

Abstract
Hypertriglyceridemic pancreatitis (HTGP) typically occurs in patients with an underlying dyslipidemia (such as type I, IV or V dyslipidemia) and in the presence of a secondary condition, such as inadequately controlled diabetes, excess alcohol consumption or medication use. Although the symptoms of HTGP are similar to those of acute pancreatitis from other etiologies, HTGP is often associated with greater clinical severity and rate of complications. Therefore, accurate diagnosis of HTGP is essential so that patients receive the appropriate treatment. Novel therapies that aim to reduce the incidence of pancreatitis in this patient population are now available or in development. Understanding the etiology, pathophysiology and clinical characteristics of HTGP will enable future development of therapeutic agents to treat HTGP.
Keywords: Acute pancreatitis, dyslipidemia, hypertriglyceridemic pancreatitis, lipoprotein lipase deficiency, clinical management
Introduction
Hypertriglyceridemia (HTG) is one of the major causes of acute pancreatitis (AP), accounting for up to 10% of all cases.1–3 Although the precise mechanism by which HTG causes AP (termed hypertriglyceridemic pancreatitis (HTGP)) is not fully understood, both HTG (by causing an excess of free fatty acids (FFAs)) and elevated chylomicrons are thought to increase plasma viscosity, which may induce ischemia in pancreatic tissue and trigger organ inflammation.1,2,4,5
Several types of HTG expose patients to an increased risk of AP. Familial lipoprotein lipase deficiency (LPLD) is a genetic disorder characterized by severe HTG and chylomicronemia associated with recurrent AP episodes.6 Excess alcohol consumption may cause increased serum triglyceride levels in patients with underlying HTG that increases the risk of AP.5,7 HTGP may also occur in some pregnant women as a result of altered hormone levels that trigger elevations in serum cholesterol and triglycerides, and can lead to severe complications both for mother and fetus.5,8,9
Patients with HTGP initially exhibit symptoms that are similar to those observed in patients with AP from other etiologies. However, the clinical severity and the rate of complications with HTGP are generally higher.10 Therefore, an accurate diagnosis is essential for delivery of the most appropriate treatment, as well as for prevention of disease recurrence.
This review discusses the etiology, epidemiology, pathophysiology and clinical characteristics of HTGP, and provides an overview of the currently available and possible future treatments.
Classification of HTG and definition of HTGP
Primary HTG
According to the Fredrickson classification, primary (inherited) HTG can be caused by five types of hyperlipidemia.11 Patients with type I, IV and V hyperlipidemia have an increased risk of HTGP.5 In type I hyperlipidemia, mainly the chylomicron metabolism is affected, resulting in high levels of circulating triglyceride-rich chylomicrons (chylomicronemia). LPLD is a rare autosomal recessive disorder caused by homozygosity or compound heterozygosity for mutations in the lipoprotein lipase (LPL) gene on chromosome 8.12 LPL is a key enzyme involved in the metabolism of triglyceride-rich lipoproteins such as chylomicrons and very-low-density lipoproteins (VLDLs). Patients with LPLD present with severe HTG and chylomicronemia, and are at higher risk of pancreatitis than patients with HTG from other causes.12 Mutations in genes encoding LPL co-factors and maturation proteins such as lipase maturation factor-1, glycosyl-phosphatidyl-inositol-anchored HDL-binding protein-1, apolipoprotein (Apo) C-II and Apo A-V can cause chylomicronemia and are associated with clinical manifestations resembling LPLD.13 Therefore, full gene sequencing of LPL and the four co-factor genes is often the preferred method in establishing the diagnosis in patients with suspected LPLD.13 Type IV (familial combined hyperlipidemia) is characterized by elevated VLDL levels, and results from mutations in several genes with an environmental effect.13,14 Type V hyperlipidemia (familial HTG), in which both chylomicrons and VLDL are elevated, is caused by alterations of genes that induce a reduction in catabolism. In patients with type IV or V hyperlipidemia, elevated triglycerides are affected by environmental factors, including obesity, excessive alcohol consumption and diet.13,14 Both type IV and type V hyperlipidemia are autosomal-dominant disorders that are more prevalent than type I hyperlipidemia.5
Secondary HTG
Causes of secondary (acquired) HTG include inadequately controlled diabetes, pregnancy, obesity, excess alcohol consumption and medications.5 Secondary HTG alone does not usually represent a risk factor for AP; however, in patients with an underlying dyslipidemia, a secondary cause of HTG may result in clinically significant increases in triglyceride levels that could lead to AP.5
Definition of HTGP
The diagnosis of AP is based on the revised Atlanta classification criteria.15 In general, serum triglyceride levels > 1000 mg/dl are thought to be necessary to induce HTGP, however, there is no clear threshold above which HTG is known to trigger AP.2 The diagnosis of HTGP is therefore definitive when serum triglyceride levels are > 1000 mg/dl at clinical onset. The diagnosis remains probable if the increase of serum triglyceride levels is < 1000 mg/dl, and the presence of other cofactors should also be investigated.
Etiology of HTGP
The most common causes of AP are gallstones, alcoholism and HTG.1,2 Although the risk of AP in patients with elevated serum triglycerides has not been clearly defined, high chylomicron levels are known to be necessary to trigger AP.1 In a population-based study, the risk of AP increased by 4% with every 100 mg/dl increase in triglycerides (after adjustment for covariates and exclusion of patients hospitalized for chronic pancreatitis, gallstones, alcohol-related disorders, renal failure and other biliary disorders).16 Similarly, in a prospective cohort study, the risk of AP was significantly increased in patients with triglycerides ≥ 145 mg/dl compared with patients with triglycerides ≤ 75 mg/dl (adjusted hazard ratio 1.55, 95% confidence interval 1.09–2.21).17 Moreover, in a study in patients with a history of AP, 50% of those who experienced HTGP exhibited impaired clearance of exogenous triglycerides.18
Epidemiology of HTGP
In a retrospective study of patients with severe HTG (triglycerides > 1000 mg/dl), the incidence of AP was 20%.14 In another prospective study, the incidence of AP was higher among patients with severe HTG than those with lower triglyceride levels, with AP being reported in 16% of patients with triglyceride levels > 1772 mg/dl versus 3% of those with triglyceride levels between 886 mg/dl and 1771 mg/dl.19 In other prospective and retrospective studies among patients with AP, the incidence of HTGP was 2.3%–10%.20–22
In younger patients (aged < 50 years), HTGP appears to be more prevalent in males than females, while biliary AP seems to affect a greater percentage of older patients (aged > 70 years).23
AP during pregnancy has an estimated incidence of 1 in 1000–10,000 pregnancies, and the incidence of familial HTGP during pregnancy is estimated to be 1 in 25,000 pregnancies.24 In one study in pregnant female patients with AP, HTGP was reported in 30% of patients.8
Pathophysiology of HTGP
Two theories have been proposed to explain the pathophysiology of HTGP.1,2,5 First, excess triglycerides are transported as triglyceride-rich lipoproteins (chylomicrons), which are hydrolyzed in the vascular bed of the pancreas. This releases high levels of FFAs that exceed the binding capacity of plasma albumin, and unbound FFAs self-aggregate into micellar structures with detergent properties. These toxic structures can cause damage to platelets, the vascular endothelium and acinar cells, which results in ischemia and acidosis. Acidosis further increases FFA toxicity by activation of trypsinogen, which triggers AP.1,2,5 In the second theory, elevated levels of chylomicrons, the largest among lipoproteins, cause an increase plasma viscosity.1,2,5 Plasma hyperviscosity leads to capillary plugging and ischemia, which enhances acidosis and eventually triggers AP.1,2,5 It is likely that both mechanisms contribute to the development of HTGP (Figure 1).
Figure 1.
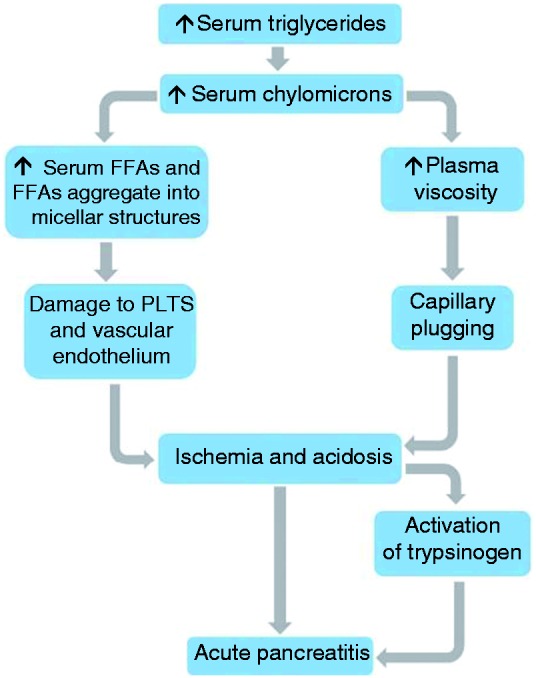
Possible mechanisms involved in the pathophysiology of hypertriglyceridemic pancreatitis (HTGP).
FFA: free fatty acids; PLTS: platelets.
Only a small proportion of patients with HTG develop AP.4 HTGP generally occurs in patients with HTG and one or more secondary factors, including inadequately controlled diabetes, alcoholism, pregnancy, medications and genetic abnormalities.4 Indeed, a genetic analysis of patients with HTG by Chang and colleagues identified two genetic mutations in the cystic fibrosis transmembrane conductance regulator and the tumor necrosis factor promoter that may contribute to HTGP in some patients.25
HTGP in particular conditions
LPLD
LPLD is a rare autosomal recessive form of primary HTG (type I hyperlipidemia) caused by mutations in the LPL gene, located on chromosome 8.6 LPL is expressed in adipose and skeletal and cardiac muscle tissues, where it hydrolyzes triglycerides in chylomicrons and VLDL to release FFAs for energy production in tissues. Patients with LPLD have impaired chylomicron plasma clearance, which results in elevated serum triglycerides and chylomicronemia. In addition, patients with LPLD typically have childhood onset, very severe HTG with episodes of abdominal pain, eruptive cutaneous xanthoma and hepatosplenomegaly. One of the more severe symptoms of LPLD is recurrent AP.6
Pregnancy
HTGP during pregnancy is largely related to familial HTG.10 Severe familial HTG can cause deregulation of lipid levels in pregnant women, particularly during the third trimester, and is known to be associated with AP during pregnancy.24
Elevated estrogen levels during pregnancy lead to reduced LPL activity and increased VLDL synthesis, which results in increased chylomicron and triglyceride levels.24 Triglyceride levels normally reach a peak during the third trimester that is approximately two-fold higher than pre-pregnancy levels.5,24 These elevated triglyceride levels contribute to fetal growth and development,8 and have no consequences for most women. However, in those with an underlying dyslipidemia, it may lead to severe HTG and chylomicronemia that precipitates AP.5
Although most cases of HTGP during pregnancy occur in women with existing dyslipidemia, pregnancy can be a contributing factor in otherwise healthy women. Cases of HTGP during pregnancy have been reported in which there was no history of lipid abnormality, alcohol use or drug intake.25,26
HTGP is often difficult to diagnose in pregnant women owing to changes in biochemical and hematological parameters that occur during pregnancy that can influence the interpretation of diagnostic tests.9,24 Nonetheless, HTGP during pregnancy is a severe condition that can lead to complications, including maternal and fetal death.8,9,25 Early diagnosis followed by prompt management of the pancreatitis and serum triglyceride levels may improve maternal and fetal outcomes.9,25
Uncontrolled diabetes
HTGP is common in patients with inadequately controlled type 1 or 2 diabetes.5 In patients with type 1 diabetes, decreased insulin production results in a marked reduction in LPL activity, which leads to HTG. Hyperinsulinemia and insulin resistance in patients with type 2 diabetes enhances triglyceride production and decreases plasma triglyceride clearance.5
Alcohol consumption
Alcohol consumption on its own does not appear to increase plasma triglycerides to levels that would trigger AP.5 However, in combination with a high-fat diet in a patient with an underlying dyslipidemia, alcohol use may exacerbate HTG and trigger AP.5,7 Studies have indicated that moderate to high alcohol consumption can lead to elevated serum triglycerides, mainly due to increased VLDL secretion.27,28 Alcohol competes for oxidation with FFAs, which results in increased FFA availability for triglyceride synthesis and increased hepatic VLDL secretion that may lead to HTG and chylomicronemia.5 However, given that alcohol abuse is commonly associated with AP,29 the diagnosis of HTGP may be overlooked in some alcoholic patients.
Other at-risk populations
As discussed above, some genetic polymorphisms are associated with HTGP, although very little is known about the role of these genetic factors and how they may increase susceptibility to HTGP.30
Several cases of drug-induced HTGP have been reported. HTGP was reported in patients on estrogen therapy in two case studies and one observational study.31–33 Other case studies have reported HTGP in patients on olanzapine therapy (triglycerides: 416.2 mg/dl), β-blockers (triglycerides: 1009 mg/dl), protease inhibitors (triglycerides: 1778.4 mg/dl), tamoxifen (triglycerides: 920 mg/dl) and isotretinoin (triglycerides: 1501.3 mg/dl).34–38 The probable mechanism of development of HTGP reported in these studies was direct elevation of serum triglycerides. However, it should be noted that because these reports are based on case studies, the level of evidence for causality between the drugs and HTGP is low.
Clinical characteristics
Diagnosis
The clinical presentation of HTGP is similar to AP from other causes,1,2,39 and includes epigastric pain, increased serum levels of pancreatic enzymes and typical AP findings on imaging.4 Among patients with AP, a diagnosis of HTGP should be based on secondary risk factors (e.g. inadequately controlled diabetes, pregnancy or alcohol consumption), physical examination findings (e.g. eruptive xanthomas or signs of metabolic syndrome) and results of biochemical assays.4 Patient history of diabetes and alcohol and medication use, as well as family history of dyslipidemia, should be carefully evaluated.5
It can be difficult to identify HTG as the etiology in AP, as mild to moderate increases in serum triglycerides are commonly observed in the early phases of AP, regardless of cause.4,40 In patients with AP, the presence of lactescent serum is a strong indicator of HTGP.4,5 As triglycerides rapidly decrease within the first 48 hours, serum triglyceride levels should be determined as early as possible after the onset of AP to ensure that a diagnosis of HTGP is not delayed or missed completely.1,2,4 Although serum amylase and/or lipase levels are elevated in patients with AP, serum levels of pancreatic enzymes can be falsely reduced in patients with HTG, most likely due to assay interference, which may also delay diagnosis.4,5 Therefore, patients with suspected HTGP may be referred to a lipid specialist in order to establish and treat the potential causes of HTG as soon as possible.2
Prognosis
The average mortality rate of AP is approximately 2% but can be as high as 20%–30% in patients with persistent organ failure.40,41 Early diagnosis and adequate medical intervention generally results in improved outcomes.39 Detection of milder cases of AP through improved diagnostic tests has also led to progressive decreases in the incidence of case fatality from AP; however, the population mortality rate remains unchanged.39 While HTGP is often associated with a higher severity or a higher complication rate than AP from other etiologies, there is no correlation between the severity of HTG and mortality.10,20 Furthermore, there is a negative correlation between recurrent AP and mortality.39 Although the reasons for this remain unclear, it has been postulated that a naïve pancreas may be more susceptible to severe injury than a pancreas that has experienced previous AP episodes.39 After recovery, the serum triglyceride levels appear to correlate with the risk of recurrence. Indeed, reduction of triglyceride levels at follow-up shows a significant clinical benefit in reducing the relapse rates of pancreatitis in patients with severe hypertriglyceridemia.42 The risk of pancreatitis recurrence has been found to increase by 4% for every 100 mg/dl increase in serum triglyceride concentration.16,42
Treatment and management
Several treatment strategies have been proposed for the management of HTGP (Table 1). Initial treatment of HTGP is similar to that of AP caused by other etiologies, and includes bowel rest, fasting, intravenous hydration and analgesics.1,4,5 As triglyceride levels normalize within 24–48 hours in most patients, estimation of serum triglycerides should be undertaken as early as possible after AP onset when HTGP is suspected.1,4,5
Table 1.
Available treatment options for hypertriglyceridemic acute pancreatitis.
| Treatment option | Activity and mechanism(s) | Treatment duration | Patient selection | Efficacy |
|---|---|---|---|---|
| Diet restriction | Reduction of TG intake | Acute and long-term | All patients | +/– |
| Apheresis | Reduction in circulating chylomicrons | Acute | Patient with severe HTG | + |
| Insulin | Regulation of LPL synthesis from adipose and muscle cells | Acute | Patients with type 1 diabetes | +/– |
| UFH | Release of endothelial LPL, followed by transient serum TG reduction | Acute | All patients | +/– |
| Fibrates | Increase in LPL activity and reduction in TG synthesis | Acute and long-term | All patients except those with LPLD | +/– |
| Antisense Apo-B and Apo-CIII inhibitors | Reduction in Apo mRNA | Long-term | Patients with severe HTG | ++ |
Apo-B, apolipoprotein B; Apo-CIII, apolipoprotein CIII; HTG, hypertriglyceridemia; LPL, lipoprotein lipase; LPLD, lipoprotein lipase deficiency; mRNA, messenger RNA; TG, triglycerides; UFH, unfractionated heparin
In patients with severe HTGP, apheresis can rapidly reduce serum chylomicron and triglyceride levels. However, the procedure has limited availability, is very expensive and there is limited evidence that it reduces HTGP-related morbidity or mortality.2,4,43
Insulin is known to activate LPL, which causes increased chylomicron degradation, and can therefore be used to lower serum triglycerides.2 It can concurrently control hyperglycemia in patients with inadequately controlled diabetes, although its use is not limited to these patients.2,4 Unfractionated heparin also stimulates the release of LPL from endothelial cells, but as the increase in LPL activity is transient and usually followed by a rapid depletion in LPL plasma stores, its use remains controversial.1,2,4
Fibrates (e.g. gemfibrozil), used for the treatment of primary HTG, can lower serum triglycerides via increased LPL and decreased hepatic triglyceride synthesis.5 However, these drugs are associated with adverse effects, including myalgia, elevated liver enzymes and AP.5 Furthermore, fibrates are ineffective in the management of chylomicronemia in patients with LPLD,12,44 and given that LPL gene mutations are associated with insulin resistance,45 insulin therapy is also inappropriate in these patients.
Strict dietary restriction of fat intake can play an important role in the management of HTGP.5 However, in patients with LPLD, compliance with dietary interventions may not decrease the risk of AP.12
Novel therapies for HTG include antisense oligonucleotides against messenger RNA (mRNA) for apolipoprotein B (mipomersen, Genzyme, USA) and C3 (volanesorsen, ISIS 304801, Akcea Therapeutics, Cambridge, MA, USA), small-molecule inhibitors of microsomal triglyceride transfer protein (lomitapide, Aegerion Pharmaceuticals, Cambridge, MA, USA) and diacylglycerol acyltransferase-1 (pradigastat, Novartis, Switzerland), a monoclonal antibody against angiopoietin-like protein 3 (REGN1500, Regeneron Pharmaceuticals, Tarrytown, NY, USA), agonists of peroxisome proliferator-activated receptors and gene therapy for LPLD (alipogene tiparvovec, uniQure N.V., Amsterdam, Netherlands).46 A meta-analysis of eight randomized, controlled trials involving patients with dyslipidemia showed that treatment with mipomersen significantly reduced serum triglyceride levels compared with placebo ( p < 0.00001).47 Another study in three patients with familial chylomicronemia reported that treatment with volanesorsen was associated with a 56%–86% reduction in triglycerides.48 A single-arm, open-label, phase 3 study in patients with homozygous familial hypercholesterolemia reported a 31% reduction in triglycerides after 78 weeks’ treatment with lomitapide.49 Similarly, an open-label clinical study in patients with LPL deficiency reported a 41% and 70% reduction in triglycerides in patients treated with pradigastat 20 mg and pradigastat 40 mg, respectively.50 Also, an open-label trial determining the safety and efficacy of alipogene tiparvovec reported a statistically significant reduction in triglyceride levels of patients with LPLD during weeks 3–12 ( p = 0.0009) and a > 40% reduction in triglycerides 3–12 weeks after treatment.44
In addition to medication, prevention of recurrent AP can be achieved through alcohol abstinence, weight loss, drug discontinuation (in cases of drug-induced HTGP), adequate control of diabetes, dietary restriction of fat intake and administration of fish oil supplements.5
Conclusions
Factors that lead to HTGP include LPLD, inadequately controlled diabetes, excess alcohol consumption and pregnancy in patients with underlying dyslipidemia. Although the symptoms of HTGP are similar to those of AP from other etiologies, clinical outcomes appear to be more severe. An accurate diagnosis is therefore essential in order that patients receive the most appropriate treatment.
Further knowledge of the etiology, pathophysiology and clinical characteristics of HTGP is needed for the continued development of novel therapeutic agents for HTGP. Identification of novel triglyceride-lowering agents is a research focus. Several therapies are now available on the market or in late-phase clinical trials, including antisense oligonucleotides against mRNA for apolipoproteins for patients with severe HTG.
Acknowledgments
We would like to thank Cécile Duchesnes, PhD, and Sarah Greig, PhD, of Springer Healthcare Communications, for medical writing assistance in preparing the outline and subsequent drafts of this manuscript. This assistance was funded by Chiesi Farmaceutici S.p.A., Italy.
Declaration of conflicting interests
Luca Frulloni has received grants from Chiesi Pharmaceutical for delivering lectures and is a member of the advisory board of Chiesi Pharmaceutical. Nicolò de Pretis and Antonio Amodio have nothing to declare.
Funding
Medical writing assistance for this manuscript was funded by Chiesi Farmaceutici S.p.A., Italy.
References
- 1.Valdivielso P, Ramírez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med 2014; 25: 689–694. [DOI] [PubMed] [Google Scholar]
- 2.Ewald N, Hardt PD, Kloer HU. Severe hypertriglyceridemia and pancreatitis: Presentation and management. Curr Opin Lipidol 2009; 20: 497–504. [DOI] [PubMed] [Google Scholar]
- 3.Papachristou GI, Machicado JD, Stevens T, et al. Acute Pancreatitis Patient Registry to Examine Novel Therapies in Clinical Experience (APPRENTICE): An international, multicenter consortium for the study of acute pancreatitis. Ann Gastroenterol 2017; 30: 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scherer J, Singh VP, Pitchumoni CS, et al. Issues in hypertriglyceridemic pancreatitis: An update. J Clin Gastroenterol 2014; 48: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yadav D, Pitchumoni CS. Issues in hyperlipidemic pancreatitis. J Clin Gastroenterol 2003; 36: 54–62. [DOI] [PubMed] [Google Scholar]
- 6.Burnett JR, Hooper AJ and Hegele RA. Familial lipoprotein lipase deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds) GeneReviews®. Seattle, WA: University of Washington, Seattle, 1999 (updated 2017 June 22), http://www.ncbi.nlm.nih.gov/pubmed/20301485. [PubMed]
- 7.Pownall HJ, Ballantyne CM, Kimball KT, et al. Effect of moderate alcohol consumption on hypertriglyceridemia: A study in the fasting state. Arch Intern Med 1999; 159: 981–987. [DOI] [PubMed] [Google Scholar]
- 8.Jin J, Yu YH, Zhong M, et al. Analyzing and identifying risk factors for acute pancreatitis with different etiologies in pregnancy. J Matern Fetal Neonatal Med 2015; 28: 267–271. [DOI] [PubMed] [Google Scholar]
- 9.Xu Q, Wang S, Zhang Z. A 23-year, single-center, retrospective analysis of 36 cases of acute pancreatitis in pregnancy. Int J Gynaecol Obstet 2015; 130: 123–126. [DOI] [PubMed] [Google Scholar]
- 10.Tsuang W, Navaneethan U, Ruiz L, et al. Hypertriglyceridemic pancreatitis: Presentation and management. Am J Gastroenterol 2009; 104: 984–991. [DOI] [PubMed] [Google Scholar]
- 11.Fredrickson DS, Lees RS. A system for phenotyping hyperlipoproteinemia. Circulation 1965; 31: 321–327. [DOI] [PubMed] [Google Scholar]
- 12.Gaudet D, de Wal J, Tremblay K, et al. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl 2010; 11: 55–60. [DOI] [PubMed] [Google Scholar]
- 13.Brahm A, Hegele RA. Hypertriglyceridemia. Nutrients 2013; 5: 981–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas 2008; 37: 13–2. [DOI] [PubMed] [Google Scholar]
- 15.Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62: 102–111. [DOI] [PubMed] [Google Scholar]
- 16.Murphy MJ, Sheng X, MacDonald TM, et al. Hypertriglyceridemia and acute pancreatitis. JAMA Intern Med 2013; 173: 162–164. [DOI] [PubMed] [Google Scholar]
- 17.Lindkvist B, Appelros S, Regner S, et al. A prospective cohort study on risk of acute pancreatitis related to serum triglycerides, cholesterol and fasting glucose. Pancreatology 2012; 12: 317–324. [DOI] [PubMed] [Google Scholar]
- 18.Domínguez-Muñoz JE, Jünemann F, Malfertheiner P. Hyperlipidemia in acute pancreatitis. Cause or epiphenomenon? Int J Pancreatol 1995; 18: 101–106. [DOI] [PubMed] [Google Scholar]
- 19.Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: A retrospective cohort study. Lipids Health Dis 2011; 10: 157–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson F, Thomson SR, Clarke DL, et al. Dyslipidaemic pancreatitis clinical assessment and analysis of disease severity and outcomes. Pancreatology 2009; 9: 252–257. [DOI] [PubMed] [Google Scholar]
- 21.Cavallini G, Frulloni L, Bassi C, et al. Prospective multicentre survey on acute pancreatitis in Italy (ProInf-AISP): Results on 1005 patients. Dig Liver Dis 2004; 36: 205–211. [DOI] [PubMed] [Google Scholar]
- 22.Charlesworth A, Steger A, Crook MA. Acute pancreatitis associated with severe hypertriglyceridaemia; a retrospective cohort study. Int J Surg 2015; 23(Pt A): 23–27. [DOI] [PubMed] [Google Scholar]
- 23.Zheng Y, Zhou Z, Li H, et al. A multicenter study on etiology of acute pancreatitis in Beijing during 5 years. Pancreas 2015; 44: 409–414. [DOI] [PubMed] [Google Scholar]
- 24.Ducarme G, Maire F, Chatel P, et al. Acute pancreatitis during pregnancy: A review. J Perinatol 2014; 34: 87–94. [DOI] [PubMed] [Google Scholar]
- 25.Gupta N, Ahmed S, Shaffer L, et al. Severe hypertriglyceridemia induced pancreatitis in pregnancy. Case Rep Obstet Gynecol 2014; 2014: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Athyros VG, Giouleme OI, Nikolaidis NL, et al. Long-term follow-up of patients with acute hypertriglyceridemia-induced pancreatitis. J Clin Gastroenterol 2002; 34: 472–475. [DOI] [PubMed] [Google Scholar]
- 27.Pownall HJ. Dietary ethanol is associated with reduced lipolysis of intestinally derived lipoproteins. J Lipid Res 1994; 35: 2105–2113. [PubMed] [Google Scholar]
- 28.Taskinen MR, Välimäki M, Nikkilä EA, et al. Sequence of alcohol-induced initial changes in plasma lipoproteins (VLDL and HDL) and lipolytic enzymes in humans. Metabolism 1985; 34: 112–119. [DOI] [PubMed] [Google Scholar]
- 29.Clemens DL, Schneider KJ, Arkfeld CK, et al. Alcoholic pancreatitis: New insights into the pathogenesis and treatment. World J Gastrointest Pathophysiol 2016; 7: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang YT, Chang MC, Su TC, et al. Association of cystic fibrosis transmembrane conductance regulator (CFTR) mutation/variant/haplotype and tumor necrosis factor (TNF) promoter polymorphism in hyperlipidemic pancreatitis. Clin Chem 2008; 54: 131–138. [DOI] [PubMed] [Google Scholar]
- 31.Goldenberg NM, Wang P, Glueck CJ. An observational study of severe hypertriglyceridemia, hypertriglyceridemic acute pancreatitis, and failure of triglyceride-lowering therapy when estrogens are given to women with and without familial hypertriglyceridemia. Clin Chim Acta 2003; 332: 11–19. [DOI] [PubMed] [Google Scholar]
- 32.Abraham M, Mitchell J, Simsovits D, et al. Hypertriglyceridemic pancreatitis caused by the oral contraceptive agent Estrostep. J Intensive Care Med 2015; 30: 303–307. [DOI] [PubMed] [Google Scholar]
- 33.Castro MR, Nguyen TT, O’Brien T. Clomiphene-induced severe hypertriglyceridemia and pancreatitis. Mayo Clin Proc 1999; 74: 1125–1128. [DOI] [PubMed] [Google Scholar]
- 34.Chapman SJ, Woolley IJ, Visvanathan K, et al. Acute pancreatitis caused by tipranavir/ritonavir-induced hypertriglyceridaemia. AIDS 2007; 21: 532–533. [DOI] [PubMed] [Google Scholar]
- 35.Durrington PN, Cairns SA. Acute-pancreatitis: A complication of beta-blockade. Br Med J (Clin Res Ed) 1982; 284: 1016–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waage C, Carlsson H, Nielsen EW. Olanzapine-induced pancreatitis: A case report. JOP 2004; 5: 388–391. [PubMed] [Google Scholar]
- 37.Alagozlu H, Cindoruk M, Unal S. Tamoxifen-induced severe hypertriglyceridaemia and acute pancreatitis. Clin Drug Investig 2006; 26: 297–302. [DOI] [PubMed] [Google Scholar]
- 38.Flynn WJ, Freeman PG, Wickboldt LG. Pancreatitis associated with isotretinoin-induced hypertriglyceridemia. Ann Intern Med 1987; 107: 63–63. [DOI] [PubMed] [Google Scholar]
- 39.Yadav D, Lowenfels AB. Trends in the epidemiology of the first attack of acute pancreatitis: A systematic review. Pancreas 2006; 33: 323–330. [DOI] [PubMed] [Google Scholar]
- 40.Gan SI, Edwards AL, Symonds CJ, et al. Hypertriglyceridemia-induced pancreatitis: A case-based review. World J Gastroenterol 2006; 12: 7197–7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forsmark CE, Vege SS, Wilcox CM. Acute pancreatitis. N Engl J Med 2016; 375: 1972–1981. [DOI] [PubMed] [Google Scholar]
- 42.Christian JB, Arondekar B, Buysman EK, et al. Determining triglyceride reductions needed for clinical impact in severe hypertriglyceridemia. Am J Med 2014; 127: 36–44. e31. [DOI] [PubMed] [Google Scholar]
- 43.Costantini N, Mameli A, Marongiu F. Plasmapheresis for preventing complication of hypertriglyceridemia: A case report and review of literature. Am J Ther 2016; 23: e288–e291. [DOI] [PubMed] [Google Scholar]
- 44.Gaudet D, Méthot J, Déry S, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: An open-label trial. Gene Ther 2013; 20: 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hölzl B, Iglseder B, Sandhofer A, et al. Insulin sensitivity is impaired in heterozygous carriers of lipoprotein lipase deficiency. Diabetologia 2002; 45: 378–384. [DOI] [PubMed] [Google Scholar]
- 46.Gryn SE, Hegele RA. Novel therapeutics in hypertriglyceridemia. Curr Opin Lipidol 2015; 26: 484–491. [DOI] [PubMed] [Google Scholar]
- 47.Panta R, Dahal K, Kunwar S. Efficacy and safety of mipomersen in treatment of dyslipidemia: A meta-analysis of randomized controlled trials. J Clin Lipidol 2015; 9: 217–225. [DOI] [PubMed] [Google Scholar]
- 48.Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014; 371: 2200–2206. [DOI] [PubMed] [Google Scholar]
- 49.Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013; 381: 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyers CD, Tremblay K, Amer A, et al. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipids Health Dis 2015; 14: 8–8. [DOI] [PMC free article] [PubMed] [Google Scholar]