

The cancer-causing HPV E6 oncoproteins all possess a PDZ binding motif at their extreme carboxy termini. Depending upon whether this motif is phosphorylated, E6 can recognize PDZ domain-containing proteins or members of the 14-3-3 family of proteins. We show here that DNA damage response pathways directly signal to the E6 PBM, resulting in Chk1- and Chk2-driven phosphorylation. This phosphorylation is particularly pronounced following treatment of cells with a variety of different chemotherapeutic drugs. A direct functional consequence of this signaling is to confer an enhanced ability upon E6 to inhibit p53 transcriptional activity in a proteasome-independent but phosphorylation-dependent manner. These results are the first example of DNA damage signaling pathways regulating PBM-PDZ interactions and provide the mechanistic link between E6 PBM function and perturbation of p53 activity.
KEYWORDS: HPV, E6, PBM, DNA damage, Chk1/Chk2, p53
ABSTRACT
The presence of a PDZ binding motif (PBM) in the human papillomavirus (HPV) E6 oncoprotein appears to be a characteristic marker of high oncogenic potential and confers interaction with a number of different cellular PDZ domain-containing substrates. The E6 PBM is also subject to phosphorylation, resulting in inhibition of E6 PDZ binding activity and instead allowing E6 to associate with 14-3-3 proteins. In this study, we analyzed the conditions under which the E6 PBM is phosphorylated. We demonstrate that in normal cycling cells, the levels of E6 phosphorylation are very low. However, following exposure of cells to oxidative stress or the induction of DNA damage, there is a striking increase in the levels of E6 phosphorylation. Depending on the specific stimulus, this phosphorylation of E6 can involve the ATM/ATR pathway and is performed primarily through Chk1, although the Chk2 pathway is also involved indirectly through activation of protein kinase A (PKA). To understand the biological relevance of these phospho-modifications of E6, we analyzed their effects upon the ability of E6 to inhibit p53 transcriptional activity. We show that an intact E6 phospho-acceptor site plays an essential role in the ability of E6 to inhibit p53 transcriptional activity on a subset of p53-responsive promoters in a manner that is independent of E6's ability to direct p53 degradation. These results are, to our knowledge, the first example of a DNA damage response controlling PBM-PDZ recognition. This study also provides links between the DNA damage response, the regulation of E6 PBM function, and the inhibition of p53 activity and begins to explain how HPV-infected cells remain within the cell cycle, despite activation of DNA damage response pathways during productive virus infections.
IMPORTANCE The cancer-causing HPV E6 oncoproteins all possess a PDZ binding motif at their extreme carboxy termini. Depending upon whether this motif is phosphorylated, E6 can recognize PDZ domain-containing proteins or members of the 14-3-3 family of proteins. We show here that DNA damage response pathways directly signal to the E6 PBM, resulting in Chk1- and Chk2-driven phosphorylation. This phosphorylation is particularly pronounced following treatment of cells with a variety of different chemotherapeutic drugs. A direct functional consequence of this signaling is to confer an enhanced ability upon E6 to inhibit p53 transcriptional activity in a proteasome-independent but phosphorylation-dependent manner. These results are the first example of DNA damage signaling pathways regulating PBM-PDZ interactions and provide the mechanistic link between E6 PBM function and perturbation of p53 activity.
INTRODUCTION
Human papillomaviruses (HPVs) are the causative agents of many different types of cancer, with cervical cancers being the most important (1). While over 200 different HPV types have been described, only a small subset of 12 so-called high-risk HPV types cause cancer. These include HPV-16 and HPV-18, which account for approximately 80% of all cervical cancers (2). These viruses encode two oncoproteins, E6 and E7, that are responsible for the initiation, progression, and maintenance of cancer development. Inhibiting the expression or function of either protein results in inhibition of transformed cell growth and the induction of cell death by apoptosis (3, 4). Therefore, both viral oncoproteins represent excellent targets for therapeutic intervention in HPV-induced malignancies.
Both E6 and E7 are highly multifunctional, with each protein interacting with a large number of cellular partners, some of which have been shown to play a role in the ability of E6 and E7 to bring about cell transformation and malignancy in different experimental settings (5, 6). In the case of E7, its critical partners include the pRb family of pocket proteins (7, 8). These interactions result in inhibition of pRb function, which is brought about in part through recruitment of a cullin ubiquitin ligase, and the subsequent degradation of pRb (9, 10). Recent studies have also shown that E7 can recruit another cellular ubiquitin ligase, p600, to target other cellular substrates for proteasome-mediated degradation (11–13). In the case of E6, a major interacting partner is the p53 tumor suppressor. This is also inactivated through E6-induced proteasome-mediated degradation, an activity that also involves the recruitment of a cellular ubiquitin ligase, E6AP (14, 15). Thus, in many HPV-containing cell lines and tumors, p53 levels are very low but not absent, indicating there may be additional mechanisms by which HPVs might inhibit p53 activity (15–19).
A further important class of E6-interacting partners includes substrates containing PDZ domains (20). The high-risk, potentially cancer-causing HPV E6 oncoproteins possess a PDZ binding motif (PBM) at their extreme carboxy termini that is absent from most benign HPV E6 oncoproteins (21, 22). Through this motif, E6 has been shown to target a variety of different cellular PDZ domain-containing substrates (23). These interactions appear to be important both for the viral life cycle and for the ability of the virus to bring about cell transformation and to contribute to cancer development in transgenic mice (20, 24, 25). Several studies have also shown that an intact PBM appears to play an important role in viral episome maintenance (26, 27). Furthermore, one study suggested a possible connection between E6 PBM function and p53 inactivation, since loss of p53 function was found to rescue episomal maintenance in cells where the E6 PBM was mutated (28, 29). Intriguingly, during the HPV life cycle, amplification of the viral genome also involves activation of the DNA damage response (DDR), primarily through activation of ATM/ATR (30–33). While much attention has been placed on how components of this pathway contribute to different aspects of the viral life cycle, many questions still remain, in particular, how HPV overcomes the potentially inhibitory activities of the pathway.
Recent studies have shown that the E6 PBM has a dual function, with a phospho-acceptor site embedded within the PBM; once phosphorylated, E6 switches from interaction with PDZ domain-containing proteins and instead associates with members of the 14-3-3 family (34). Based largely on in vitro studies, the phosphorylation of the E6 PBM has been thought to be due to either protein kinase A (PKA) or AKT activity, depending upon the precise amino acid sequence of the E6 PBM. However, regardless of the kinase, phosphorylation of E6 was found to inhibit PDZ recognition and promote interaction with 14-3-3 (34, 35). This was particularly intriguing, considering the potential link between the E6 PBM activity, p53 function (28, 29), and the known requirement for certain 14-3-3 isoforms for optimal p53 activity. Thus, 14-3-3σ has been shown to play a critical role in regulating p53 subcellular distribution (36), while 14-3-3γ and 14-3-3ε have been implicated in p53's transcriptional activation of certain promoters (37). Interestingly, both 14-3-3ε and 14-3-3γ appear to be bound particularly strongly by the phosphorylated HPV-16 and HPV-18 E6 oncoproteins (35).
Despite the above dissection of E6 PBM function, nothing is known about when E6 is phosphorylated in vivo. Here, we show that in the normal cell cycle, E6 is only very weakly phosphorylated. However, following the induction of a stress response, the level of E6 phosphorylation increases dramatically, and this is particularly pronounced following induction of a DDR. Intriguingly, retention of an intact PBM and phospho-acceptor site within E6 correlates with the ability of E6 to inhibit p53's transcriptional activation of a subset of p53-responsive promoters, thereby linking regulation of the E6 PBM function with inhibition of p53 activity.
RESULTS
Differential phosphorylation of HPV E6 during the cell cycle.
Since phosphorylation plays a major role in influencing E6 substrate specificity, we first wanted to ascertain at what point in the cell cycle E6 is normally phosphorylated. To do this, HPV-18-containing HeLa cells were synchronized using a double-thymidine block, and following release, cell extracts were made at different points in the cell cycle, as verified by fluorescence-activated cell sorter (FACS) analysis (Fig. 1A). The extracts were then analyzed by Western blotting using a specific antibody raised against HPV-18 E6 phosphorylated on T156 within the E6 PBM (35). The results shown in Fig. 1B demonstrate very low levels of E6 phosphorylation in asynchronously growing cells, with a slight increase in the levels of phospho-E6 in the G1 population and undetectable levels of phosphorylation of E6 in the S and G2/M phases of the cell cycle. We also performed an analysis of the E6 phospho-status in cells that were arrested in G2/M by nocodazole treatment. The results, also shown in Fig. 1B, demonstrate surprisingly high levels of E6 phosphorylation in these G2/M phase-arrested cells, indicating that nocodazole can also induce E6 phosphorylation. This contrasts with the results from the double-thymidine block, where no phosphorylation of E6 was observed in the G2/M population of cells. In order to determine whether there is phosphorylation of E6 when the cells reenter G1, the assay was repeated, and the cells were followed for an extended time. As can be seen in Fig. 1C, the cells reentered G1 10 to 12 h after release from the double-thymidine block. However, in contrast to the G1-arrested cells, no phosphorylation of E6 was detectable when the cells reentered G1 (Fig. 1D). These results suggest that in normal cycling cells the levels of E6 phosphorylation are very low and that only following cell growth arrest, albeit when induced through different means, does E6 become phosphorylated. These results suggest that there may be multiple kinases capable of phosphorylating E6, depending upon the precise stimulus.
FIG 1.

HPV-18 E6 oncoprotein is only weakly phosphorylated during the normal cell cycle. (A) FACS analysis of HeLa cells synchronized by double- thymidine block and subsequently released and harvested at different times to obtain cells in different phases of the cell cycle. y axis, number of cells/channel; x axis, fluorescence intensity of designated parameter. (B) Western blot analysis of HeLa cell extracts from asynchronous (Ays) and synchronized HeLa cells in G1, S, M1, and M2 phases using antibodies against phosphorylated E6 (18-pE6) and total E6; α-actinin was used as a loading control. (C) FACS analysis of HeLa cells synchronized by double-thymidine block and subsequently released and harvested at different times to obtain cells in different phases of the cell cycle. y axis, number of cells/channel; x axis, fluorescence intensity of designated parameter. (D) Western blot analysis of HeLa cell extracts from G1, S, M1, M2, and the next G1 phases using antibodies against phosphorylated E6 (18-pE6) and total E6; α-actinin was used as a loading control.
The above-mentioned results indicate that significant levels of E6 phosphorylation are obtained only in growth-arrested cells. We therefore investigated whether the nocodazole-induced E6 phosphorylation patterns might be part of a stress response. Indeed, nocodazole has been implicated in induction of oxidative stress (38) and DNA damage (39, 40) in the cell. One of the potential mechanisms is inhibition of MTH1 (41), which leads to the incorporation of oxidized nucleotides into the DNA and subsequent DNA damage (42). To ascertain whether the phosphorylation of E6 following exposure to nocodazole might be an oxidative-stress response, we performed a series of analyses of nocodazole-treated HeLa cells, using hydrogen peroxide (H2O2) treatment as a positive control for the induction of oxidative stress. The E6 phospho-status in these cell extracts was then analyzed by Western blotting. The results are shown in Fig. 2A and demonstrate a strong increase in the levels of E6 phosphorylation in response to H2O2 and nocodazole treatment. To determine whether this increase in E6 phosphorylation was due to induction of oxidative stress, we treated the cells with N-acetyl-cysteine (NAC), an antioxidant, following the exposure to H2O2 and nocodazole. As can be seen in Fig. 2A, H2O2- and nocozadole-induced E6 phosphorylation was significantly reduced upon treatment with NAC, suggesting an involvement of oxidative-stress response kinases in E6 phosphorylation. Interestingly, we also found that blocking protein synthesis using cycloheximide (CHX) resulted in significantly elevated levels of phospho-E6, although, as expected, the total level of E6 was reduced under these conditions (Fig. 2A). However, in this case, NAC did not reduce the phospho-E6 levels, suggesting that the cycloheximide-induced phosphorylation of E6 is not an oxidative-stress response, but rather, that another stress response pathway(s) might be involved.
FIG 2.

HPV-18 E6 is phosphorylated as a result of cellular stress responses. (A) Representative Western blot analysis of HeLa cells either untreated (UN) or treated with H2O2 for 4 h, nocodazole for 15 h, or CHX for 4 h in the presence or absence of the antioxidant NAC. The blots were probed with antibodies against total HPV-18 E6 and HPV-18 E6 phospho-T156 (18-pE6); α-actinin was used as a loading control. (B) Relative quantification of Western blots was done using Image J software, where the ratio of the net intensity of pE6 to the net actinin loading control was calculated. The histogram shows the results from at least three independent experiments, statistically quantified by Student's t test; the error bars indicate standard errors of the mean. *, P = 0.05; ns, nonsignificant.
Potential involvement of DDR kinases in regulation of E6 phosphorylation.
The above-mentioned results suggest that one source of signaling through oxidative stress could be by stimulation of a DDR. Accordingly, we compared the known amino acid recognition motifs of a number of DDR kinases, including ATM, ATR, Chk1, and Chk2, with the sequence of the HPV-18 E6 PBM. As can be seen in Fig. 3A, HPV-18 E6 has a good match with the consensus site for Chk1. In order to determine whether E6 could be phosphorylated by either Chk1 or Chk2, we performed an in vitro kinase assay using the purified kinases with purified HPV-18 E6 glutathione S-transferase (GST) fusion protein in the presence of radiolabeled ATP. The results shown in Fig. 3B demonstrate that HPV-18 E6 is a very good substrate for phosphorylation by Chk1, but not by Chk2. Furthermore, the lack of any phosphorylation of the HPV-18 E6 T156E mutant indicates that Chk1 phosphorylates E6 within the PBM at residue T156.
FIG 3.
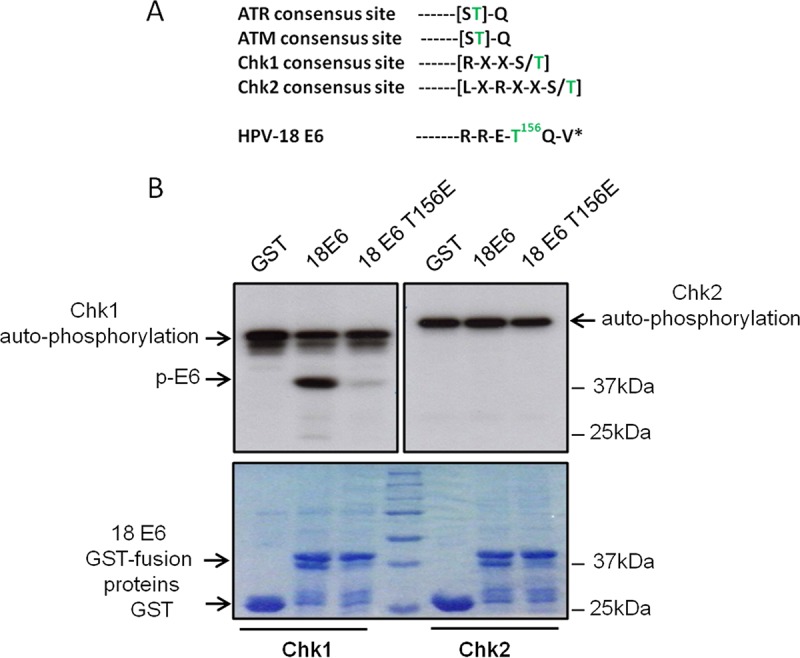
HPV-18 E6 is phosphorylated by the DNA damage response kinase Chk1. (A) Consensus phosphorylation motifs recognized by ATR, ATM, Chk1, and Chk2 compared with the sequence of the carboxy terminus of HPV-18 E6. *, carboxy terminus of E6; green text, serine/threonine phospho-acceptor. (B) In vitro phosphorylation assay using purified GST fusion proteins of wild-type HPV-18 E6 and the HPV-18 E6 T156E mutant incubated with Chk1 or Chk2 in the presence of [γ-32P]ATP. (Top) Autoradiograms. (Bottom) Coomassie-stained SDS-PAGE gels. The arrows indicate phospho-E6, the GST E6 fusion protein, the GST control, and the autophosphorylated Chk1 and Chk2.
To ascertain whether any of these DDR kinases might indeed be involved in stress-responsive E6 phosphorylation in vivo, we treated HeLa cells with specific inhibitors of ATR, ATM, Chk1, and Chk2 kinases for 15 h and then exposed the cells to either nocodazole or H2O2. The results obtained are shown in Fig. 4 and demonstrate that under nonstressed conditions, the levels of E6 phosphorylation are, again, very low. Following exposure to nocodazole, E6 was clearly phosphorylated, but this was greatly decreased following inhibition of the Chk1 kinase, suggesting that it is primarily responsible for phosphorylating E6 after exposure to nocodazole (Fig. 4B). In contrast, in the case of H2O2, the greatest inhibition of phosphorylation occurred following treatment with the Chk2 inhibitor (Fig. 4C). This was rather surprising, considering the low level of phosphorylation of E6 by Chk2 observed in vitro (Fig. 3). However, previous studies had indicated that one downstream kinase activated by DDR kinases was PKA (43–45). Since we had previously shown that HPV-18 E6 was a very good substrate for PKA in vitro (34, 35, 46), we repeated the analysis but also treated cells with H89 to block PKA activity in the H2O2-exposed cells. The results shown in Fig. 4D demonstrate a marked inhibition of H2O2-induced phosphorylation of E6 following inhibition of either PKA or Chk2. These results indicate that PKA is most likely responsible for phosphorylating E6 following the activation of Chk2 in response to an H2O2 oxidative-stress-induced DDR.
FIG 4.

Different DDR pathways lead to phosphorylation of E6 in response to different stresses. (A to C) HeLa cells were treated with specific inhibitors of ATR (ATRi), ATM (ATMi), Chk1 (Chk1i), or Chk2 (Chk2i) for 15 h and then with dimethyl sulfoxide (DMSO) as a control (A) or treated with nocodazole (B) or H2O2 (C). Western blots were probed with antibodies against total HPV-18 E6 and HPV-18 E6 phospho-T156. α-Actinin was used as a loading control. (D) The assay was repeated with H2O2, additionally treating the cells with a specific PKA inhibitor, H89, as well as Chk1i and Chk2i. The pE6 levels were detected by Western blotting, and total E6 is also shown. α-Actinin was used as a loading control. (E) HeLa cells were treated with H2O2, together with the specific PKA inhibitor H89 and the DDR kinase inhibitors, as indicated. The pChk2 levels were analyzed to check the efficacy of ATMi and ATRi, and pE6 levels were also analyzed in the presence or absence of the various inhibitors. Also shown are total E6 levels. α-Actinin was used as a loading control.
In order to further confirm Chk2 activation after exposure to H2O2, we analyzed the levels of phosphorylated Chk2 (Thr68). Indeed, H2O2 treatment led to a significant increase in pChk2 levels, and inhibition of ATM and ATR also reduced pChk2 levels. However, there was a striking increase in E6 phosphorylation when ATM and Chk1 were inhibited (Fig. 4C to E). In order to investigate whether this increase in E6 phosphorylation following inhibition of ATM and Chk1 was also mediated by PKA, the assay was repeated in the presence of the PKA inhibitor H89. The results shown in Fig. 4E demonstrate that increased phosphorylation of E6 following inhibition of ATM or Chk1 is mediated largely by PKA. This observation supports previous studies showing that Chk1 inhibition causes replication stress and activates the ATR pathway (47–49) and that ATR signals activate Chk2 in response to H2O2 treatment (50), which subsequently activates PKA (44).
Having shown that DDR response kinases play an important role in regulating E6 phosphorylation, we were interested to know whether DDR modulators might also affect E6 phosphorylation. Since various chemotherapeutic agents have been shown to indirectly modulate the DDR pathways (51), it was of interest to know what effect they might have on E6 phosphorylation. We used various chemotherapeutic agents that are known to target molecules involved in the DNA replication and DNA repair pathways (listed in Fig. 5A). HeLa cells were exposed to the different chemotherapeutic drugs, and the cells were then harvested and the levels of E6 phosphorylation were ascertained by Western blotting. At the same time, the levels of p53 expression were also analyzed. The results of the analysis, shown in Fig. 5B, demonstrate a number of interesting points. Inhibition of topoisomerase, ribonucleotide reductase, and DNA replication all provoked a pronounced increase in the levels of E6 phosphorylation, suggesting that multiple DDRs can signal to E6. In contrast, while PARP inhibitors have been documented to accumulate double-strand DNA breaks, resulting in the accumulation of DNA damage in the cell (52, 53), PARP inhibition did not induce E6 phosphorylation in HeLa cells, and this appeared to be related to the very low level of DDR activation in these cells following PARP inhibition (data not shown). Finally, none of the drugs induced an increase in p53 levels, consistent with E6's ability to target p53 for degradation (14, 54), and in a subset of cases, p53 levels were also further reduced. This is consistent with previous studies showing that induction of certain DDR pathways can also increase the levels of E6-induced degradation of p53 (55).
FIG 5.

Exposure to DNA damage-inducing chemotherapeutic agents can induce E6 phosphorylation. (A) Chemotherapeutic agents used and their cellular targets. (B) HeLa cells were treated with the indicated drugs for 15 h prior to harvesting and analysis by Western blots probed with antibodies against total HPV-18 E6, HPV-18 pE6, and p53; α-actinin was used as a loading control. (C) The assay was repeated with cisplatin, additionally including specific kinase inhibitors, and the Western blot was probed with antibodies against total HPV-18 E6 and HPV-18 pE6; α-actinin was used as a loading control.
Having found that multiple DDR-inducing agents could increase the levels of E6 phosphorylation, we then wanted to determine whether Chk1 or Chk2 was responsible. To do this, we chose to analyze cisplatin. HeLa cells were exposed to cisplatin in the presence of different DDR kinase inhibitors. The cells were then harvested, and the levels of E6 phosphorylation were analyzed by Western blotting. The results shown in Fig. 5C demonstrate that the majority of E6 phosphorylation that occurs following exposure to cisplatin is Chk1 dependent.
Taken together, the above-mentioned results indicate that various forms of DDR result in phosphorylation of E6. This can be mediated by Chk1 in response to an oxidative-stress response following treatment with nocodazole and a DDR following treatment with cisplatin. However, in the case of an oxidative-stress response induced by H2O2, phosphorylation of E6 occurs through a Chk2-activated pathway that requires downstream PKA to phosphorylate E6 directly.
The HPV-18 E6 PBM perturbs p53 transcriptional-transactivation activity.
Having shown that the HPV E6 PBM can be phosphorylated by a number of stress response kinases in response to various cellular stresses, the next question was, what is the biological result of the E6 phosphorylation? Interestingly, a number of recent studies with the HPV-16 and HPV-18 genomes in organotypic systems have shown that the E6 PBM is required for HPV episomal maintenance and, furthermore, that p53 loss can partly rescue this episome loss following the mutation of the E6 PBM (28, 29). It is also known that HPVs activate the ATM/ATR DNA repair pathway and that this is necessary for HPV genome amplification (30, 31, 56). Taken together, these studies suggest a probable link between phosphorylation of the E6 PBM and perturbation of p53 activity. One potential common link between these observations is 14-3-3, since this family of proteins is linked to the regulation of p53 transcriptional-transactivation activity during cell cycle checkpoint control (57–59).
Therefore, we initiated a series of studies to investigate whether the E6 PBM has any effect on p53 transcriptional transactivation on a variety of different promoters—PUMA, p21, Mdm2, and BAX—using Renilla luciferase as a transfection efficiency control. Since a major function of HPV-18 E6 is the degradation of p53 through the ubiquitin proteasome pathway (14, 54), it was necessary to perform our studies in the presence of the proteasome inhibitor CBZ (MG132). The p53-null H1299 cells were transfected with the appropriate reporter constructs, together with p53 and different HPV-18 E6 expression plasmids. In the first analysis, we focused on wild-type HPV-18 E6 and the HPV-18 E6ΔPBM mutant. After 24 h, cells were harvested and luciferase activity was measured using the dual-luciferase assay system; the results are shown in Fig. 6. As can be seen, there are some striking differences between the promoters in how the E6 PBM can modulate p53 transcriptional activity following proteasome inhibition. In all cases, in the absence of proteasome inhibition, the wild type and the E6ΔPBM mutant show similar abilities to inhibit p53 transcriptional activity. Following proteasome inhibition, the wild-type E6 retains the ability to inhibit p53 transcriptional activity. In contrast, for the E6ΔPBM mutant, this holds true only for the BAX promoter (Fig. 6C), where both wild-type and mutant E6 retain inhibition of p53 transcriptional activity in the presence of proteasome inhibitors. In the case of p21 (Fig. 6A), the E6ΔPBM mutant allowed a modest increase in p53 transcriptional activity, but this was even more apparent with PUMA (Fig. 6B) and Mdm2 (Fig. 6D), where lack of a PBM allowed a marked increase in the level of p53 transcriptional activity in the presence of proteasome inhibitors.
FIG 6.

The HPV-18 E6 PBM contributes to inhibition of p53 transcriptional-transactivation activity. H1299 cells were transfected with the indicated promoter constructs upstream of a luciferase promoter—p21-Luc (A), PUMA-Luc (B), BAX-Luc (C), or Mdm2-Luc (D)—together with p53 and either wild-type or mutant HPV-18 E6 in the presence (CBZ) or absence (UN) of the proteasome inhibitor CBZ. The histograms show the results from at least three independent experiments quantified using Student's t test; the error bars indicate standard errors of the mean. Also shown are the P values (*, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, nonsignificant) for the changes in relative luciferase activity.
In order to elucidate whether this effect was linked to the ability of E6 to be phosphorylated and was not due to an overall defect in PDZ interaction, we included the HPV-18 E6 R153G mutant, which we have shown previously to be defective in the phospho-recognition motif but which retains PDZ binding potential (34). The results for the Mdm2 promoter are shown in Fig. 6D and demonstrate that it has the ability to block p53 transcription similar to that of the wild-type E6 but that, following addition of proteasome inhibitors, this activity is compromised and there is a dramatic increase in p53 transcriptional activity, comparable to that seen with the E6ΔPBM mutant.
Taken together, these results demonstrate that HPV-18 E6 can inhibit p53 transcriptional activity in a manner that is PBM- and phospho-site dependent, indicating that phosphorylation of E6 within the PBM in response to a variety of DDR signals can directly link PBM function to inhibition of p53 transcriptional activity.
DISCUSSION
The high-risk HPV E6 oncoproteins are characterized by the presence of a carboxy-terminal PBM, embedded within which is a phospho-acceptor site, phosphorylation of which inhibits PDZ binding activity and confers interaction with 14-3-3 proteins (34, 35). Thus, this site offers the potential of fine-tuning E6's activity with respect to a number of different interaction partners, both within the normal viral life cycle and during the development of malignancy. Previously, we had shown that AKT and PKA could both potentially phosphorylate E6 within the PBM in vitro (34, 35), but we had little evidence about which kinases normally phosphorylate endogenously expressed E6 in cells derived from cervical cancers. Here, we show that E6 is normally only very weakly phosphorylated, if at all, during the normal cell cycle. However, following induction of a variety of stress response kinases, and in particular those associated with the DDR, there is a dramatic increase in the levels of E6 phosphorylation. The bulk of this phosphorylation appears to be mediated via Chk1, and this therefore directly links regulation of the E6 PBM activity to induction of the DDR, which plays a critical role in the normal viral life cycle and in the development of HPV-induced malignancy.
While HPV-16 and HPV-18 E6 proteins were previously shown to be good substrates for phosphorylation by both AKT and PKA, we had little evidence as to when endogenously expressed E6 was normally phosphorylated within the cell cycle. We therefore first proceeded to monitor HPV-18 E6 phosphorylation in HeLa cells using two different approaches: double-thymidine block and release to follow changes through the cell cycle and nocodazole treatment to analyze levels of E6 phosphorylation in G2/M. To our surprise we found that thymidine block and release resulted in weak E6 phosphorylation in G1/S-arrested cells, while nocodazole treatment induced high levels of phosphorylation of E6 in G2/M-arrested cells. Since no phosphorylation was observed when the cells cycled normally through G2/M and back into G1 after release from a double-thymidine block, this suggested that phosphorylation of E6 might be linked more to a stress response than to normal progression through the cell cycle. Since in our previous unpublished investigations we had been unable to block the nocodazole-induced phosphorylation of E6 using PKA inhibitors (data not shown), we reasoned that another pathway of E6 phosphorylation might be operational. Indeed, nocodazole has been implicated in the induction of oxidative-stress responses and also in induction of a DDR (38–40). In agreement with this, we found that induction of an oxidative-stress response by either H2O2 or nocodazole was responsible for the very high levels of E6 phosphorylation that were observed following exposure to these agents. Intriguingly, cycloheximide also induces very high levels of E6 phosphorylation, but this appears to be mediated by a different stress response pathway. Since protein synthesis inhibition has been shown to activate several kinases (60–62), it is possible that E6 can be phosphorylated by a variety of other stress response kinases. This result also argues for caution in using cycloheximide as a means of monitoring E6 stability over time, as currently we have no information on what this phosphorylation might do to E6 stability. However, previous studies have suggested a biphasic half-life for E6, and this could well be linked to changes in E6 phosphorylation status during the course of these assays (63, 64). It is worth noting that the detection of E6 phosphorylation is dependent upon the use of a very well-characterized phospho-specific antibody, which appears to be very highly specific for the phosphorylated form of HPV-18 E6 (34, 35). Unfortunately, we have not been able to detect phospho-peptides of HPV-18 E6 using standard proteomic approaches.
Having found that oxidative-stress responses were responsible for inducing E6 phosphorylation, we next investigated which DDR pathways might also be involved. We noticed a very high degree of homology within the E6 PBM to a Chk1 consensus recognition site, and indeed, using in vitro assays, we found that E6 was a very good substrate for phosphorylation by Chk1 at residue T156 within the E6 PBM. In contrast, the closely related Chk2 kinase appeared to be incapable of phosphorylating HPV-18 E6 in vitro, further demonstrating the specificity of these assays. In vivo, the situation was rather more complicated, with different kinases being responsible for E6 phosphorylation, depending upon the specific stimulus. Thus, both H2O2- and nocodazole-induced phosphorylation of E6 involves oxidative-stress signaling, as we demonstrated by blocking it with NAC, but the kinases involved are different. In the case of nocodazole, Chk1 inhibition clearly blocked E6 phosphorylation, strongly supporting the in vitro kinase assays. However, for H2O2, Chk2 appeared to be involved, despite the fact that it appeared incapable of phosphorylating E6 in vitro. Previous studies have shown that the DDR pathway and Chk2 can also activate PKA (43, 44), which we know from our earlier results could also be responsible for phosphorylating E6. Indeed, this does seem to be the case, where H2O2-induced phosphorylation of E6 is blocked by treatment with either a Chk2 inhibitor or the PKA inhibitor H89, suggesting that PKA is the kinase responsible for phosphorylating E6 in response to an H2O2-induced oxidative-stress-induced DDR.
We then extended these studies to investigate whether clinically relevant inducers of a DDR might also induce E6 phosphorylation. To do this, we assessed cisplatin as an agent capable of intercalating DNA and blocking DNA replication, plus inhibitors of topoisomerase, ribonucleotide reductase, and PARP. Interestingly, cisplatin treatment, and also blocking topoisomerase or ribonucleotide reductase, all induced a high level of E6 phosphorylation in HeLa cells. In contrast, blocking PARP, which has also been linked to induction of DDR, had no discernible effect upon the levels of E6 phosphorylation, which seems to be related to the very low level of induction of DDR in HeLa cells following PARP inhibition (data not shown). Further analysis also demonstrated that, at least in the case of cisplatin treatment, the induction of E6 phosphorylation was largely mediated by Chk1.
When E6 is phosphorylated in the PBM, it blocks PDZ recognition and instead confers interaction with 14-3-3 family members. Several previous studies have shown a critical requirement for the activation of DDR pathways in the HPV life cycle, with ATM/ATR in particular playing essential roles in genome amplification (30, 33, 56). Recent studies have also indicated a potential link between the E6 PBM and HPV genome maintenance, with inactivation of p53 appearing to be able to compensate for loss of PBM function (28). This, therefore, raised the obvious question as to whether the E6 PBM might have an additional function linking it to inactivation of p53. To investigate this, we analyzed a panel of different p53-responsive promoters and asked whether an intact PBM could contribute to the ability of E6 to block p53 transcriptional activity in both the presence and absence of proteasome inhibition. We found a quite complex picture, where certain promoters, such as BAX, are inhibited by E6 regardless of the presence of an intact PBM. In contrast, in the cases of Mdm2 and PUMA, the E6 PBM appears to play an essential role in inhibiting p53 transcriptional activity under conditions of proteasome inhibition. Furthermore, using the HPV-18 E6 R153G mutant, which has been shown previously to be defective for phosphorylation but retains PDZ binding activity (35), we found that it was also defective in its ability to block p53 transcriptional activity on the Mdm2 promoter in the presence of proteasome inhibitors. While proteasome inhibition could result in the formation of aberrant complexes consisting of p53, E6AP, and E6, which in turn could affect p53 transcriptional activity (18), the lack of activity observed using the R153G mutant argues against this and instead strongly suggests that the major role of the PBM in this activity is retention of the ability to be phosphorylated. These studies allow us to propose a model, shown in Fig. 7, in which a variety of different cellular stresses can induce phosphorylation of E6 within the PBM, either directly by Chk1 or indirectly through PKA, which in turn can directly contribute to the ability of E6 to block p53 transcriptional activation of a subset of p53-responsive genes. Obviously, whether this directly involves 14-3-3 or another, as yet undetermined mechanism requires further investigation.
FIG 7.

Phosphorylation of the HPV E6 PBM links cellular stress response signaling to E6 inhibition of p53 transcriptional activity. The model shows how the E6 PBM can become phosphorylated following exposure to various stress-inducing conditions. Our results demonstrate the involvement of at least two major pathways. The DDR pathway through Chk1 activation leads directly to E6 phosphorylation, and Chk2 activation results in E6 phosphorylation via PKA. Other, unknown cellular kinases are also likely to be involved, for instance, those induced following cycloheximide treatment. Our studies provide compelling evidence of a direct link from the induction of oxidative-stress-induced DDR to the phospho-regulation of HPV E6 oncoproteins. Finally, we propose that phosphorylation of E6 contributes to the inhibition of p53 transcriptional activity, possibly through a mechanism involving 14-3-3 proteins.
In summary, the above-described studies provide compelling evidence of a direct link from the induction of oxidative stress and induction of DDR to the phospho-regulation of high-risk HPV E6 oncoproteins and their ability to perturb p53 transcriptional activity on a subset of p53-responsive promoters. This is the first example of PBM-PDZ recognition being regulated by DDR kinases and begins to explain how HPV can make use of the ATM/ATR pathway that is induced during viral infection while at the same time overcoming some of the deleterious effects that would also be expected. These results also raise the intriguing possibility that some HPV types may be more responsive to certain chemotherapeutic strategies, depending upon the specific HPV type and the sequence of the E6 PBM. Future studies will aim to further clarify these possibilities.
MATERIALS AND METHODS
Cell culture.
HeLa (ATCC) and H1299 (ATCC) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/ml), and glutamine (300 μg/ml). The HeLa cells were transfected with small interfering RNA (siRNA) against the appropriate genes using Lipofectamine RNAiMax transfection reagent (Invitrogen), as recommended by the manufacturer. The samples were analyzed 72 h posttransfection.
Plasmids and transfections.
The p53-null H1299 cells were transfected with appropriate plasmids by the calcium-phosphate precipitation method. The plasmids used were as follows: p21-Luciferase, BAX-Luciferase, MDM2-Luciferase, PUMA-luciferase, and Renilla luciferase (kind gifts from Giannino Del Sal).
The GST, HPV-18 E6 GST, and HPV-18 E6 T156E GST fusion proteins have been described previously (34, 35). FLAG-p53, pGWI-18 E6, pCDNA 18 E6 R153G, and pGWI-18 E6 ΔPBM have also been described previously (34, 65, 66).
Inhibitors, chemicals, reagents, and antibodies.
The inhibitors and chemicals used for experiments were as follows: 5 μM AZ-20 ATR inhibitor, 5 μM KU-60019 ATM inhibitor, 100 nM UCN01 Chk1 inhibitor, 5 μM Chk2 inhibitor, 500 μM H2O2, 5 mM NAC, 100 μg/ml cycloheximide, 200 ng/ml nocodazole, 2.5 mM thymidine, 10 μM H-89 PKA inhibitor, and 20 nM MG-132 (all from Sigma-Aldrich).
The chemotherapeutic reagents used were as follows: 5 μM teniposide, 50 to 100 μM cisplatin as indicated, 10 μM triapine/3-AP, and 10 μM etoposide (all from Sigma-Aldrich).
The antibodies used were as follows: rabbit anti-α-actinin (Santa Cruz), mouse monoclonal anti-p53 (Santa Cruz), and rabbit polyclonal pChk2 (Thr68) (Novus Biologicals). Rabbit polyclonal HPV-18 E6 phospho-specific antibody and mouse anti-HPV-18 E6 (Arbor Vita) have been described previously (34, 35).
Luciferase assay.
H1299 cells were transfected with appropriate plasmids expressing the luciferase reporters, and cell lysates were collected 24 h posttransfection in lysis buffer provided by the manufacturer. The luciferase assay was performed using the dual-luciferase reporter assay system (Promega), according to the manufacturer's instructions. The firefly luciferase and Renilla luciferase readings were taken using a TD20/20 luminometer by Turner Designs.
In vitro phosphorylation assays.
The Chk1 and Chk2 kinase assays were performed using the Chk1 and Chk2 kinase assay system (Promega). The GST fusion proteins were incubated with the respective kinases for 1 h at room temperature with kinase buffer plus 2.5 μCi [γ-32P]ATP, according to the manufacturer's instructions. The samples were then analyzed by SDS-PAGE and autoradiography.
Cell synchronization and FACS analysis.
The HeLa cells were treated with 2.5 mM thymidine for 15 h, washed thoroughly with 1× PBS, and supplemented with complete medium. After 9 h, a second dose of thymidine was added to block the cells at the G1/S phase. The cells were washed with PBS after 15 h, and the medium was replaced. The cells were then harvested at different time points as follows: 0 h (G1/S phase), 3 h (S phase), 8 h (M1 phase), 9 to 10 h (M2 phase), and 12 h (the next G1 phase). The cell cycle phase analysis was done using propidium iodide staining and FACS analysis with a FACScalibur Cell Sorter (Becton Dickinson).
Western blotting.
Cells were harvested in SDS sample buffer, and samples were run on SDS-PAGE. Following incubation with primary antibody, the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Dako) were used, followed by enhanced chemiluminescence (ECL) detection according to the manufacturer's instructions.
ACKNOWLEDGMENTS
We are very grateful to Giannino Del Sal for the p53 reporter constructs and to the Arbor Vita Corporation for generously providing the anti-E6 antibody 399.
This work was supported by a research grant from the Associazione Italiana per la Ricerca sul Cancro (grant no. 18578). Jayashree Thatte is the recipient of an ICGEB Arturo Falaschi Fellowship and is registered with the Open University, United Kingdom.
We declare that we have no conflict of interest.
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers - a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.International Agency for Research on Cancer, Working Group on the Evaluation of the Carcinogenic Risks to Humans. 2012. A review of human carcinogens: biological agents. IARC monographs on the evaluation of carcinogenic risks to humans, volume 100 B, p 255–313. [Google Scholar]
- 3.Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. 2003. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 4.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. 2003. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol Ther 8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani F, Banks L. 2001. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 20:7874–7887. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- 6.Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. 2001. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 7.Lee JO, Russo AA, Pavletich NP. 1998. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 8.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 9.Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. 2007. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol 81:9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. 2001. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol 75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. 2005. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci U S A 102:11492–11497. doi: 10.1073/pnas.0505337102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szalmas A, Tomaic V, Basukala O, Massimi P, Mittal S, Konya J, Banks L. 2017. The PTPN14 tumor suppressor is a degradation target of human papillomavirus E7. J Virol 91:e00057-17. doi: 10.1128/JVI.00057-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White EA, Munger K, Howley PM. 2016. High-risk human papillomavirus E7 proteins target PTPN14 for degradation. mBio 7:e01530-. doi: 10.1128/mBio.01530-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 15.Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 10:4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekaric P, Shamanin VA, Luo J, Androphy EJ. 2007. hAda3 regulates p14ARF-induced p53 acetylation and senescence. Oncogene 26:6261–6268. doi: 10.1038/sj.onc.1210462. [DOI] [PubMed] [Google Scholar]
- 17.Thomas MC, Chiang CM. 2005. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell 17:251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Thomas M, Massimi P, Banks L. 1996. HPV-18 E6 inhibits p53 DNA binding activity regardless of the oligomeric state of p53 or the exact p53 recognition sequence. Oncogene 13:471–480. [PubMed] [Google Scholar]
- 19.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30:(Suppl 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 20.Banks L, Pim D, Thomas M. 2012. Human tumour viruses and the deregulation of cell polarity in cancer. Nat Rev Cancer 12:877–886. doi: 10.1038/nrc3400. [DOI] [PubMed] [Google Scholar]
- 21.Lee SS, Weiss RS, Javier RT. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A 94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. 1997. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 23.Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A 94:11612–11616. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. 2003. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J Virol 77:6957–6964. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spanos WC, Geiger J, Anderson ME, Harris GF, Bossler AD, Smith RB, Klingelhutz AJ, Lee JH. 2008. Deletion of the PDZ motif of HPV16 E6 preventing immortalization and anchorage-independent growth in human tonsil epithelial cells. Head Neck 30:139–147. doi: 10.1002/hed.20673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee C, Laimins LA. 2004. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J Virol 78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delury CP, Marsh EK, James CD, Boon SS, Banks L, Knight GL, Roberts S. 2013. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J Virol 87:9463–9472. doi: 10.1128/JVI.01234-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenz LD, Rivera Cardona J, Lambert PF. 2013. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog 9:e1003717. doi: 10.1371/journal.ppat.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brimer N, Vande Pol SB. 2014. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J Virol 88:3027–3030. doi: 10.1128/JVI.02360-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong S, Cheng S, Iovane A, Laimins LA. 2015. STAT-5 regulates transcription of the topoisomerase IIbeta-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. mBio 6:e02006-. doi: 10.1128/mBio.02006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong S, Laimins LA. 2013. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog 9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spriggs CC, Laimins LA. 2017. Human papillomavirus and the DNA damage response: exploiting host repair pathways for viral replication. Viruses 9:E232. doi: 10.3390/v9080232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boon SS, Banks L. 2013. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J Virol 87:1586–1595. doi: 10.1128/JVI.02074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boon SS, Tomaic V, Thomas M, Roberts S, Banks L. 2015. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J Virol 89:1579–1586. doi: 10.1128/JVI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee MH, Lozano G. 2006. Regulation of the p53-MDM2 pathway by 14-3-3 sigma and other proteins. Semin Cancer Biol 16:225–234. doi: 10.1016/j.semcancer.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Rajagopalan S, Sade RS, Townsley FM, Fersht AR. 2010. Mechanistic differences in the transcriptional activation of p53 by 14-3-3 isoforms. Nucleic Acids Res 38:893–906. doi: 10.1093/nar/gkp1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Signoretto E, Honisch S, Briglia M, Faggio C, Castagna M, Lang F. 2016. Nocodazole induced suicidal death of human erythrocytes. Cell Physiol Biochem 38:379–392. doi: 10.1159/000438638. [DOI] [PubMed] [Google Scholar]
- 39.Dalton WB, Nandan MO, Moore RT, Yang VW. 2007. Human cancer cells commonly acquire DNA damage during mitotic arrest. Cancer Res 67:11487–11492. doi: 10.1158/0008-5472.CAN-07-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orth JD, Loewer A, Lahav G, Mitchison TJ. 2012. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell 23:567–576. doi: 10.1091/mbc.e11-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawamura T, Kawatani M, Muroi M, Kondoh Y, Futamura Y, Aono H, Tanaka M, Honda K, Osada H. 2016. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci Rep 6:26521. doi: 10.1038/srep26521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, Saleh A, Gokturk C, Baranczewski P, Svensson R, Berntsson RP, Gustafsson R, Stromberg K, Sanjiv K, Jacques-Cordonnier MC, Desroses M, Gustavsson AL, Olofsson R, Johansson F, Homan EJ, Loseva O, Brautigam L, Johansson L, Hoglund A, Hagenkort A, Pham T, Altun M, Gaugaz FZ, Vikingsson S, Evers B, Henriksson M, Vallin KS, Wallner OA, Hammarstrom LG, Wiita E, Almlof I, Kalderen C, Axelsson H, Djureinovic T, Puigvert JC, Haggblad M, Jeppsson F, Martens U, Lundin C, Lundgren B, Granelli I, Jensen AJ, Artursson P, Nilsson JA, Stenmark P, Scobie M, Berglund UW, Helleday T. 2014. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 508:215–221. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- 43.Marazita MC, Ogara MF, Sonzogni SV, Marti M, Dusetti NJ, Pignataro OP, Canepa ET. 2012. CDK2 and PKA mediated-sequential phosphorylation is critical for p19INK4d function in the DNA damage response. PLoS One 7:e35638. doi: 10.1371/journal.pone.0035638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bensimon A, Aebersold R, Shiloh Y. 2011. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett 585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 45.Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. 2004. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol 6:138–145. doi: 10.1038/ncb1092. [DOI] [PubMed] [Google Scholar]
- 46.Kuhne C, Gardiol D, Guarnaccia C, Amenitsch H, Banks L. 2000. Differential regulation of human papillomavirus E6 by protein kinase A: conditional degradation of human discs large protein by oncogenic E6. Oncogene 19:5884–5891. doi: 10.1038/sj.onc.1203988. [DOI] [PubMed] [Google Scholar]
- 47.Gagou ME, Zuazua-Villar P, Meuth M. 2010. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 21:739–752. doi: 10.1091/mbc.e09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi S, Toledo LI, Fernandez-Capetillo O, Bakkenist CJ. 2011. CGK733 does not inhibit ATM or ATR kinase activity in H460 human lung cancer cells. DNA Repair 10:1000–1001. doi: 10.1016/j.dnarep.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Gao G, Chen L, Li J, Deng X, Zhao QS, Huang C. 2014. Hydrogen peroxide/ATR-Chk2 activation mediates p53 protein stabilization and anti-cancer activity of cheliensisin A in human cancer cells. Oncotarget 5:841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cossar LH, Schache AG, Risk JM, Sacco JJ, Jones NJ, Lord R. 2017. Modulating the DNA damage response to improve treatment response in cervical cancer. Clin Oncol (R Coll Radiol) 29:626–634. doi: 10.1016/j.clon.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Jelinic P, Levine DA. 2014. New insights into PARP inhibitors' effect on cell cycle and homology-directed DNA damage repair. Mol Cancer Ther 13:1645–1654. doi: 10.1158/1535-7163.MCT-13-0906-T. [DOI] [PubMed] [Google Scholar]
- 53.Booth L, Cruickshanks N, Ridder T, Dai Y, Grant S, Dent P. 2013. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol Ther 14:458–465. doi: 10.4161/cbt.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 55.Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L. 2011. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol 85:3120–3127. doi: 10.1128/JVI.02004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M. 2013. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol 87:951–964. doi: 10.1128/JVI.01943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterman JL, Shenk JL, Halazonetis TD. 1995. The dihedral symmetry of the p53 tetramerization domain mandates a conformational switch upon DNA binding. EMBO J 14:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. 1998. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet 19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 59.Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 2008. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res 36:5983–5991. doi: 10.1093/nar/gkn598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai CL, Shi J, Chen Y, Iqbal K, Liu F, Gong CX. 2013. Inhibition of protein synthesis alters protein degradation through activation of protein kinase B (AKT). J Biol Chem 288:23875–23883. doi: 10.1074/jbc.M112.445148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. 1995. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol 15:4930–4938. doi: 10.1128/MCB.15.9.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oksvold MP, Pedersen NM, Forfang L, Smeland EB. 2012. Effect of cycloheximide on epidermal growth factor receptor trafficking and signaling. FEBS Lett 586:3575–3581. doi: 10.1016/j.febslet.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 63.Kranjec C, Tomaic V, Massimi P, Nicolaides L, Doorbar J, Banks L. 2016. The high-risk HPV E6 target scribble (hScrib) is required for HPV E6 expression in cervical tumour-derived cell lines. Papillomavirus Res 2:70–77. doi: 10.1016/j.pvr.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grossman SR, Mora R, Laimins LA. 1989. Intracellular localization and DNA-binding properties of human papillomavirus type 18 E6 protein expressed with a baculovirus vector. J Virol 63:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pim D, Thomas M, Javier R, Gardiol D, Banks L. 2000. HPV E6 targeted degradation of the discs large protein: evidence for the involvement of a novel ubiquitin ligase. Oncogene 19:719–725. doi: 10.1038/sj.onc.1203374. [DOI] [PubMed] [Google Scholar]
- 66.Thomas M, Matlashewski G, Pim D, Banks L. 1996. Induction of apoptosis by p53 is independent of its oligomeric state and can be abolished by HPV-18 E6 through ubiquitin mediated degradation. Oncogene 13:265–273. [PubMed] [Google Scholar]