

This question of whether fungi undergo apoptosis-like programmed cell death can be separated into two questions. One question is about applying the term “apoptosis” to fungi, and the other is a more challenging question of whether fungi have evolved mechanisms that inflict self-injury.
KEYWORDS: apoptosis, cell death, cell-autonomous, fungi, mycoptosis, necroptosis, programmed cell death, yeast, pathogenic fungi, MLKL
ABSTRACT
This question of whether fungi undergo apoptosis-like programmed cell death can be separated into two questions. One question is about applying the term "apoptosis" to fungi, and the other is a more challenging question of whether fungi have evolved mechanisms that inflict self-injury. The answers to both questions depend on the definitions applied to “apoptosis” and “programmed cell death.” Considering how these and other cell death terms originated and are currently defined for animals, some confusion arises when the terms are applied to fungi. While it is difficult to defend the concept of fungal apoptosis, the more interesting issue is whether fungi will eventually be found to encode programmed or extemporaneous self-destructive processes, as suggested by intriguing new findings.
PERSPECTIVE
The possibility that fungi, bacteria, parasites, and other microorganisms undergo programmed cell death (PCD) has been an emerging issue for many years (1, 2). Nowhere is this debate more intense than for fungi, but no consensus has been reached. Resolution currently rests on the fulfillment of divergent nonstandardized criteria. Aside from the inherent value and intrigue of new science, interest in this topic is spurred by the prospects for novel therapeutics and possible curtailment of the animal and plant devastations ascribed to fungi, such as annihilation of amphibian species and European and North American trees (3, 4). Taking the reverse perspective, knowledge about fungal cell death might help guide the preservation of diverse fungal species as world food resources, medicines, and the critical terrestrial ecosystems that we depend on (5–7). However, the relevant intracellular molecular events that occur in microbes as they die are not known.
DERIVATION OF THE TERM “PROGRAMMED CELL DEATH”
Initiating discussions about fungal cell death is challenged by the lack of a vocabulary with generally agreed-upon definitions. The definition of “programmed cell death” also holds layers of complexity. The concept of physiological cell death arose in the 19th and 20th centuries when investigators observed the systematic disappearance of cells in various developing animal models (8). With the discovery of lysosomes in 1955 (9), some researchers in the cell death field became occupied with the idea that leakage of hydrolases from these “suicide bags” was to blame, while others argued that unleashed hydrolases were a consequence rather than a cause of cell death (10). These ideas have been revisited and extended more recently both in human disease (11) and in yeast cell death (12).
In pursuit of the components that control developmental cell death, Richard Lockshin and his PhD adviser Carroll Williams at Harvard published a series of papers in 1964 to 1965 applying the term “programmed cell death” to describe the dying process of larval abdominal muscle cells in newly emerged adult American silkmoths (13–17). Lockshin explains that this word choice was a metaphor, “a felicitous turn of phrase designed to exploit the trendiness of the then-nascent computer era”—hence “programmed,” analogous to developmental programs (18). By eliminating some potential causes of cell death, Lockshin and Williams also conceptualized the death as a "cell-autonomous process," meaning that the cell destined to die is capable of facilitating its own death (with or without triggers from neighboring cells) (13).
The accumulation of knowledge in the subsequent two to three decades from studies of cancer, virus infections, and, most notably, the worm Caenorhabditis elegans finally produced unequivocal evidence of an evolutionarily conserved, genetically encoded program of self-elimination (19–23). Identification of cell death genes in worms by the use of genetics (Nobel Prize, 2002), combined with the disease relevance revealed by mammalian models, was soon solidified by in vitro reconstitutions and crystal structures (24–26). Perhaps most salient was the discovery of worm ced-3, a gene required for developmental elimination of cells (19). Its sequence homology with the gene encoding the mammalian protease that activates proinflammatory cytokine interleukin-1β (IL-1β) (IL-1β-converting enzyme [ICE]), now designated caspase-1 (mediator of nonapoptotic death by pyroptosis), revealed that worm CED-3 likely causes cell-autonomous programmed cell death via its proteolytic activity (27, 28). The trail quickly led to identification of mammalian caspase-3, the mediator of mammalian apoptosis (29). Another key worm homologue, human BCL-2 (C. elegans CED-9), was a novel oncogene of unknown function (30–32) with viral equivalents (E1B-19k and BHRF1) (21, 33). BCL-2 became the first known antiapoptotic protein and the first apoptosis designer drug target and was approved by the FDA for cancer therapy in 2016 (34, 35). A large field of human/animal programmed cell death research sprang up in the 1990s and captured all the major meeting venues and continues through the present. This field focuses on metazoan cell death, leaving room for emergence of a new field of fungal cell death marked by the establishment in 2002 of IMYA (International Meeting on Yeast Apoptosis [a name change is under consideration]), which continues to grow (36, 37).
Programmed cell death, subsequently dubbed "PCD," is currently applied to all types of physiological cell death, encompassing both developmental and nondevelopmental processes required to sustain health (38). A newer term, “regulated cell death” (RCD), has gained usage as recommended by the Nomenclature Committee on Cell Death (NCCD) (39). RCD includes PCD and further encompasses nonphysiological/pathological cell death, though these terms are often used interchangeably, in part because the same death mechanisms that occur physiologically can also occur in disease states or be induced therapeutically.
Given that the terms PCD and RCD were developed to describe cell death in metazoans, some confusion arises when the terms are applied to single-cell species (though fungi typically form multicellular structures). Despite advice against the coining of neologisms from the NCCD and a new parallel guideline specifically intended for yeast that pushes the concept of yeast RCD (40), new nomenclature might be useful (e.g., to conduct literature searches on fungal cell death versus infected host cell death). The term “phenoptosis” (programmed death of an entire organism, multicellular or unicellular) (41, 42) is an appealing descriptor, or perhaps another derivative of the word apoptosis such as “fungitosis” or “mycoptosis” to specify fungal death should be adopted. First, consider how the existing terms arose and why they are thus not readily applicable to microbes.
CRITERIA FOR CLASSIFICATION AS “PROGRAMMED CELL DEATH”
When one bacterial cell releases a toxin that kills a different bacterial cell, should this be considered programmed cell death? It depends. Few would doubt that bacterial killer toxins were selected during evolution for the ability to kill their target cells and in this way could be considered to represent a type of nonautonomous cell death “program.” However, the death mechanisms involved do not necessarily qualify as “programmed cell death” unless additional criteria are met. The burden of proof requires the dying cell to contribute actively to its own death (i.e., in a cell-autonomous manner). If the dying cell makes no contribution to its own death, this death can be considered equivalent to murder—the cell was simply hammered by forces beyond its control (Fig. 1A). An analogous example from mammals might be perforin, which is released from cytotoxic lymphocytes and forms pores in target cell membranes (43). (The NCCD unfortunately refers to death by assault as “accidental” cell death [ACD] [39].) A possible variant of death by assault is the complete absence of any death mechanisms or processes; in such a case, death is defined simply as the absence of life (life simply stops, like a car that runs out of gas). This way of thinking was once prominent but has faded with the accumulation of knowledge about death (Fig. 1A). However, even if there is compelling evidence that a dying cell is capable of contributing to its own death, this is still not sufficient to qualify as “programmed cell death.”
FIG 1 .
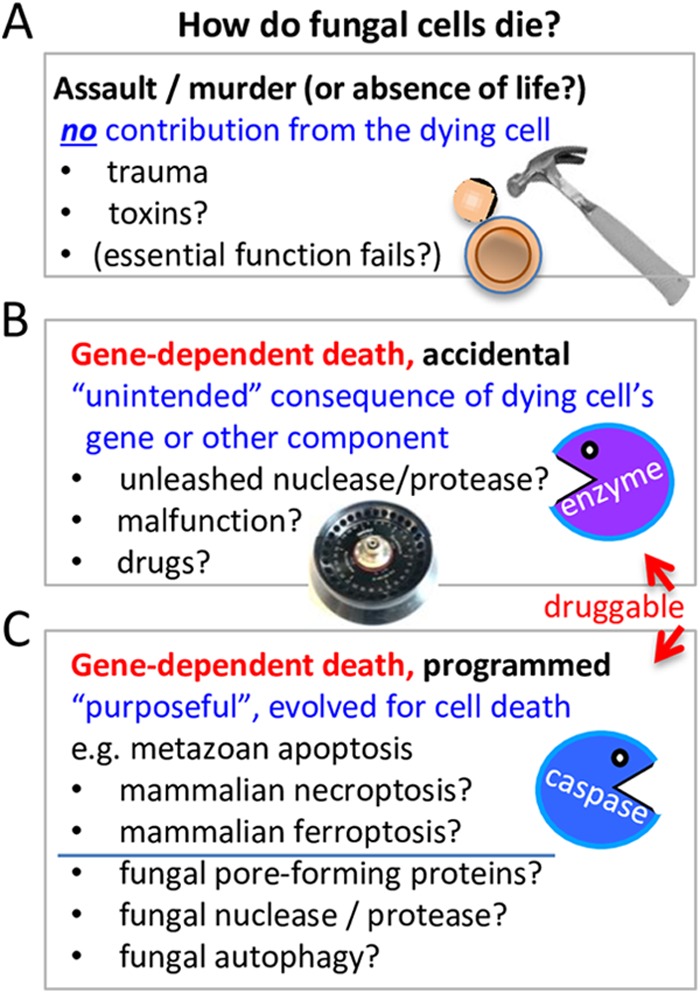
Three broad categories of cell death can be distinguished based on the level of participation by the dying cell. (A) Cell death that occurs without any contributions from the cell that dies is analogous to death by assault or murder (i.e., death is not dependent on a gene or other component originating in the cell destined to die), or, potentially, a cell could expire (like a car running out gas), though enthusiasm for the latter idea has waned in recent years. (B and C) A gene product or other component of the dying cell can contribute to its demise (gene-dependent, cell-autonomous death) in two conceptually distinct processes: accidental/extemporaneous cell death occurring via mechanisms not selected by evolution (B) and programmed cell death occurring via mechanisms selected during evolution (C). Cell death in all three categories (A to C) can be induced by conditions/factors (hammer, toxin, death receptor ligand, growth factor withdrawal, irradiation, sunlight, etc.) external to the cell that dies, while only cell death in categories B and C can also be triggered by events inside the cell, for example, events analogous to those involving the centrifuge rotor (B) or inherent errors in replication (C). These definitions differ somewhat from the NCCD definitions (39).
There is one more hurdle. Theoretically, a microbial toxin could trigger two conceptually distinct types of events in the target cell destined to die; both events require contributions by the dying cell, but only one represents programmed cell death. The distinction between them depends on the answer to the following question: did a gene product or other component originating from the dying cell "purposely" contribute to its own death? If yes, the death fits the long-held definition of “programmed cell death” as currently interpreted (39).
PROGRAMMED VERSUS GENE-DEPENDENT CELL (OR CENTRIFUGE) DEATH
To illustrate that final distinction between programmed and nonprogrammed cell death, take the case of the laboratory centrifuge. Let the centrifuge represent the dying cell, and the centrifuge rotor represent the death-promoting gene product encoded by the dying cell. If the rotor fractures while spinning and wrecks the centrifuge beyond repair, this would be analogous to a cell’s protein actively contributing to the cell's own death. In this case, the centrifuge/cell-killing function of the rotor/protein (e.g., an unleashed protease or nuclease) was not a feature of the rotor/protein design. Cell death analogous to this centrifuge example can be classified as cell-autonomous and gene-dependent death (i.e., a gene or other component originating from the dying cell helps promote self-elimination). Here I refer to this type of cell death as accidental or extemporaneous (metaphorically, “unintended cell death”) (Fig. 1B). However, if the centrifuge rotor had been designed by its engineers to destroy the centrifuge when its time was up, this would be considered programmed death (metaphorically, “deliberate cell death”) (Fig. 1C). Thus, cell-autonomous mechanisms that evolved for cell death, or that can be reasonably anticipated as such, are generally considered to be programmed death mechanisms. Note, however, that the rotor/protein has another “day job” function in the centrifuge/healthy cell.
Importantly, both types of gene-dependent death (programmed and nonprogrammed) are potentially druggable; thus, the question of their evolutionary origins seems less important. Furthermore, distinguishing between accidental/unintended death and deliberate/programmed death may not be feasible experimentally, even in animals. Therefore, a more readily testable definition would be useful. We ask whether the death of a cell is dependent on its own gene products or other components (e.g., proteases, reactive oxygen species [ROS], oxidized lipids, unfolded proteins, etc.), regardless of whether such pro-death functions arose through a selection process during evolution (implying a purposeful death) and regardless of whether they wholly or partly cause cell death. For simplicity, we refer to this as “gene-dependent cell death,” without any expectations or requirements that the process would be programmed or regulated (Fig. 1B and 1C). Death-promoting factors can be either direct executors of death (e.g., mammalian caspase-3) or indirect contributors to death (e.g., death signaling pathways). However, applying the term “gene-dependent death” to fungi still requires further considerations.
CAN WE EXTRAPOLATE FROM ANIMAL KNOCKOUT DATA TO FUNGAL KNOCKOUT DATA?
The evidence suggesting that yeast undergo gene-dependent cell death (PCD, RCD) relies heavily on genetic approaches and the analysis of mutants that are resistant or sensitive to death stimuli. The same powerful genetic approach was used in the landmark C. elegans studies that defined the apoptosis pathway (19), but there is a critical difference. In sharp contrast to yeast, the worm cell death is physiologically relevant by definition—it occurs during normal development. Analogous physiological model systems are limited in availability for the laboratory workhorse Saccharomyces cerevisiae, though there are reports of efforts in this direction (population dynamics, failed mating, quorum sensing, virus infection, sporulation, aging, colony differentiation, and many others) (44–51).
This limitation raises a looming question. Is it reasonable to conclude that a gene has pro-death activity if deletion of that gene confers death resistance to the cell, or could there be unrelated explanations? This is a troublesome question. In fact, researchers in the larger (non-cell death) yeast genetics field are adamant about this issue in my personal experience, and they have an excellent point worthy of consideration. They argue that the improved survival is likely explained by reasons other than the loss of a death-promoting gene. Consider the death-resistant strains of S. cerevisiae lacking either YCA1/MCA1 (metacaspase related to mammalian caspases) or DNM1/DRP-1 (conserved dynamin-like mitochondrial fission factor implicated as an accessory to mammalian cell death) (52–54). In both cases, enzymatic activity is required for the death function. However, if these knockout strains also have heightened defenses or stress responses simply as a consequence of their gene deficiencies, this could potentially account for their ability to survive stress better than the wild type. The same applies to metazoans.
This criticism is difficult to overcome, and yet it has its own caveats. Testing all known adaptive stress responses cannot resolve the criticism; there could always be another untested compensatory mechanism. Conversely, even if adaptive stress responses are elevated, and even if these adaptations partly contribute to death resistance, it remains possible that the gene in question had evolved in part to inflict self-harm. Thus, the possibility of the existence of a cell death mechanism cannot be dismissed because stress responses are heightened, just as a cell death mechanism cannot be inferred from the absence of detectable stress responses. Adaptive responses and pro-death functions are not mutually exclusive possibilities and can even be expected to co-occur. For example, careful analyses revealed that depletion of mammalian apoptosis regulators alter antioxidant defenses at the steady state (55).
Other potential confounders for studying fungal cell death include the secondary mutations affecting cell death that frequently arise in gene knockout strains (56) and the day job functions of most, if not all, pro-death factors that could also significantly impact susceptibility to stress (57–59). Our studies of animal caspases and other pro-death factors in the nervous system revealed alternative and nonapoptotic roles (60–62). Antiapoptotic proteins also have day jobs. How the effects of antiapoptotic mammalian BCL-xL on mitochondria and cellular energetics in healthy cells (57, 58, 63) are related to its antiapoptotic role (BH3/BAX-inhibition) is not yet known. How are we to address all these issues for fungi?
WHAT IS ACCEPTABLE EVIDENCE OF PROGRAMMED FUNGAL CELL DEATH?
Cell death mechanisms could potentially have evolved in single-cell species to enable them to respond to inevitable pathogens (47) or to live in the differentiated communities of a simple colony harboring layers with distinct transcriptional profiles and death-susceptible subpopulations that likely benefit the community (50, 51, 64). However, except for a few salient examples supporting the existence of fungal cell death pathways (below), the evidence is limited thus far. Based on the history of the metazoan PCD field, the idea of the existence of PCD mechanisms in fungi will remain controversial and lack general acceptance until molecularly defined fungal death pathways are compellingly demonstrated. This includes identification of the direct effectors of fungal cell death and the biochemical mechanisms involved. Until then, claims that fungi undergo gene-dependent PCD/RCD may be overstated without further clarification. Admittedly, the bar is higher now than it was for mammalian PCD in the mid-20th century.
REASONS TO KEEP OR ABANDON THE TERM “APOPTOSIS” FOR FUNGI
The application of the term "apoptosis" to fungi is difficult to defend by any definition and has been decisively rejected (2, 65–67), and yet studies on “fungal apoptosis” continue to populate the literature (40, 68, 69). This may be an extension of the early metazoan and nonmetazoan literature that was published when "apoptosis" was the only relevant word available and served as a blanket term to describe any type of regulated cell death. However, this practice has declined in recent years to separate apoptosis from several other molecularly defined nonapoptotic death pathways in metazoans (notably necroptosis, ferroptosis, and pyroptosis).
Thus, apoptosis is now more narrowly defined, demanding consideration of authors’ intentions before citing their earlier works as evidence of “apoptosis” as currently defined. The current working definition of mammalian “apoptosis” is caspase-3-mediated cell death (39), and yet fungi do not encode true caspases. Therefore, "apoptosis" applied to fungi presumably has a different definition. Although a defensible consensus definition has not emerged, several criteria have been reported (40). Related fungal metacaspases appear not to behave like mammalian caspases or to be regulated like caspases (70–72). Thus, the use of promiscuous mammalian caspase reporters (e.g., FITC-VAD-FMK) as evidence of “caspase-like” activities in fungi currently lacks rigorous justification (73), and newer better reagents may be helpful (74). However, these facts do not in any way rule out a role for fungal metacaspases or other proteases in cell death, and the possibility remains open.
Is the term “yeast apoptosis” justifiable in other ways? Some justify the use of the term "apoptosis" (or "apoptosis-like") by restricting its use to morphological and biophysical features of dying fungal cells without further implications. Indeed, the word "apoptosis" was put forth in a landmark paper in 1972 (75) without any direct evidence for programmed cell death, supported only by the distinct morphological features of occasional cells in normal human tissues and in rat liver at delayed times after ischemic injury, though the claims have not been free of controversy (76). However, unlike fungi, the apoptotic features of mammalian cells are now recognized as the handiwork of caspase-3, including chromatin condensation, membrane blebbing, and phosphatidylserine exposure on the cell surface to facilitate engulfment into phagocytic cells (where some cells may finish dying) (77–80). Accordingly, the field has advanced. Importantly, apoptosis-like features of mammalian cells not caused by caspases are no longer classified as a type of apoptosis in the latest NCCD guidelines (39). New biochemical or molecular validations may eventually justify the use of the term “apoptosis-like” for fungal death, though it is not presently clear if apoptosis-like fungal death is more like apoptosis than like any other known death mechanism. Perhaps the best justification of the use of the term "fungal apoptosis" is according to the original definition of apoptosis—for use not as a morphological term but to conceptualize the idea of a deliberate cell-autonomous death mechanism. However, an explicit explanation of this otherwise retired definition will be required to avoid the inevitable misunderstandings that can compromise a field’s credibility.
CONSERVED DEATH PROGRAMS IN FUNGI AND HUMANS?
The new focus is on mammalian cell death by programmed necrosis resulting in plasma membrane rupture. One potentially conserved necrotic cell death mechanism is represented by the proposed N-terminal pore-forming domain of mammalian MLKL, mediator of necroptosis (81). The HET or HeLo-like (HELL) domains of filamentous fungi that mediate cell death upon fusion of two incompatible cells (heterokaryon incompatibility) (82, 83) have an MLKL-like structure prediction (e.g., Phyre2). A different form of death occurs in conidia of the agriculturally important plant pathogen Magnaporthe oryzae (rice blast) during germination (84). Deletion of any of 16 conserved autophagy genes causes conidia to remain alive, blocking both appressorium formation and pathogenicity (85), raising the possibility of autophagy-dependent cell death (ADCD) (86, 87). However, more studies are needed to rule out indirect effects of autophagy (not true ADCD), for example, by degrading an inhibitor of the primary death effector (39). One study suggested that fungi may also undergo iron-dependent ferroptosis, which is thought to result from loss of membrane integrity as a consequence of lipid peroxidation (88), conceivably shared across many species.
Fungi may also encode inhibitors of programmed cell death. Conserved BIR (baculovirus inhibitor of apoptosis repeat) domains that potently suppress cell death were first identified in insect virus genomes and later in humans and Drosophila (89). Interestingly, BIR-containing proteins of the plant-pathogenic fungus Botrytis cinerea and the human-pathogenic fungus Aspergillus fumigatus were found to suppress fungal cell death and increase virulence (68, 90). Although the mechanisms are not known, the suggestion that BIR1 suppresses caspase-dependent apoptosis-like fungal cell death is debatable (73), and with rare exceptions, BIR-containing proteins are not direct caspase inhibitors (72, 91). Continuing to stretch mammalian cell death nomenclature to accommodate fungi requires changing ingrained assumptions, and the field may be better served by new nomenclature to convey new discoveries.
ACKNOWLEDGMENTS
Insightful suggestions were generously provided by Tobias Hohl, Abdel Aouacheria, Richard Lockshin, Zdena Palkova, Rollie Clem, Lorenzo Galluzzi, Kyle Cunningham, Zach Stolp, Jason Huska, and Madhura Kulkarni.
This effort was supported by U.S. NIH grants RO1 NS083373, R21 NS096677, and RO1 GM077875.
Footnotes
Citation Hardwick JM. 2018. Do fungi undergo apoptosis-like programmed cell death? mBio 9:e00948-18. https://doi.org/10.1128/mBio.00948-18.
Contributor Information
Deborah A. Hogan, Geisel School of Medicine at Dartmouth.
Judith Berman, Tel Aviv University.
REFERENCES
- 1.Ameisen JC. 2002. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 9:367–393. doi: 10.1038/sj/cdd/4400950. [DOI] [PubMed] [Google Scholar]
- 2.Green DR, Fitzgerald P. 2016. Just so stories about the evolution of apoptosis. Curr Biol 26:R620–R627. doi: 10.1016/j.cub.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pounds JA, Bustamante MR, Coloma LA, Consuegra JA, Fogden MP, Foster PN, La Marca E, Masters KL, Merino-Viteri A, Puschendorf R, Ron SR, Sánchez-Azofeifa GA, Still CJ, Young BE. 2006. Widespread amphibian extinctions from epidemic disease driven by global warming. Nature 439:161–167. doi: 10.1038/nature04246. [DOI] [PubMed] [Google Scholar]
- 4.Loo JA. 2009. Ecological impacts of non-indigenous invasive fungi as forest pathogens. Biol Invasions 11:81–96. doi: 10.1007/s10530-008-9321-3. [DOI] [Google Scholar]
- 5.Boa E. 2004. Wild edible fungi, a global overview of their use and importance to people. Non-wood forest products 17. Food and Agriculture Organization of the United Nations, Rome, Italy: http://www.fao.org/docrep/007/y5489e/y5489e00.htm#Contents. [Google Scholar]
- 6.Heilmann-Clausen J, Barron ES, Boddy L, Dahlberg A, Griffith GW, Nordén J, Ovaskainen O, Perini C, Senn-Irlet B, Halme P. 2015. A fungal perspective on conservation biology. Conserv Biol 29:61–68. doi: 10.1111/cobi.12388. [DOI] [PubMed] [Google Scholar]
- 7.Bagchi R, Gallery RE, Gripenberg S, Gurr SJ, Narayan L, Addis CE, Freckleton RP, Lewis OT. 2014. Pathogens and insect herbivores drive rainforest plant diversity and composition. Nature 506:85–88. doi: 10.1038/nature12911. [DOI] [PubMed] [Google Scholar]
- 8.Clarke PG, Clarke S. 1996. Nineteenth century research on naturally occurring cell death and related phenomena. Anat Embryol 193:81–99. doi: 10.1007/BF00214700. [DOI] [PubMed] [Google Scholar]
- 9.Appelmans F, Wattiaux R, De Duve C. 1955. Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem J 59:438–445. doi: 10.1042/bj0590438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders JW., Jr. 1966. Death in embryonic systems. Science 154:604–612. doi: 10.1126/science.154.3749.604. [DOI] [PubMed] [Google Scholar]
- 11.Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lønborg A, Vindeløv SD, Hanahan D, Arenz C, Ejsing CS, Kirkegaard T, Rohde M, Nylandsted J, Jäättelä M. 2013. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24:379–393. doi: 10.1016/j.ccr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Kim A, Cunningham KW. 2015. A LAPF/phafin1-like protein regulates TORC1 and lysosomal membrane permeabilization in response to endoplasmic reticulum membrane stress. Mol Biol Cell 26:4631–4645. doi: 10.1091/mbc.E15-08-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lockshin RA, Williams CM. 1964. Programmed cell death–II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 10:643–649. doi: 10.1016/0022-1910(64)90034-4. [DOI] [Google Scholar]
- 14.Lockshin RA, Williams CM. 1965. Programmed cell death–I. Cytology of degeneration in the intersegmental muscles of the Pernyi silkmoth. J Insect Physiol 11:123–133. doi: 10.1016/0022-1910(65)90099-5. [DOI] [PubMed] [Google Scholar]
- 15.Lockshin RA, William CM. 1965. Programmed cell death–III. Neural control of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 11:601–610. doi: 10.1016/0022-1910(65)90142-3. [DOI] [PubMed] [Google Scholar]
- 16.Lockshin RA, Williams CM. 1965. Programmed cell death–IV. The influence of drugs on the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 11:803–809. doi: 10.1016/0022-1910(65)90159-9. [DOI] [PubMed] [Google Scholar]
- 17.Lockshin RA, Williams CM. 1965. Programmed cell death–V. Cytolytic enzymes in relation to the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 11:831–844. doi: 10.1016/0022-1910(65)90186-1. [DOI] [PubMed] [Google Scholar]
- 18.Lockshin RA. 2016. Programmed cell death 50 (and beyond). Cell Death Differ 23:10–17. doi: 10.1038/cdd.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis HM, Horvitz HR. 1986. Genetic control of programmed cell death in the nematode C. elegans. Cell 44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 20.Graninger WB, Seto M, Boutain B, Goldman P, Korsmeyer SJ. 1987. Expression of Bcl-2 and Bcl-2-Ig fusion transcripts in normal and neoplastic cells. J Clin Invest 80:1512–1515. doi: 10.1172/JCI113235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White E, Grodzicker T, Stillman BW. 1984. Mutations in the gene encoding the adenovirus early region 1B 19,000-molecular-weight tumor antigen cause the degradation of chromosomal DNA. J Virol 52:410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clem RJ, Fechheimer M, Miller LK. 1991. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 23.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. 1993. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- 24.Newmeyer DD, Farschon DM, Reed JC. 1994. Cell-free apoptosis in Xenopus egg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell 79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 25.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. 1994. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 26.Minn AJ, Vélez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. 1997. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature 385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 27.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. 1993. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 28.Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. 1993. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell 75:653–660. doi: 10.1016/0092-8674(93)90486-A. [DOI] [PubMed] [Google Scholar]
- 29.Fernandes-Alnemri T, Litwack G, Alnemri ES. 1994. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem 269:30761–30764. [PubMed] [Google Scholar]
- 30.Hengartner MO, Ellis RE, Horvitz HR. 1992. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 31.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. 1984. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 32.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. 1985. Involvement of the bcl-2 gene in human follicular lymphoma. Science 228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 33.Cleary ML, Smith SD, Sklar J. 1986. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 34.Vaux DL, Cory S, Adams JM. 1988. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 35.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF. 2016. Targeting BCL2 with Venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Côrte-Real M. 2004. The second international meeting on yeast apoptosis (IMYA 2), Smolenice, Slovak Republic, 17–20 September 2003. FEMS Yeast Res 4:887–888. doi: 10.1016/j.femsyr.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Giannattasio S, Mazzoni C. 2017. 12th international meeting on yeast apoptosis (IMYA) https://imya12.azuleon.org/. Accessed 5 2018. [Google Scholar]
- 38.Lockshin RA, Zakeri Z. 2007. Cell death in health and disease. J Cell Mol Med 11:1214–1224. doi: 10.1111/j.1582-4934.2007.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, et al. 2018. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carmona-Gutierrez D, Bauer MA, Zimmermann A, Aguilera A, Austriaco N, Ayscough K, Balzan R, Bar-Nun S, Barrientos A, Belenky P, Blondel M, Braun RJ, Breitenbach M, Burhans WC, Büttner S, Cavalieri D, Chang M, Cooper KF, Côrte-Real M, Costa V, Cullin C, Dawes I, Dengjel J, Dickman MB, Eisenberg T, Fahrenkrog B, Fasel N, Fröhlich KU, Gargouri A, Giannattasio S, Goffrini P, Gourlay CW, Grant CM, Greenwood MT, Guaragnella N, Heger T, Heinisch J, Herker E, Herrmann JM, Hofer S, Jiménez-Ruiz A, Jungwirth H, Kainz K, Kontoyiannis DP, Ludovico P, Manon S, Martegani E, Mazzoni C, Megeney LA, Meisinger C, et al. 2018. Guidelines and recommendations on yeast cell death nomenclature. Microb Cell 5:4–31. doi: 10.15698/mic2018.01.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skulachev VP. 1999. Phenoptosis: programmed death of an organism. Biochemistry (Mosc) 64:1418–1426. [PubMed] [Google Scholar]
- 42.Sukhanova EI, Rogov AG, Severin FF, Zvyagilskaya RA. 2012. Phenoptosis in yeasts. Biochemistry 77:761–775. doi: 10.1134/S0006297912070097. [DOI] [PubMed] [Google Scholar]
- 43.Voskoboinik I, Whisstock JC, Trapani JA. 2015. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 15:388–400. doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 44.Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, Diaspro A, Dossen JW, Gralla EB, Longo VD. 2004. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J Cell Biol 166:1055–1067. doi: 10.1083/jcb.200404002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Severin FF, Hyman AA. 2002. Pheromone induces programmed cell death in S. cerevisiae. Curr Biol 12:R233–R235. doi: 10.1016/S0960-9822(02)00776-5. [DOI] [PubMed] [Google Scholar]
- 46.Váchová L, Palková Z. 2005. Physiological regulation of yeast cell death in multicellular colonies is triggered by ammonia. J Cell Biol 169:711–717. doi: 10.1083/jcb.200410064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanovska I, Hardwick JM. 2005. Viruses activate a genetically conserved cell death pathway in a unicellular organism. J Cell Biol 170:391–399. doi: 10.1083/jcb.200503069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eastwood MD, Cheung SW, Lee KY, Moffat J, Meneghini MD. 2012. Developmentally programmed nuclear destruction during yeast gametogenesis. Dev Cell 23:35–44. doi: 10.1016/j.devcel.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Weinberger M, Mesquita A, Caroll T, Marks L, Yang H, Zhang Z, Ludovico P, Burhans WC. 2010. Growth signaling promotes chronological aging in budding yeast by inducing superoxide anions that inhibit quiescence. Aging (Albany, NY) 2:709–726. doi: 10.18632/aging.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cáp M, Stěpánek L, Harant K, Váchová L, Palková Z. 2012. Cell differentiation within a yeast colony: metabolic and regulatory parallels with a tumor-affected organism. Mol Cell 46:436–448. doi: 10.1016/j.molcel.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Sťovíček V, Váchová L, Palková Z. 2012. Yeast biofilm colony as an orchestrated multicellular organism. Commun Integr Biol 5:203–205. doi: 10.4161/cib.18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madeo F, Herker E, Maldener C, Wissing S, Lächelt S, Herlan M, Fehr M, Lauber K, Sigrist SJ, Wesselborg S, Fröhlich KU. 2002. A caspase-related protease regulates apoptosis in yeast. Mol Cell 9:911–917. doi: 10.1016/S1097-2765(02)00501-4. [DOI] [PubMed] [Google Scholar]
- 53.Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basañez G, Hardwick JM. 2004. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev 18:2785–2797. doi: 10.1101/gad.1247904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. 2016. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540:139–143. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kowaltowski AJ, Fiskum G. 2005. Redox mechanisms of cytoprotection by Bcl-2. Antioxid Redox Signal 7:508–514. doi: 10.1089/ars.2005.7.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teng X, Dayhoff-Brannigan M, Cheng WC, Gilbert CE, Sing CN, Diny NL, Wheelan SJ, Dunham MJ, Boeke JD, Pineda FJ, Hardwick JM. 2013. Genome-wide consequences of deleting any single gene. Mol Cell 52:485–494. doi: 10.1016/j.molcel.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. 2011. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol 195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, Sharfi H, Zhai B, Rodriguez-Mias R, Luithardt H, Cantley LC, Daley GQ, Asara JM, Gygi SP, Wagner G, Liu CF, Yuan J. 2011. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell 146:607–620. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, Alavian KN, Pontarelli F, Roelofs BA, Hickman JA, Hardwick JM, Zukin RS, Jonas EA. 2012. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci 15:574–580. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. 1999. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med 5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- 61.Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, Mandir AS, Dawson TM, Dike S, Sappington AL, Kerr DA, Jonas EA, Kaczmarek LK, Hardwick JM. 2003. BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell 4:575–585. doi: 10.1016/S1534-5807(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 62.Tang HL, Tang HM, Fung MC, Hardwick JM. 2015. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci Rep 5:9015. doi: 10.1038/srep09015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB III, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. 2009. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol 184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Podholová K, Plocek V, Rešetárová S, Kučerová H, Hlaváček O, Váchová L, Palková Z. 2016. Divergent branches of mitochondrial signaling regulate specific genes and the viability of specialized cell types of differentiated yeast colonies. Oncotarget 7:15299–15314. doi: 10.18632/oncotarget.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim H, Kim A, Cunningham KW. 2012. Vacuolar H+-ATPase (V-ATPase) promotes vacuolar membrane permeabilization and nonapoptotic death in stressed yeast. J Biol Chem 287:19029–19039. doi: 10.1074/jbc.M112.363390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Váchová L, Palková Z. 2007. Caspases in yeast apoptosis-like death: facts and artefacts. FEMS Yeast Res 7:12–21. doi: 10.1111/j.1567-1364.2006.00137.x. [DOI] [PubMed] [Google Scholar]
- 67.Aouacheria A, Rech de Laval V, Combet C, Hardwick JM. 2013. Evolution of Bcl-2 homology motifs: homology versus homoplasy. Trends Cell Biol 23:103–111. doi: 10.1016/j.tcb.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shlezinger N, Irmer H, Dhingra S, Beattie SR, Cramer RA, Braus GH, Sharon A, Hohl TM. 2017. Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death. Science 357:1037–1041. doi: 10.1126/science.aan0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gonçalves AP, Heller J, Daskalov A, Videira A, Glass NL. 2017. Regulated forms of cell death in fungi. Front Microbiol 8:1837. doi: 10.3389/fmicb.2017.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hill SM, Nyström T. 2015. The dual role of a yeast metacaspase: what doesn’t kill you makes you stronger. Bioessays 37:525–531. doi: 10.1002/bies.201400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee RE, Brunette S, Puente LG, Megeney LA. 2010. Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc Natl Acad Sci U S A 107:13348–13353. doi: 10.1073/pnas.1006610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Minina EA, Coll NS, Tuominen H, Bozhkov PV. 2017. Metacaspases versus caspases in development and cell fate regulation. Cell Death Differ 24:1314–1325. doi: 10.1038/cdd.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aouacheria A, Cunningham KW, Hardwick JM, Palková Z, Powers T, Severin FF, Váchová L. 2018. Comment on “Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death”. Science 360:eaar6910. doi: 10.1126/science.aar6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poreba M, Groborz K, Navarro M, Snipas SJ, Drag M, Salvesen GS. 2018. Caspase selective reagents for diagnosing apoptotic mechanisms. Cell Death Differ doi: 10.1038/s41418-018-0110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anonymous. 1972. Apoptosis. Lancet 300:1011–1012. doi: 10.1016/S0140-6736(72)92414-2. [DOI] [PubMed] [Google Scholar]
- 77.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 78.Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y. 1999. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 401:168–173. doi: 10.1038/43678. [DOI] [PubMed] [Google Scholar]
- 79.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. 2001. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 80.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. 2014. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 81.Shan B, Pan H, Najafov A, Yuan J. 2018. Necroptosis in development and diseases. Genes Dev 32:327–340. doi: 10.1101/gad.312561.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Daskalov A, Habenstein B, Sabaté R, Berbon M, Martinez D, Chaignepain S, Coulary-Salin B, Hofmann K, Loquet A, Saupe SJ. 2016. Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled programmed cell death is related to necroptosis. Proc Natl Acad Sci U S A 113:2720–2725. doi: 10.1073/pnas.1522361113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Daskalov A, Heller J, Herzog S, Fleissner A, Glass NL. 2017. Molecular mechanisms regulating cell fusion and heterokaryon formation in filamentous fungi. Microbiol Spectr 5. doi: 10.1128/microbiolspec.FUNK-0015-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veneault-Fourrey C, Barooah M, Egan M, Wakley G, Talbot NJ. 2006. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science 312:580–583. doi: 10.1126/science.1124550. [DOI] [PubMed] [Google Scholar]
- 85.Kershaw MJ, Talbot NJ. 2009. Genome-wide functional analysis reveals that infection-associated fungal autophagy is necessary for rice blast disease. Proc Natl Acad Sci U S A 106:15967–15972. doi: 10.1073/pnas.0901477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu T, Nicolson S, Denton D, Kumar S. 2015. Distinct requirements of autophagy-related genes in programmed cell death. Cell Death Differ 22:1792–1802. doi: 10.1038/cdd.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang H, Baehrecke EH. 2015. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol 25:376–387. doi: 10.1016/j.tcb.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. 2014. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clem RJ. 2015. Viral IAPs, then and now. Semin Cell Dev Biol 39:72–79. doi: 10.1016/j.semcdb.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 90.Shlezinger N, Minz A, Gur Y, Hatam I, Dagdas YF, Talbot NJ, Sharon A. 2011. Anti-apoptotic machinery protects the necrotrophic fungus Botrytis cinerea from host-induced apoptotic-like cell death during plant infection. PLoS Pathog 7:e1002185. doi: 10.1371/journal.ppat.1002185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Byers NM, Vandergaast RL, Friesen PD. 2016. Baculovirus inhibitor-of-apoptosis op-IAP3 blocks apoptosis by interaction with and stabilization of a host insect cellular IAP. J Virol 90:533–544. doi: 10.1128/JVI.02320-15. [DOI] [PMC free article] [PubMed] [Google Scholar]