9, 345–350. 10.1021/acsmedchemlett.7b00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Young R. J. Molecular Property Design: Does Everyone Get It?. ACS Med. Chem. Lett. 2015, 6, 722–725. 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keserü G. M.; Makara G. M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 2009, 8, 203–212. 10.1038/nrd2796. [DOI] [PubMed] [Google Scholar]
- Gu H.; Chio T. I.; Lei Z.; Staples R. J.; Hirschi J. S.; Bane S. Formation of hydrazones and stabilized boron–nitrogen heterocycles in aqueous solution from carbohydrazides and ortho-formylphenylboronic acids. Org. Biomol. Chem. 2017, 15, 7543–7548. 10.1039/c7ob01708a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gütschow M.; Kuerschner L.; Neumann U.; Pietsch M.; Löser R.; Koglin N.; Eger K. 2-(Diethylamino)thieno[1,3]oxazin-4-ones as Stable Inhibitors of Human Leukocyte Elastase. J. Med. Chem. 1999, 42, 5437–5447. 10.1021/jm991108w. [DOI] [PubMed] [Google Scholar]
- Tummino P. J.; Copeland R. A. Residence Time of Receptor–Ligand Complexes and Its Effect on Biological Function. Biochemistry 2008, 47, 5481–5492. 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15, 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Vauquelin G. Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br. J. Pharmacol. 2016, 173, 2319–2334. 10.1111/bph.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.