

Abstract
Background
The natural history of adult liver disease due to α1-antitrypsin deficiency (A1AD) remains poorly understood.
Objective
We investigated whether heterozygosity for the Z-allele predisposes for the development of clinically significant portal hypertension (CSPH). Moreover, we aimed to non-invasively assess the prevalence of liver fibrosis and hepatic steatosis in adults with A1AD treated by pulmonologists.
Methods
SERPINA1 rs28929474 (Z-allele) was genotyped in 315 patients with CSPH (hepatic venous pressure gradient ≥10 mmHg; cases) and 248 liver donors (controls). In addition, 31 adults with A1AD (Pi*ZZ/Pi*SZ) and 11 first-degree relatives (Pi*MZ/Pi*MS) underwent liver stiffness and controlled attenuation parameter (CAP) measurement.
Results
Heterozygosity for the Z-allele was observed in 6.7% of patients with CSPH and 2.8% of liver donors. Thus, harboring the Z-allele was associated with increased odds of CSPH (odds ratio: 2.47; 95% confidence interval: 1.03–5.9; P = 0.042). Among Pi*ZZ/Pi*SZ patients, 23%/3% had liver stiffness values indicative of liver fibrosis ( ≥F2/ ≥F3). Interestingly, 65%/52% of Pi*ZZ/Pi*SZ patients had CAP values indicative of hepatic steatosis ( ≥S1/ ≥S2).
Conclusions
Heterozygosity for the Z-allele predisposes for the development of CSPH, confirming its role as a genetic (co)factor in liver disease. Pi*ZZ/SZ patients rarely develop liver fibrosis ≥F3 during adulthood; however, liver fibrosis ≥F2 is common. Elevated CAP values hint at underlying hepatic steatosis, which might promote liver fibrosis progression.
Keywords: Alpha 1-antitrypsin deficiency, portal hypertension, liver fibrosis, hepatic steatosis, transient elastography, controlled attenuation parameter
Key points
1. Summarize the established knowledge on this subject
α1-antitrypsin deficiency (A1AD) is one of the most common genetic disorders, since about 1:25 European Caucasians harbor at least one Z-allele and about 1:2000 are homozygous for the Z-allele (Pi*ZZ).
While the pulmonary manifestation of A1AD has been intensively studied, the natural history of adult liver disease due to A1AD remains poorly understood.
2. What are the significant and/or new findings of this study?
In a genetic sub-study (case-control design), heterozygosity for the Z-allele predisposed for the development of clinically significant portal hypertension, confirming its role as a genetic (co)factor.
Moreover, we evaluated thoroughly characterized α1-antitrypsin-deficient patients treated by pulmonologists (Pi*ZZ/Pi*SZ), as well as their first-degree relatives (Pi*MZ/Pi*MS), by transient elastography with controlled attenuation parameter (CAP) measurement.
Pi*ZZ/SZ patients rarely develop fibrosis ≥F3; however, about one-quarter had fibrosis ≥F2, which warrants the use of non-invasive methods in this population.
Elevated CAP values were observed in about two-thirds of Pi*ZZ/SZ patients and hint at underlying hepatic steatosis, which might promote liver fibrosis progression.
Introduction
α1-antitrypsin deficiency (A1AD) is a consequence of misfolding and intracellular polymerization of mutant α1-antitrypsin within the endoplasmic reticulum (ER) of hepatocytes (toxic gain-of-function), which induces a proinflammatory and fibrogenic response by nuclear factor-κB activation. Moreover, A1AD increases the sensitivity to ER stress in the presence of concomitant liver diseases.1
In contrast, the lack of α1-antitrypsin in the lung (loss-of-function) leads to early-onset panlobular basal emphysema, the typical manifestation of A1AD.
A1AD is one of the most common genetic disorders, since about 1:25 European Caucasians harbor at least one Z-allele (SERPINA1 rs28929474[T]). About 1:2000 is homozygous for the Z-allele (Pi*ZZ). While the pulmonary manifestation of A1AD has been intensively studied, the natural history of adult liver disease due to A1AD remains poorly understood.
In 1986, Eriksson and co-workers observed cirrhosis in about one-third of autopsied patients with the Z phenotype (i.e. mostly Pi*ZZ genotype).2 Based on these and other observations, A1AD is considered a separate aetiological entity.1 However, until recently, liver biopsy was the only method for assessing the extent of hepatic involvement. Since liver biopsy is limited by its invasiveness, it is mostly performed in patients with findings suggestive of advanced chronic liver disease (ACLD), and thus, biopsy series in these referral populations might substantially overestimate the risk of liver fibrosis/cirrhosis. Population-based surveys, on the other hand, only capture the tip of the iceberg (e.g. patients who have already progressed to ACLD), rather than providing information on the complete spectrum of sub-clinical liver involvement.3,4 Promising treatment approaches are currently moving from preclinical to clinical development.5 Thus, it is of high relevance that we improve our understanding of the natural history of this condition and identify patients who have already developed significant liver fibrosis, in order to facilitate the design of such clinical trials.
Besides Pi*ZZ patients, heterozygosity for the Z-allele might be a genetic (co)factor for the development of ACLD.6–9 However, this association has not been observed throughout all aetiologies/cohorts, and thus, more data is required to facilitate genetic counselling.10,11
In a genetic sub-study (case-control design), we investigated whether heterozygosity for the Z-allele predisposes for the development of ACLD, as indicated by the presence of clinically significant portal hypertension (CSPH).
Moreover, we assessed thoroughly characterized A1AD patients treated by pulmonologists (Pi*ZZ/Pi*SZ) as well as their first-degree relatives (Pi*MZ/Pi*MS) by transient elastography (TE) with controlled attenuation parameter (CAP) measurement, a well-validated non-invasive method for the assessment of liver fibrosis and hepatic steatosis.
Patients and methods
Sub-studies
In sub-study I, the SERPINA1 rs28929474(T) (Z-allele) was genotyped in 315 patients who were diagnosed with CSPH at the Medical University of Vienna between 2004 and 2014 (cases) and 248 donors from liver transplantations performed at the Medical University of Innsbruck (controls).12
In sub-study II, 31 adults with A1AD (Pi*ZZ/Pi*SZ) and 11 first-degree relatives (Pi*MZ/Pi*MS) underwent liver stiffness and CAP measurement within a prospective study in 2015/2016. Patients were identified based on a registry of subjects with A1AD treated by pulmonologists and directly contacted by the investigators.13
Please see the Supplementary material for information on hepatic venous pressure gradient (HVPG) measurement and SERPINA1 genotyping (sub-study I), as well as liver stiffness and CAP measurement, laboratory tests, the short form (36) health survey (SF-36), and the alcohol use disorders identification test (AUDIT) (sub-study II).
Results
Sub-study I
Characteristics of patients with CSPH
Most patients were male (73.9% (232/314)) with a median age of 52.7 (16) years. Hepatitis C (47.8% (150/314)), alcoholic liver disease (31.2% (98/314)), and non-alcoholic fatty liver disease (NAFLD) (7.3% (23/314)) were the most common aetiologies. The prevalence of diabetes was 19.4% (61/314). The patients had a median HVPG of 17 (8) mmHg and a median model for end-stage liver disease score of 10 (6.8) points.
After excluding one Pi*ZZ patient who underwent liver transplantation, 6.7% (21/314) of patients with CSPH were heterozygous for the Z-allele.
Except for aetiology of liver disease, a comparison of patients with and without the Z-allele revealed no statistically significant differences (Table 1). Interestingly, the Z-allele was overrepresented in patients with cryptogenic cirrhosis when compared to patients with known aetiology (33.3% (3/9) vs. 5.9% (18/305); P = 0.017).
Table 1.
Comparison of patients with clinically significant portal hypertension with and without the Z-allele.
| Parameter | All, n = 314 | No Z-allele n = 293 | Z-allele n = 21 | P-value |
|---|---|---|---|---|
| Age, years | 52.7 (16) | 52.8 (16) | 51.8 (18.2) | 0.652 |
| Sex | ||||
| Male | 232 (73.9%) | 216 (73.7%) | 16 (76.2%) | 0.803 |
| Females | 82 (26.1%) | 77 (26.3%) | 5 (23.8%) | |
| Aetiology | ||||
| Hepatitis C | 150 (47.8%) | 143 (48.8%) | 7 (33.3%) | |
| Alcoholic liver disease | 98 (31.2%) | 90 (30.7%) | 8 (38.1%) | |
| NAFLD | 23 (7.3%) | 22 (7.5%) | 1 (4.8%) | |
| Other | 34 (10.8%) | 32 (11%) | 2 (9.5%) | |
| Unknown | 9 (2.9%) | 6 (2%) | 3 (14%) | |
| Diabetes | 61 (19.4%) | 59 (20.1%) | 2 (9.5%) | 0.39 |
| HVPG, mmHg | 17 (8) | 17 (8) | 19 (9) | 0.632 |
| MELD score, points | 10 (6.8) | 10 (6.78) | 10 (6) | 0.393 |
| Platelet count, G × L−1 | 100 (73) | 98.5 (68) | 122 (100) | 0.118 |
| AST, U × L−1 | 65 (56) | 65 (56) | 53 (78) | 0.565 |
| ALT, U × L−1 | 46 (50) | 46 (53) | 42 (37) | 0.772 |
| GGT, U × L−1 | 129 (150) | 125 (152) | 149 (90) | 0.131 |
| Albumin, g × L−1 | 35.5 (8.3) | 35.7 (8.3) | 34.1 (10.2) | 0.612 |
| Triglycerides, mg × dL−1 | 87 (53) | 86 (52) | 86 (74) | 0.482 |
| Cholesterol, mg × dL−1 | 144 (52) | 144 (52) | 165 (41) | 0.055 |
| LDL, mg × dL−1 | 81 (39.2) | 80.8 (38.8) | 81.8 (30.6) | 0.162 |
| HDL, mg × dL−1 | 41 (21) | 41 (20) | 51 (23) | 0.063 |
| Glucose, mg × dL−1 | 108 (38) | 108 (37) | 110 (45) | 0.996 |
| CRP, mg × L−1 | 0.34 (0.7) | 0.34 (0.7) | 0.345 (0.79) | 0.955 |
| vWF antigen, % | 324 (151) | 323 (153) | 338 (120) | 0.142 |
NAFLD: non-alcoholic fatty liver disease; HVPG: hepatic venous pressure gradient; MELD: model for end-stage liver disease; AST: aspartate transaminase; ALT: alanine transaminase; GGT: gamma-glutamyltransferase; LDL: low-density lipoprotein; HDL: high-density lipoprotein; CRP: C-reactive protein; vWF: von Willebrand factor.
Comparison of the proportions of patients heterozygous for the Z-allele
Among liver donors, 2.8% (7/248) were heterozygous for the Z-allele (P = 0.049 vs. CSPH), which is similar to the expected frequency in the Austrian population (Figure 1). Harboring the Z-allele increased the odds of CSPH (odds ratio: 2.47, 95% confidence interval: 1.03–5.9; P = 0.042).
Figure 1.
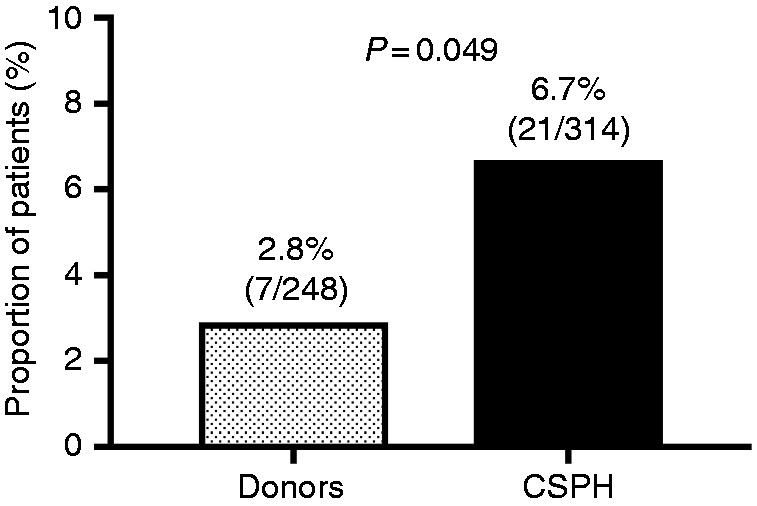
Comparison of the proportion of patients heterozygous for the Z-allele between patients with clinically significant portal hypertension (CSPH; cases) and liver donors (controls).
Sub-study II
Enrollment
Forty-two A1AD index patients treated by pulmonologists (Pi*ZZ/SZ) were informed about the study. Among these, 31 patients (73.8%) agreed to participate in sub-study II. In addition, 11 first-degree relatives (Pi*MZ/MS) were enrolled.
Patient characteristics
Please see Table 2 and the Supplementary material for patient characteristics.
Table 2.
Comparison of subjects with significant liver fibrosis ( ≥F2), or without (F0/F1), as defined by a liver stiffness cut-off of 7.1 kPa.
| Parameter | All, n = 42 | Liver fibrosis F0/F1, n = 34 | Liver fibrosis ≥F2, n = 8 | P-value |
|---|---|---|---|---|
| Age, years | 54.2 (27.5) | 49.1 (27.7) | 63.7 (15.4) | 0.21 |
| Sex | ||||
| Male | 25 (59.5%) | 16 (47.1%) | 7 (87.5%) | 0.114 |
| Females | 17 (40.5%) | 18 (52.9%) | 1 (12.5%) | |
| BMI, kg × m−2 | 25.1 ± 0.6 | 24.5 ± 0.7 | 27.6 ± 1.4 | 0.049 |
| <25 kg × m−2 | 21 (50%) | 18 (52.9%) | 3 (37.5%) | 0.061 |
| 25-29.9 kg × m−2 | 16 (38.1%) | 14 (41.2%) | 2 (25%) | |
| ≥30 kg × m−2 | 5 (11.9%) | 2 (5.9%) | 3 (37.5%) | |
| Group | ||||
| Treated by pulmonologists | 31 (73.8%) | 24 (70.6%) | 7 (87.5%) | 0.419 |
| First-degree relatives | 11 (26.2%) | 10 (29.4%) | 1 (12.5%) | |
| SERPINA1 genotype | ||||
| ZZ | 28 (66.7%) | 21 (61.8%) | 7 (87.5%) | 0.486 |
| MZ | 10 (23.8%) | 9 (26.5%) | 1 (12.5%) | |
| SZ | 3 (7.1%) | 3 (8.8%) | 0 (0%) | |
| MS | 1 (2.4%) | 1 (2.9%) | 0 (0%) | |
| CAP, dB × m−1a | 260 ± 9 | 253 ± 10 | 285 ± 18 | 0.148 |
| ≥248 dB × m−1 ( ≥S1)a | 25 (59.5%) | 18 (53%) | 7 (87.5%) | 0.078 |
| ≥268 dB × m−1 ( ≥S2)a | 18 (42.9%) | 12 (35.3%) | 6 (75%) | 0.056 |
| ≥280 dB × m−1 (S3)a | 15 (35.7%) | 10 (29.4%) | 5 (62.5%) | 0.11 |
| Platelet count, G × L−1 | 209 (82) | 221 (83) | 174 (23) | < 0.001 |
| Prothrombin time, % | 90.8 ± 2.6 | 92.3 ± 3 | 84.5 ± 4.2 | 0.242 |
| AP, U × L−1 | 67.9 ± 2.2 | 67.5 ± 2.4 | 69.9 ± 5.4 | 0.668 |
| AST, U × L−1 | 27.1 ± 1.5 | 26.5 ± 1.4 | 29.8 ± 5.0 | 0.397 |
| ALT, U × L−1 | 24 (24) | 22.5 (25) | 27 (26) | 0.498 |
| GGT, U × L−1 | 31 (19) | 27 (19) | 45 (62) | 0.013 |
| Bilirubin, mg × dL−1 | 0.62 (0.43) | 0.63 (0.41) | 0.58 (0.52) | 0.888 |
| Albumin, g × L−1 | 44.1 (4.4) | 44.1 (4.6) | 43.9 (4.0) | 0.440 |
| Triglycerides, mg × dL−1 | 90 (64) | 80 (60) | 94.5 (233) | 0.352 |
| Cholesterol, mg × dL−1 | 198 ± 6 | 199.6 ± 7 | 191 ± 11 | 0.579 |
| LDL, mg × dL−1 | 113 ± 5 | 114 ± 6 | 110 ± 9 | 0.756 |
| HDL, mg × dL−1 | 63.1 ± 2.6 | 66.2 ± 2.9 | 50.1 ± 4.1 | 0.014 |
| HbA1c, % | 5.3 (0.6) | 5.3 (0.6) | 5.4 (1) | 0.63 |
| HOMA-IR, mg × µU × dL−1 × mL−1 | 2.16 (2.11) | 2.01 (1.99) | 3.31 (2.61) | 0.107 |
| hsCRP, mg × L−1 | 0.13 (0.243) | 0.13 (0.25) | 0.09 (0.1) | 0.218 |
| vWF antigen, % | 136.5 (63) | 136.5 (59) | 153.5 (120) | 0.368 |
| α1-antitrypsin, mg × dL−1 | 73.7 (68.3) | 73.9 (69.3) | 66.6 (59.9) | 0.519 |
| SF-36 PCS score, points | 38.9 ± 2 | 38.9 ± 2.2 | 39.1 ± 4.8 | 0.978 |
| SF-36 MCS score, points | 55.5 (8.6) | 56.0 (9.4) | 55.2 (8.4) | 1 |
| AUDIT, points | 3 (4) | 3 (4) | 1.5 (4) | 0.32 |
| < 8 points | 39 (92.9%) | 31 (91.2%) | 8 (100%) | 0.609 |
| ≥8 points | 3 (7.1%) | 3 (8.8%) | 0 (0%) | |
Available in 41 patients.
BMI: body mass index; CAP: controlled attenuation parameter; AP: alkaline phosphatase; AST: aspartate transaminase; AST: alanine transaminase; GGT: gamma-glutamyltransferase; LDL: low-density lipoprotein; HDL: high-density lipoprotein; HbA1c: glycated hemoglobin; HOMA-IR: homeostatic model assessment of insulin resistance; hsCRP: high-sensitivity C-reactive protein; vWF: von Willebrand factor; SF-36: short form (36) health survey; PCS: physical component summary; MCS: mental component summary; AUDIT: alcohol use disorders identification test.
Liver stiffness
Liver stiffness was comparable between Pi*ZZ/Pi*SZ patients (5.77 ± 0.3 kPa) and their first-degree relatives (Pi*MZ/MS; 5.36 ± 0.6 kPa; P = 0.518; Figure 2).
Figure 2.

Comparison of (a) liver stiffness and (b) controlled attenuation parameter (CAP) values between Pi*ZZ/SZ patients and their first-degree relatives (Pi*MZ/MS).
Among Pi*ZZ/Pi*SZ patients, 22.6% (7/31) and 3.2% (1/31) had liver stiffness values indicative of liver fibrosis ≥F2 and ≥F3, respectively (Figure 3).
Figure 3.
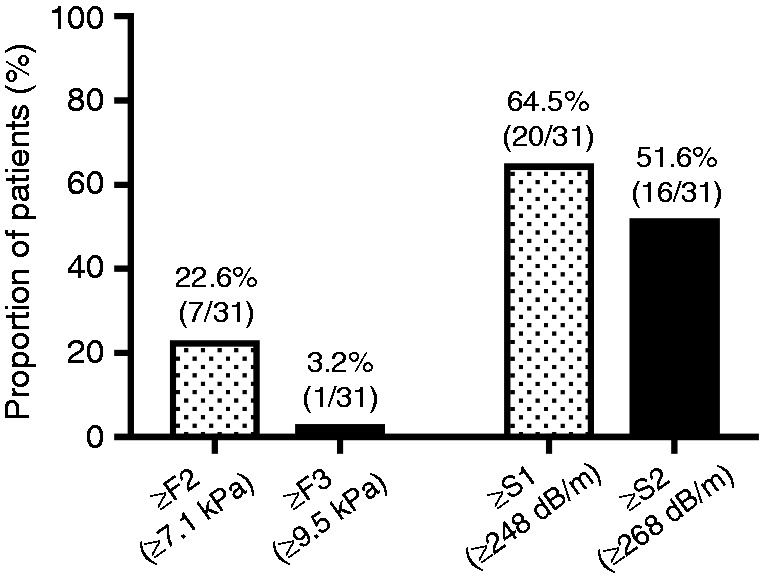
Prevalence of significant and advanced fibrosis (left) as well as controlled attenuation parameter (CAP) values indicative of hepatic steatosis ≥S1 and ≥S2 (right) in patients with α1-antitrypsin deficiency who were treated by pulmonologists (Pi*ZZ/SZ).
Risk factors for liver fibrosis ≥F2
Combining all subjects included in sub-study II, subjects with liver fibrosis ≥F2 had higher body mass index (BMI; 27.6 ± 1.4 vs. 24.5 ± 0.7 kg × m−2; P = 0.049) and serum gamma-glutamyltransferase levels (GGT; 45 (62) vs. 27 (19) U/L; P = 0.013) (Table 2). In contrast, lower serum high-density lipoprotein (HDL) cholesterol levels (50.1 ± 4.1 vs. 66.2 ± 2.9 mg × dL−1; P = 0.014) were observed in the liver fibrosis ≥F2 group. Moreover, we observed trends towards a higher prevalence of hepatic steatosis ≥S1 (87.5% (7/8) vs. 53% (18/34); P = 0.078) and ≥S2 (75% (6/8) vs. 35.3% (12/34); P = 0.078), as well as higher HOMA-IR (homeostatic model assessment of insulin resistance) levels (3.31 (2.61) vs. 2.01 (1.99) mg × µU × dL−1 × mL−1; P = 0.107) in subjects with liver fibrosis ≥F2. As expected, subjects with liver fibrosis ≥F2 showed lower platelet counts (174 (23) vs. 221(83) G × L−1; P < 0.001).
If analysed as a continuous variable, liver stiffness tended to correlate with the same parameters (Supplementary Table 2).
CAP values
Pi*ZZ/Pi*SZ patients had numerically higher CAP values (267 ± 9 dB × m−1) than their first-degree relatives (Pi*MZ/MS; 237 ± 22 dB × m−1; P = 0.148; Figure 2).
Interestingly, 64.5% (20/31) of Pi*ZZ/Pi*SZ patients had CAP values indicative of hepatic steatosis ≥S1 and 51.6% (16/31) showed values suggestive of ≥S2 (Figure 3).
CAP correlated positively with age and metabolic parameter such as BMI (ρ = 0.449; P = 0.003), HbA1c (ρ = 0.394; P = 0.013), and HOMA-IR (ρ = 0.474; P = 0.002) (Supplementary Table 2). We observed direct correlations with high-sensitive C-reactive protein (hsCRP; ρ = 0.418; P = 0.008) as a marker of inflammation, and von Willebrand factor antigen (vWF; ρ = 0.451; P = 0.003) as a marker of endothelial dysfunction. Moreover, CAP tended to correlate positively with liver stiffness (ρ = 0.304; P = 0.053).
Concomitant liver diseases
Overall, only three subjects (7.1% (3/42)) reported at-risk drinking (AUDIT ≥8 points); however, serum carbohydrate-deficient transferrin levels were < 2.6% in all individuals (Table 2).14 Three subjects (7.1% (3/42)) were hepatitis B surface- and core antibody-positive, while we did not observe hepatitis B surface antigen positivity. There was no evidence of other chronic liver diseases.
Please see the Supplementary material for information on hepatitis A/B vaccination and hepatitis E seroprevalence, as well as health-related quality of life.
Discussion
To assess the role of the Z-allele as a genetic (co)factor for the development of ACLD, we investigated whether heterozygosity for the Z-allele predisposes for the development of CSPH (sub-study I). Harboring the Z-allele was associated with approximately 2.5-fold increased odds of CSPH. This might be explained by the impact of A1AD on hepatic inflammation and fibrosis.1 Interestingly, the Z-allele was overrepresented in patients with cryptogenic cirrhosis (33.3%) when compared to patients with known aetiology (5.9%). This confirms a previous report of Graziadei et al., who observed a prevalence of 26.9% in patients who underwent liver transplantation for cryptogenic cirrhosis.7 Thus, heterozygosity for the Z-allele might have implications in patients without a known aetiology. This prompts SERPINA1 genotyping in patients with cryptogenic cirrhosis, as well as in patients with findings indicative of an otherwise unexplained chronic liver disease. The group of patients with cryptogenic cirrhosis might have comprised a considerable proportion of NAFLD patients (i.e. ‘burnt-out non-alcoholic steatohepatitis’); however, a previous study did not observe a link between A1AD alleles and liver fibrosis in NAFLD.15–17 Moreover, we did not observe an overrepresentation of the Z-allele in NAFLD patients.
Since sub-study I confirmed the role of the Z-allele as a genetic (co)factor for the development of ACLD, we evaluated thoroughly characterized α1-antitrypsin-deficient patients treated by pulmonologists (Pi*ZZ/Pi*SZ) as well as their first-degree relatives (Pi*MZ/Pi*MS) by TE with CAP to further investigate the relationship between A1AD genotype and liver phenotype (sub-study II).
In sub-study II, we observed a low prevalence of liver fibrosis ≥F3 ( ≥9.5 kPa; 3.2%), although about one-quarter of patients showed liver stiffness values indicative of liver fibrosis ≥F2 ( ≥7.1 kPa; 22.6%). Interestingly, the prevalence of liver fibrosis ≥F3 in our study was considerably lower than the proportion of patients with ACLD reported by previous studies.2,18
In the only prospective study performing biopsies to investigate liver involvement in Pi*ZZ patients (n = 57), Dawwas et al. observed liver fibrosis ≥F3 in 17.5% of the overall study population.18 Liver biopsies were only performed in patients with clinical, laboratory or radiological evidence of liver injury, and some patients declined liver biopsy. Thus, the authors might have even underestimated the prevalence of liver fibrosis ≥F3 in their cohort, since the diagnosis might have been missed in patients with sub-clinical liver disease who were not referred for liver biopsy. Mean/median age and BMI were very similar to our study and the authors reported a high participation rate of 89%, which limits the risk of selection bias. Therefore, the different study settings seem to be the only plausible explanation for the discrepancy in the proportion of patients with liver fibrosis ≥F3: Dawwas and co-workers recruited study participants among patients referred to the respiratory clinic of a tertiary care centre.18 In contrast, the Pi*ZZ/SZ patients included in our study were identified based on a registry of subjects with A1AD treated by pulmonologists and directly contacted by the investigators.13 Since our study was not restricted to patients treated at a tertiary care centre, our study population is presumably more representative for the overall population of Pi*ZZ/SZ patients.
Another recently published study used acoustic radiation force impulse (ARFI) elastography, a non-invasive method that is less commonly applied than TE.19,20 Importantly, ARFI was performed in only 39.5% of the study population, which puts this study at particularly high risk of selection bias. Moreover, the proportion of study participants with liver stiffness values suggestive of liver fibrosis ≥F2 was comparable between Pi*ZZ (25%) and Pi*MM subjects (22%), which questions the reliability of the measurements. Of note, study participants were fasted for only 3 h and liver stiffness assessed by ARFI elastography has been shown to remain numerically increased, even 3 h after a meal intake.21
Kim and colleagues performed liver biopsies in a small series (n = 11) of Pi*ZZ patients to investigate the value of magnetic resonance elastography for liver fibrosis.22 The proportion of patients with liver fibrosis ≥F2 was 18.2%, which is very similar to our study. However, the small sample size and the strict in- and exclusion criteria substantially limit the conclusions that can be drawn from this study. Nevertheless, its results support the use of non-invasive methods in patients with A1AD, since the diagnostic performance in A1AD was comparable to NAFLD patients who served as a control group.
Interestingly, in our study, about two-thirds of Pi*ZZ/Pi*SZ patients had CAP values indicative of hepatic steatosis ≥S1. This is in line with the histological results of Dawwas et al., which observed hepatic steatosis in 87.5% of patients who underwent liver biopsy (i.e. the sub-group of patients with evidence of liver injury).18 Importantly, subjects with liver fibrosis ≥F2 had higher BMI and GGT levels, and also tended to show higher CAP values. Of note, a recent meta-analysis based on individual patient data demonstrated that high CAP is not associated with a systematic overestimation of liver fibrosis by TE.23 CAP itself was linked to a series of metabolic parameters such as BMI, HbA1c and HOMA-IR. Moreover, we observed direct correlations with hsCRP and vWF. To summarize these findings, hepatic steatosis, which can be assessed non-invasively by CAP, seemed to be associated with more advanced liver disease, and also several cardiovascular risk factors. These findings underline that, similar to NAFLD, lifestyle interventions might be of high relevance in patients with A1AD. As clinical studies on specific treatment approaches are still ongoing, it appears reasonable to counsel A1AD patients with hepatic steatosis in analogy to the NAFLD population,24 although some measures might be complicated by the underlying pulmonary manifestation.
Except for three subjects reporting at-risk drinking (however, none of them had liver stiffness values indicative of liver fibrosis ≥F2), there was no evidence of concomitant liver diseases.
Interestingly, less than one-third of Pi*ZZ/Pi*SZ patients were vaccinated against hepatitis A/B (Supplementary material). Even in the sub-group of patients on α1-antitrypsin augmentation therapy, only half of the patients were vaccinated, although vaccination is recommended by the label since it is a plasma derivative. Thus, it will require the combined efforts of pulmonologists and hepatologists to increase vaccination rates in this patient population.
Several limitations should be considered when interpreting the findings of our study. First, not all contacted patients (73.8%) participated in our study. However, most patients declined to participate for logistical reasons (e.g. distance to the study center). Moreover, liver stiffness/CAP measured by TE has not been validated against liver biopsy in patients with A1AD. However, its diagnostic value has been confirmed in most aetiologies of chronic liver disease, including NAFLD.24–26 All relevant confounding factors for the TE-based assessment of liver fibrosis induce false-high liver stiffness results leading to an overestimation of liver fibrosis stage.27 Thus, they cannot explain the very low prevalence of liver fibrosis ≥F3 observed in our study. Since sample size limits the conclusions of our study, collaborative efforts are required to perform large international studies.28,29 However, it can be assumed, that multicenter studies will not be able to characterize subjects in the same detail as our monocenter study.
In conclusion, heterozygosity for the Z-allele predisposes for the development of CSPH, confirming its role as a genetic (co)factor in liver disease. Pi*ZZ/SZ patients rarely develop liver fibrosis ≥F3 during adulthood. However, liver fibrosis ≥F2 is common, which warrants the use of non-invasive methods to identify patients at risk for the development of ACLD. High BMI and GGT values identify patients with an increased pretest probability of having liver fibrosis ≥F2. Elevated CAP values hint at underlying hepatic steatosis, which might promote liver fibrosis progression.
Supplementary Material
Author contributions
All authors contributed either to research design (MM and TR), and/or the acquisition (genotyping: BS, HZ, and PF; phenotyping: all authors), analysis (MM) or interpretation (all authors) of data. MM drafted the manuscript, which was then critically revised by all other authors. All authors approved the submitted version of the manuscript.
Declaration of conflicting interests
The authors declare that there are no conflicting interests relevant to this work.
Funding
This work was supported by the Joseph Skoda Award of the Austrian Society of Internal Medicine, awarded to MM.
Informed consent
Except for liver donors (requirement of informed consent was waived by the ethics committee of the Medical University of Innsbruck), all subjects were consented for genetic testing.
Ethics approval
All sub-studies were conducted in accordance with the Declaration of Helsinki as reflected by the a priori approval of the ethics committees of the Medical Universities of Vienna (numbers 1964/2014 and 1526/ 2017) and Innsbruck (number UN4218).
References
- 1.Lomas DA, Hurst JR, Gooptu B. Update on alpha-1 antitrypsin deficiency: new therapies. J Hepatol 2016; 65: 413–424. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. New Engl J Med 1986; 314: 736–739. [DOI] [PubMed] [Google Scholar]
- 3.Clark VC, Dhanasekaran R, Brantly M, et al. Liver test results do not identify liver disease in adults with alpha(1)-antitrypsin deficiency. Clin Gastroenterol Hepatol 2012; 10: 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowlus CL, Willner I, Zern MA, et al. Factors associated with advanced liver disease in adults with alpha1-antitrypsin deficiency. Clin Gastroenterol Hepatol 2005; 3: 390–396. [DOI] [PubMed] [Google Scholar]
- 5.Teckman JH. Emerging concepts and human trials in alpha-1-antitrypsin deficiency liver disease. Semin Liver Dis 2017; 37: 152–158. [DOI] [PubMed] [Google Scholar]
- 6.Eigenbrodt ML, McCashland TM, Dy RM, et al. Heterozygous alpha 1-antitrypsin phenotypes in patients with end stage liver disease. Am J Gastroenterol 1997; 92: 602–607. [PubMed] [Google Scholar]
- 7.Graziadei IW, Joseph JJ, Wiesner RH, et al. Increased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiency. Hepatology 1998; 28: 1058–1063. [DOI] [PubMed] [Google Scholar]
- 8.Fischer HP, Ortiz-Pallardo ME, Ko Y, et al. Chronic liver disease in heterozygous alpha1-antitrypsin deficiency PiZ. J Hepatol 2000; 33: 883–892. [DOI] [PubMed] [Google Scholar]
- 9.Goltz D, Hittetiya K, Vossing LM, et al. Alpha(1)-antitrypsin PiMZ heterozygosity has an independent aggravating effect on liver fibrosis in alcoholic liver disease. Virchows Arch 2014; 465: 539–546. [DOI] [PubMed] [Google Scholar]
- 10.Cacciottolo TM, Gelson WT, Maguire G, et al. Pi*Z heterozygous alpha-1 antitrypsin states accelerate parenchymal but not biliary cirrhosis. Eur J Gastroenterol Hepatol 2014; 26: 412–417. [DOI] [PubMed] [Google Scholar]
- 11.Kok KF, te Morsche RH, van Oijen MG, et al. Prevalence of genetic polymorphisms in the promoter region of the alpha-1 antitrypsin (SERPINA1) gene in chronic liver disease: a case control study. BMC Gastroenterol 2010; 10: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkenstedt A, Auer C, Glodny B, et al. Patatin-like phospholipase domain-containing protein 3 rs738409-G in recipients of liver transplants is a risk factor for graft steatosis. Clin Gastroenterol Hepatol 2013; 11: 1667–1672. [DOI] [PubMed] [Google Scholar]
- 13.Huber F, Schmid-Scherzer K, Wantke F, et al. Alpha1-antitrypsin deficiency in Austria: analysis of the Austrian Alpha1-international-registry database. Wien Klin Wochenschr 2010; 122: 390–396. [DOI] [PubMed] [Google Scholar]
- 14.Staufer K, Andresen H, Vettorazzi E, et al. Urinary ethyl glucuronide as a novel screening tool in patients pre- and post-liver transplantation improves detection of alcohol consumption. Hepatology 2011; 54: 1640–1649. [DOI] [PubMed] [Google Scholar]
- 15.Maheshwari A, Thuluvath PJ. Cryptogenic cirrhosis and NAFLD: are they related?. Am J Gastroenterol 2006; 101: 664–668. [DOI] [PubMed] [Google Scholar]
- 16.Valenti L, Dongiovanni P, Piperno A, et al. Alpha 1-antitrypsin mutations in NAFLD: high prevalence and association with altered iron metabolism but not with liver damage. Hepatology 2006; 44: 857–864. [DOI] [PubMed] [Google Scholar]
- 17.Powell EE, Cooksley WG, Hanson R, et al. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 1990; 11: 74–80. [DOI] [PubMed] [Google Scholar]
- 18.Dawwas MF, Davies SE, Griffiths WJ, et al. Prevalence and risk factors for liver involvement in individuals with PiZZ-related lung disease. Am J Respir Crit Care Med 2013; 187: 502–508. [DOI] [PubMed] [Google Scholar]
- 19.Mostafavi B, Diaz S, Tanash HA, et al. Liver function in alpha-1-antitrypsin deficient individuals at 37 to 40 years of age. Medicine 2017; 96: e6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bota S, Herkner H, Sporea I, et al. Meta-analysis: ARFI elastography versus transient elastography for the evaluation of liver fibrosis. Liver Int 2013; 33: 1138–1147. [DOI] [PubMed] [Google Scholar]
- 21.Popescu A, Bota S, Sporea I, et al. The influence of food intake on liver stiffness values assessed by acoustic radiation force impulse elastography-preliminary results. Ultrasound Med Biol 2013; 39: 579–584. [DOI] [PubMed] [Google Scholar]
- 22.Kim RG, Nguyen P, Bettencourt R, et al. Magnetic resonance elastography identifies fibrosis in adults with alpha-1 antitrypsin deficiency liver disease: a prospective study. Aliment Pharmacol Ther 2016; 44: 287–299. [DOI] [PubMed] [Google Scholar]
- 23.Karlas T, Petroff D, Sasso M, et al. Impact of hepatic steatosis and controlled attenuation parameter (CAP) on accuracy of fibrosis staging using transient elastography. J Hepatol 2017; 66: S674–S675. [Google Scholar]
- 24.European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD) and European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016; 64: 1388–1402. [DOI] [PubMed] [Google Scholar]
- 25.European Association for Study of Liver and Asociacion Latinoamericana para el Estudio del Higado. EASL-ALEH Clinical Practice Guidelines: non-invasive tests for evaluation of liver disease severity and prognosis. J Hepatol 2015; 63: 237–264. [DOI] [PubMed] [Google Scholar]
- 26.Karlas T, Petroff D, Sasso M, et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol 2017; 66: 1022–1030. [DOI] [PubMed] [Google Scholar]
- 27.Schwabl P, Bota S, Salzl P, et al. New reliability criteria for transient elastography increase the number of accurate measurements for screening of cirrhosis and portal hypertension. Liver Int 2015; 35: 381–390. [DOI] [PubMed] [Google Scholar]
- 28.Strnad P, Buch S, Hamesch K, et al. Heterozygous carriage of the alpha1-antitrypsin Z variant rs28929474 predisposes to the development of cirrhosis in the presence of alcohol misuse and non-alcohol-related fatty liver disease. J Hepatol 2017; 66: S177. [Google Scholar]
- 29.Hamesch K, Scholten D, Spivak I, et al. Multi-center study of liver disease in alpha1-antitrypsin deficiency: non-invasive evaluation of liver fibrosis in homozygous PiZZ patients. J Hepatol 2017; 66: S176–S177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.