

Much remains unknown about how TREX1 influences HIV-1 replication: whether it targets full-length viral DNA versus partial intermediates, how intracellular TREX1 protein levels correlate with ISG induction, and whether TREX1 digestion of cytoplasmic DNA and subsequent cGAS pathway activation affects both initial and subsequent cycles of infection. To answer these questions, we experimentally varied the intracellular level of TREX1 and showed that this strongly determines the innate immunogenicity of HIV-1. In addition, several lines of evidence, including time-of-addition experiments with drugs that impair reverse transcription or capsid integrity, showed that the pathogen-associated molecular patterns sensed after viral entry contain DNA, are TREX1 and cGAS substrates, and are derived from incomplete reverse transcriptase (RT) products. In contrast, the experiments demonstrate that full-length integration-competent viral DNA is immune to TREX1. Treatment approaches that reduce TREX1 levels or facilitate release of DNA intermediates may advantageously combine enhanced innate immunity with antiviral effects.
KEYWORDS: HIV-1, PF74, TREX1, cGAS, capsid, innate immunity, reverse transcription
ABSTRACT
TREX1 has been reported to degrade cytosolic immune-stimulatory DNA, including viral DNA generated during HIV-1 infection; but the dynamic range of its capacity to suppress innate immune stimulation is unknown, and its full role in the viral life cycle remains unclear. A main purpose of our study was to determine how the intracellular level of TREX1 affects HIV-1 activation and avoidance of innate immunity. Using stable overexpression and CRISPR-mediated gene disruption, we engineered a range of TREX1 levels in human THP-1 monocytes. Increasing the level of TREX1 dramatically suppressed HIV-1 induction of interferon-stimulated genes (ISGs). Productive infection and integrated proviruses were equal or increased. Knocking out TREX1 impaired viral infectivity, increased early viral cDNA, and caused 10-fold or greater increases in HIV-1 ISG induction. Knockout of cyclic GMP-AMP synthase (cGAS) abrogated all ISG induction. Moreover, cGAS knockout produced no increase in single-cycle infection, establishing that HIV-1 DNA-triggered signaling is not rapid enough to impair the initial ISG-triggering infection cycle. Disruption of the HIV-1 capsid by PF74 also induced ISGs, and this was TREX1 level dependent, required reverse transcriptase catalysis, and was eliminated by cGAS gene knockout. Thus, the intracellular level of TREX1 pivotally modulates innate immune induction by HIV-1. Partial HIV-1 genomes are the TREX1 target and are sensed by cGAS. The nearly complete lack of innate immune induction despite equal or increased viral integration observed when the TREX1 protein level is experimentally elevated indicates that integration-competent genomes are shielded from cytosolic sensor-effectors during uncoating and transit to the nucleus.
IMPORTANCE Much remains unknown about how TREX1 influences HIV-1 replication: whether it targets full-length viral DNA versus partial intermediates, how intracellular TREX1 protein levels correlate with ISG induction, and whether TREX1 digestion of cytoplasmic DNA and subsequent cGAS pathway activation affects both initial and subsequent cycles of infection. To answer these questions, we experimentally varied the intracellular level of TREX1 and showed that this strongly determines the innate immunogenicity of HIV-1. In addition, several lines of evidence, including time-of-addition experiments with drugs that impair reverse transcription or capsid integrity, showed that the pathogen-associated molecular patterns sensed after viral entry contain DNA, are TREX1 and cGAS substrates, and are derived from incomplete reverse transcriptase (RT) products. In contrast, the experiments demonstrate that full-length integration-competent viral DNA is immune to TREX1. Treatment approaches that reduce TREX1 levels or facilitate release of DNA intermediates may advantageously combine enhanced innate immunity with antiviral effects.
INTRODUCTION
The innate immune system plays a central role in controlling viral infections (1). Recognition of viral pathogen-associated molecular patterns (PAMPs) by host pattern recognition receptors (PRRs) activates signaling pathways that lead to induction of type I interferons (IFNs) and IFN-stimulated genes (ISGs), which in turn restrict viruses by various mechanisms. Viruses have counterevolved mechanisms to prevent recognition by cellular PRRs or to inhibit subsequent ISG induction. Several innate immunity systems recognize distinctive features of viral nucleic acids, e.g., double-stranded RNA (dsRNA) or 5′ triphosphates. An incoming retrovirus particle sequentially contains a variety of potential nucleic acid PAMPs, including dsRNA, RNA:DNA hybrids, single-stranded DNA (ssDNA), and double-stranded DNA (dsDNA). While the mechanisms involved in innate sensing of HIV-1 remained unknown for many years, significant strides were made in the last half-decade, including identification of PRRs and PAMPs (2). Some main findings are that induction of type I interferon is reverse transcription dependent, that viral DNA is a major PAMP (3), and that cyclic GMP-AMP (cGAMP) synthase (cGAS) and IFI16 are sensors of HIV-1 DNA in different cell types (4, 5).
TREX1, a 314-amino-acid protein encoded by a single exon, is expressed in most tissues (6, 7). The protein has an exonuclease domain at its amino terminus and an endoplasmic reticulum (ER) localization domain at its carboxyl terminus. TREX1 digests both ssDNA and dsDNA, with preferential activity on mispaired termini (6, 8). The main function of TREX1 is presently considered to be negative regulation of innate immune responses directed at or triggered by DNA. Increased levels of cytosolic DNA are observed in TREX1-deficient mouse embryonic fibroblasts (MEFs) and are associated with increased type I interferon production (9). Proposed sources of endogenous cytosolic immune-stimulatory DNA (ISD) include endogenous retroelements (9) and DNA damage/repair and replication intermediates (10). In MEFs derived from TREX1-knockout (KO) mice, interferon induction is dependent on cGAS (11, 12), a recently discovered principal sensor of cytosolic DNA (13). Furthermore, Nakaya et al. (14) reported that the Aim2 like receptors (ALRs) Aim2 and Ifi205 participated in both positive and negative regulation of the IFN/cytokine response to mouse retroelement DNAs that accumulated in TREX-deficient cells. Human loss-of-function mutations in TREX1 can cause autoimmune and autoinflammatory disorders, including Aicardi-Goutieres syndrome (AGS), which has features that mimic congenital viral infection and systemic lupus erythematosus (SLE) (12, 15, 16). Mechanisms mediating TREX1-associated autoinflammatory states are not well understood. For example, while most AGS-associated TREX1 mutations are autosomal recessive and result in loss of exonuclease activity, most SLE-associated mutant TREX1 proteins are autosomal dominant and do not affect DNase activity (17, 18). In addition, TREX1 was also reported to regulate interferon-independent ISG induction and promote lysosomal biogenesis (19), and the membrane-associated C-terminal portion of the protein has been reported to play a nuclease-independent role in limiting the accumulation of glycans in the endoplasmic reticulum (20).
Within the incoming viral core, HIV-1 reverse transcriptase (RT) copies the single-stranded genomic RNA (gRNA) into dsDNA, within a capsid-enclosed macromolecular complex termed the reverse transcription complex (RTC). The process occurs in the cytoplasm and may initiate in virions preentry. As it proceeds, the viral gRNA is copied into a DNA:RNA hybrid molecule in which the templating gRNA strand is subjected to viral RNase H degradation and then into partially dsDNA segments prior to conversion to full-length dsDNA (21). Reverse transcription is inherently inefficient, such that a small percentage of particles that enter the cell and initiate reverse transcription complete it successfully. Thus, in the majority of infecting viral particles, reverse transcription aborts prematurely, producing incomplete cDNA intermediates.
It has been reported that HIV-1 benefits from the ability of TREX1 to degrade immune-stimulatory viral DNA, thereby minimizing innate sensing (3). These studies were carried out in MEFs derived from Trex1-knockout mice, in which cytosolic total HIV DNA was increased but integrated proviruses were reduced, suggesting that most HIV DNA that accumulates in Trex1-deficient cells does not contribute to productive infection. In this study also, reexpression of TREX1 in the TREX1−/− MEFs did not reduce total HIV DNA below the level observed in wild-type MEFs, suggesting that TREX1 may not access all HIV DNA products (3). TREX1 knockdown in CD4+ cells was effective in augmenting ISG responses to HIV-1 and blocking or delaying HIV-1 infection in cervicovaginal explant tissue and humanized mice, respectively (22). In previous studies TREX1 was also shown to be important for innate sensing of a gammaretrovirus (23, 24).
Much remains unknown about how the HIV-1 replication cycle is influenced by TREX1, about how reverse transcription products are metabolized by the enzyme, and about the roles of full-length viral DNA molecules versus partial cDNA intermediates in induction of type I interferon responses. How intracellular TREX1 levels correlate with ISG induction is not known except that experimentally knocking TREX1 down (or carrying out infection in TREX1−/− MEFs) enhances HIV-relevant ISG induction, and rare congenital gene loss in humans leads to severe autoimmune syndromes. While TREX1 has been described as the most abundant exonuclease in mammalian cells, there is little information on protein levels, and the dynamic range of the protein's capacity to inhibit innate immune stimulation is not known. For example, the effects of raising TREX1 levels have not been studied. In addition, analyses of viral DNA life cycle stages have so far been done in murine cells. In the present study, we determined the effects of stably increasing and decreasing TREX1 levels on innate sensing, HIV-1 DNA levels, and productive HIV-1 infection in human cells. Using these systems, we demonstrate that overexpressing TREX1 above its already substantially bioactive levels in THP-1 cells leads to dramatic suppression of innate immune induction by HIV-1 despite the presence of equal or greater infection and integrated proviruses. Conversely, major increases in induced ISGs and decreased HIV-1 infection were observed in cells depleted of TREX1. Analyses of viral DNA forms, in conjunction with capsid disruption experiments, provide evidence that the HIV-1 DNA targets of TREX1 exonuclease activity are not complete but, rather, incomplete reverse transcription products. Collectively, these data support a model in which the PAMPs detected by cellular sensors are partial, abortive HIV-1 DNA species, the levels of which are minimized by TREX1. The data suggest that levels of TREX1 in HIV-1 target cells may have significant effects on viral transmission and persistence. Moreover, we show for the first time that HIV-1 viral DNA-mediated signaling does not act rapidly enough to affect the initial cycle of infection as neither cGAS gene knockout nor combined TREX1 and cGAS knockout in THP-1 cells increases the level of single-cycle infection.
RESULTS
Generation of THP-1 cells with increased and decreased TREX1.
Human myelomonocytic THP-1 cells were employed. THP-1 cells express TREX1 and also possess intact major functional innate immunity sensor and effector systems; they have been used widely to characterize innate responses to HIV-1 and other primate lentiviruses (25, 26). We began by engineering THP-1 lines with variant intracellular TREX1 levels (Fig. 1). The THP-1-Lucia ISG line was used unless otherwise indicated because it contains a readily monitored secreted luciferase reporter under the transcriptional control of an interferon-stimulated response element (ISRE) promoter, which serves as a quantifiable surrogate for ISG induction. To overexpress TREX1, we transduced these cells with a lentiviral vector encoding a hemagglutinin (HA) epitope-tagged human protein. The resulting cells were designated THP-1HI. Overexpression was demonstrated by quantitative reverse transcription-PCR (qRT-PCR) and immunoblotting (Fig. 1A and B). Compared to the level in parental THP-1 cells, the level of TREX1 mRNA was elevated 21-fold in THP-1HI cells (Fig. 1A), and TREX1 overexpression was detected by blotting for HA (Fig. 1B) as well as for the endogenous protein with anti-TREX1 (Fig. 1F). CRISPR/Cas9-targeted guide RNAs (gRNAs) were used to generate stable TREX1 gene knockouts, which were designated THP-1KO cells. Single-cell clones were derived by limiting dilution 3 days after electroporation of CRISPR components. For determining monoallelic or biallelic gene disruption, clones were screened by PCR amplification of the targeted region, followed by amplicon restriction enzyme digestion (Fig. 1C to E). Two THP-1 cell clones with biallelic disruption in the single TREX1 coding exon were used for subsequent experiments (THP-1KO-11 and THP-1KO-18). Amplification and sequencing of genomic DNA showed that THP-1KO-11 has a 334-bp deletion and 6-bp insertion in one allele and a 406-bp deletion in the other allele (data not shown). THP-1KO-18 has a 13-bp deletion and 2-bp insertion in one allele and a 45-bp deletion in the other allele (data not shown). Of the two indels in THP-1KO-18 (one in each allele), one is in frame, but both delete the arginine 62 residue known to be essential for catalytic activity of TREX1 (27). TREX1 deficiency in both clones was confirmed by immunoblotting for endogenous TREX1 (Fig. 1F).
FIG 1.

THP-1HI and THP-1KO cell lines. TREX1 overexpression and CRISPR/Cas9-mediated targeting of human TREX1 were done in THP-1-Lucia ISG cells (InvivoGen), which contain a secreted luciferase reporter gene under the control of a composite promoter comprised of five IFN-stimulated response elements (ISREs) fused to an ISG54 minimal promoter. (A and B) Cells were transduced with a lentiviral vector with a U3 deletion and expressing HA epitope-tagged TREX1, generating THP-1HI cells. TREX1 mRNA levels were measured by qRT-PCR and normalized to the level of GAPDH. Immunoblotting was performed with anti-HA primary antibody. (C) THP-1 cells were subjected to CRISPR/Cas9-mediated knockout with guide RNAs targeting the gene's coding exon. The human TREX1 locus on chromosome 3 and spliced mRNA are shown. Binding sites for CRISPR-A and CRISPR-B are indicated by lightning bolt symbols, the PCR primer pair (1 and 2) used for screening genomic DNA for editing is indicated by horizontal arrows, and location of the DraIII restriction enzyme site used for screening for biallelic editing at CRISPR-A's cut site is indicated by a vertical arrowhead. (D) Genomic DNA PCR with primers 1 and 2. (E) Dra III digest of genomic DNA PCRs. (F) Immunoblotting of THP-1HI and THP-1KO cells with anti-TREX1 primary antibody is shown.
Infection of THP-1 cells with HIV-1 led to ISRE induction in a multiplicity of infection (MOI)-dependent manner (Fig. 2A). Azidothymidine (zidovudine; AZT) titration (Fig. 2B), as well as use of nevirapine and use of an RT catalytic mutant virus (D185E) (Fig. 2C) showed that the extent of ISRE induction correlated precisely with reverse transcription. By comparison, the integrase strand transfer inhibitor raltegravir reduced ISRE induction only about 2-fold (Fig. 2D). In contrast to these virally triggered effects, neither RT inhibitors nor raltegravir reduced the ISRE response to transfected DNA (Fig. 2E).
FIG 2.

HIV-1 induces innate immunity in a reverse transcription- and integration-dependent manner in THP-1 cells. THP-1 cells were infected with VSV-G pseudotyped luciferase or GFP reporter viruses (HIV-1luc and HIV-1GFP). Cells were treated with HIV reverse transcriptase inhibitor, azidothymidine (AZT) or nevirapine (Nev), or the integrase strand transfer inhibitor raltegravir (Ral) for 30 min prior to infection. Cell culture supernatants were assayed for ISRE-promoted luciferase activity at 48 hpi. Note that the virally encoded firefly luciferase and the ISRE-promoted coelenterate luciferase are separately measured with substrates that have no cross-reactivity (luciferin and coelenterazine, respectively). (A) ISRE relative light unit (RLU) activity in THP-1 cells infected with HIV-1GFP at the indicated MOIs. (B) Viral infection and ISRE activities in THP-1 cells treated with AZT (dose range, 0 to 10 μM) and infected with HIV-1GFP (left panel) or HIV-1 LaiLuc (right panel). (C) Cells were treated with 5 μM nevirapine and infected with wild-type or D185E HIV-1luc as indicated. (D) ISRE activity in THP-1 cells infected with HIV-1 in the presence of 5 μM Ral. (E) THP-1 cells treated with the indicated drugs (5 μM) were transfected with double-stranded immune-stimulatory DNA (ISD), and ISRE activity was measured at 24 h posttransfection. Data in all panels are from one representative experiment of three experiments, with error bars showing standard deviations of triplicate measurements.
Cellular TREX1 levels determine innate immune induction by HIV-1.
A main purpose of the present study was to determine how intracellular TREX1 levels affect the viral life cycle. In particular, the effects of increasing TREX1 expression levels above endogenous levels on innate immune induction by HIV-1 have not been studied. We addressed this question by determining ISRE induction and representative ISG mRNA induction in THP-1HI cells. We found that the ISRE induction we had observed in experiments such as the experiment shown in Fig. 2A was strongly suppressed by stable TREX1 overexpression (Fig. 3A). This effect was quantitatively impressive. At higher viral inputs, 20- to 30-fold suppressions of ISRE induction were repeatedly observed in THP-1HI cells compared with levels in THP-1 cells. Some saturation of TREX1 enzymatic activity was suggested at higher viral inputs. When we examined specific interferons and ISGs, we observed that IFN-β mRNA induction was drastically lower in THP-1HI cells than in parental THP-1 cells 24 h after infection (Fig. 3B) and that induction of a representative ISG mRNA (IFIT1) was similarly much lower (Fig. 3C).
FIG 3.

Cellular levels of TREX1 determine interferon-stimulated gene (ISG) induction by HIV-1. THP-1 and THP-1HI cells were infected with HIV-1luc or HIV-1GFP at a range of MOIs and assayed for ISG induction by ISRE activity or qRT-PCR. (A) ISRE activity in THP-1 and THP-1HI cells infected with HIV-1luc at 48 hpi. (B and C) Interferon beta mRNA and IFIT1 mRNA levels were quantified by qRT-PCR and normalized to the level of GAPDH at 24 hpi in the indicated cells. (B) Interferon beta mRNA induction (MOI of 2). (C) IFIT1 mRNA induction (MOI of 2). (D) THP-1 and THP-1HI cells were transfected with indicated doses (in nanograms) of circular plasmid DNA, and cells were assayed for ISRE luciferase activity at 24 h posttransfection. (E) THP-1 cells and THP-1HI cells were transduced with the indicated amounts of catalytic mutant D200H TREX1-encoding lentiviral vector or an empty vector and selected with puromycin. Stably transduced cells were assayed for ISRE luciferase activity. (F) cGAS mRNA levels were measured by qRT-PCR and normalized to the level of GAPDH in THP-1 cells and THP-1HI cells. The data presented in panel A are representative of over 10 experiments, with error bars representing standard deviations of triplicate measurements. Data presented in all other panels in this figure are from one representative experiment of three experiments, with error bars representing standard deviations of triplicate measurements. ns, not significant.
It remained possible that TREX1-bound HIV-1 DNA might not be sensed efficiently by cytosolic sensor proteins or that DNA sensing in general might for some reason be defective in these cells. We therefore assessed the abilities of THP-1HI cells to sense cytosolic DNA by two experimental approaches. First, THP-1 and THP-1HI cells were transfected with ISD. We used circular plasmid DNA, which is not a target for TREX1, to prevent confounding by TREX1 degradation of linear nonviral DNA. THP-1HI cells exhibited ISRE induction to levels similar to those observed in THP-1 cells (Fig. 3D). In the second approach, THP-1HI cells were transduced with the AGS-associated dominant negative TREX1 mutant D200H. Most cases of AGS are autosomal recessive, but the D200H mutation causes autosomal-dominant AGS (28). Purified TREX1 D200H protein lacks exonuclease activity, and coincubation of the mutant enzyme and wild-type (wt) TREX1 inhibits degradation of dsDNA by wt TREX1 (28). Also, this dominant negative inhibition depends on preserved DNA binding ability. Here, THP-1 cells transduced with D200H TREX1 exhibited reduced TREX1 activity as evidenced by marked ISRE induction. D200H TREX1 expression in THP-1HI cells produced similar results (Fig. 3E). This induction was specific to D200H TREX1 since the empty vector did not increase ISRE levels. Both of the above approaches demonstrated that TREX1-overexpressing cells are capable of sensing cytosolic DNA, whether of extrinsic (Fig. 3D) or endogenous origin (Fig. 3E), and that the cytosolic DNA sensing/signaling machinery in THP-1HI cells is intact.
We also considered the possibility that cells with stable overexpression of TREX1 may acquire altered levels of cGAS. However, this was not the case. Quantitative RT-PCR analyses demonstrated that cGAS mRNA levels in THP-1HI cells were not different than those in THP-1 cells (Fig. 3F).
We then studied the effect of reducing THP-1 cell TREX1 levels on ISRE induction by HIV-1. In polar contrast to the results described above with THP-1HI cells, infection of THP-1KO-18 cells with HIV-1 expressing green fluorescent protein (HIV-1GFP) triggered increased ISRE induction compared with that in THP-1 cells (compare Fig. 4A and 3A, noting the different y axis scales in the two graphs). At an MOI of 1, a 6-fold increase in ISRE activity over that in uninfected cells was observed in THP-1 cells whereas 28-fold and 18-fold induction over levels in uninfected KO cells was observed in THP-1KO-18 (Fig. 4A) and THP-1KO-11 cells (Fig. 4D), respectively. Similarly, IFN-β mRNA induction (Fig. 4B and E) and induction of IFIT1 mRNA (Fig. 4C and F) were increased in the knockout cells. Of note, the ISRE-promoted luciferase activities in the uninfected THP-1KO cell clones were 4 to 6 times higher than those in uninfected THP-1 cells, a finding that is consistent with a known role for TREX1 in metabolism of cell-intrinsic DNA ligands (13).
FIG 4.

TREX1 depletion modulates the antiviral state of cells at baseline and ISG induction upon HIV-1 infection. THP-1 and THP-1KO cells were infected with HIV-1luc or HIV-1GFP at a range of MOIs and assayed for ISG induction by ISRE activity measurements at 48 hpi. In panels A and D, graphs to the upper right show magnifications of the low-MOI regions indicated by boxed areas in the main graphs. (A) ISRE activity in THP-1 cells and THP-1KO-18 cells, at baseline and 48 hpi are shown. (B and C) Interferon beta mRNA and IFIT1 mRNA levels were quantified by qRT-PCR and normalized to the level of GAPDH, at baseline and at 24 hpi in the indicated cells. (B) Interferon beta mRNA levels shown as fold change over levels in respective uninfected cells at 24 hpi (MOI of 0.1). *, P ≤ 0.05. (C) IFIT1 mRNA levels are shown as fold change over levels in respective uninfected cells at 24 hpi (MOI of 0.1). IFIT1 mRNA levels in uninfected THP-1KO-18 cells are expressed as fold change over levels in uninfected wild-type (wt) THP-1 cells. ***, P ≤ 0.001. (D) ISRE activity in THP-1 cells and THP-1KO-11 cells, at baseline and 48 hpi are shown. (E) Interferon beta mRNA levels shown as fold change over levels of respective uninfected cells at 24 hpi (MOI of 0.1). *, P ≤ 0.05. (F) IFIT1 mRNA levels are shown as fold change over levels of respective uninfected cells at 24 hpi (MOI of 0.1). IFIT1 mRNA in THP-1KO-11 cells at baseline are expressed as fold change over levels in uninfected wt THP-1 cells. ***, P ≤ 0.001. (G) ISRE activity in THP-1 cells and monoallelic TREX1 KO-3 and TREX1 KO-39 clones at baseline and 48 hpi are shown. Data presented in all panels in this figure are from one representative experiment of three experiments, with error bars representing standard deviations of triplicate measurements.
These findings were recapitulated in THP-1 cells stably expressing the D200H TREX1 mutant (THP-1 D200H cells). These cells can serve as a surrogate for a TREX1 deficiency state. Similarly to the observations in TREX1 knockout cells, THP-1 D200H cells exhibited elevations in baseline ISRE activity, with additional ISRE induction of 3- to 5-fold following infection with HIV (data not shown).
TREX1 haploinsufficiency.
Cells with monoallelic knockouts of TREX1 did not demonstrate increased ISRE activity in response to HIV-1 infection. This was observed in two cell clones, one with a 406-bp deletion in one allele, designated THPKO-39, and a second clone with a 1-bp insertion in one allele, designated THP-1KO-3 (Fig. 4G). This observation suggests that reduction of TREX1 below a threshold level may be needed to significantly stimulate innate immunity by HIV-1.
Correlation of HIV-1 infection and ISG induction in THP-1KO, THP-1, and THP-1HI cells.
A main unanswered question in the HIV postentry sensing field is whether full-length or partial reverse transcription products trigger innate immune system DNA sensors. We therefore quantified and correlated HIV-1 infection and viral DNA forms with ISG induction in THP-1 and THP-1HI cells (Fig. 5A and B). In these experiments, infection of THP-1HI cells with HIV-1 reporter virus expressing luciferase (HIV-1luc) or HIV-1GFP consistently resulted in equivalent or greater luciferase activities or GFP expression than observed in THP-1 cells across a range of MOIs (Fig. 5A and B, bar graphs). However, ISRE induction was drastically lower (Fig. 5A and B, line graphs). Next, we quantified two long terminal repeat (2-LTR) circles and proviral integrants, which represent end products of unintegrated and integrated complete DNA genomes after nuclear entry. The primers used for 2-LTR circles were those described by De Iaco et al. to amplify circles with perfect junctions to restrict amplification to fully double-stranded genomes and exclude circular species that may result from abortive reverse transcription events (29). These experiments confirmed the presence of late viral DNA forms in THP-1HI cells at levels equal to or greater than those in THP-1 cells (Fig. 5C and D). Therefore, elevating TREX1 protein levels does not decrease bona fide integration-competent genomes.
FIG 5.

Analysis of effects of increased TREX1 expression on interferon-stimulated gene induction and infection demonstrates shielding of complete genomes from cytoplasmic sensor effectors. The THP-1 cells and THP-1HI cells were infected with luciferase-encoding (HIV-1 LAI) or GFP-encoding (HIV-1 NL4-3) single-cycle reporter viruses at a range of MOIs. Samples were assayed for ISRE and viral luciferase activities or analyzed for percent positive GFP cells by fluorescence-activated cell sorting analysis at 48 hpi as indicated. (A) ISRE and viral luciferase activities in THP-1 and THP-1HI cells. (B) ISRE activity and percent positive GFP THP-1 and THP-1HI cells at 48 hpi. *, P ≤ 0.05; ***, P ≤0.001 (for ISRE activity in panels A and B). (C) THP-1 and THP-1HI cells were infected with HIV-1GFP (MOI of 1), and 2-LTR circles were quantified by quantitative PCR and normalized to the level of GAPDH. Data are presented as mean and standard deviation of technical triplicates from one of three independent experiments. (D) THP-1 and THP-1HI cells were infected with HIV-1GFP (MOI of 1). Proviral integrants at 24 hpi were quantified by Alu-PCR and normalized to GAPDH. Data are presented as means and standard deviations of technical triplicates from one of three independent experiments. (E and F) THP-1 and THP-1HI cells were infected with HIV-1GFP at an MOI of 0.5, and U3 and gag DNA were quantified by quantitative PCR and normalized to GAPDH. (G and H) THP-1 and THP-1HI cells were infected with HIV-1GFP at an MOI of 1, and U3 and gag DNA were quantified by quantitative PCR and normalized to GAPDH. Data are presented as means and standard deviations of technical triplicates from two independent experiments. **, P ≤ 0.01 (panels E and G).
The above results suggested that HIV DNA is degraded more efficiently in THP-1HI cells. To test this directly, we used qPCR to measure the time courses of viral DNA accumulation in THP-1 and THP-1HI cells (Fig. 5E to H). These results showed lower levels of U3 and gag DNA in THP-1HI cells at 8 to 10 h after infection. This was the case at an MOI of 0.5 (Fig. 5E and F) and MOI of 1.0 (Fig. 5G and H). In contrast, when we measured the levels of the earliest HIV-1 cDNA species—the strong-stop DNA–we did not observe any difference at any time point (data not shown). The last result suggests that the earliest products of reverse transcription are not accessible and metabolized by TREX1.
To confirm the correlations between TREX1 levels and ISG induction and HIV infection, we then infected TREX1 knockout cells with HIV-1luc and HIV-1GFP. The knockout cells demonstrated significant reduction in viral infection levels while demonstrating significant increases in ISRE induction, an effect opposite to the results shown in Fig. 5A and B. As shown in Fig. 6A, infectivity in THP-1KO-18 cells at an MOI of 0.5 was 4-fold lower than that in THP-1 cells, with a 13-fold induction in ISRE activity above that observed in THP-1 cells. Infection with HIV-1luc reproduced these effects: viral luciferase activity in THP-1KO-18 cells was 14% of that in THP-1 cells (MOI of 0.5) while associated with an 8-fold increase in ISRE luciferase activity over that in THP-1 cells (Fig. 6B). The same effects were observed in THP-1KO-11 cells (Fig. 6C and D). Measurements of gag viral DNA showed a 3-fold increase in THP-1KO-18 cells, indicating accumulation of reverse transcription products in cells deficient in TREX1 (Fig. 6E), but 2-LTR circles and proviral integrants were 2-fold reduced in THP-1KO-18 cells (Fig. 6F and G).
FIG 6.

Analysis of effects of TREX1 deficiency on interferon-stimulated gene induction and infection implicates partial HIV DNA genomes as the viral pathogen-associated molecular pattern. The THP-1 cells and THP-1KO cells were infected with luciferase- or GFP-encoding single-cycle viruses at a range of MOIs. Samples were assayed for ISRE and viral luciferase activities or analyzed for percent positive GFP cells by fluorescence-activated cell sorting analysis at 48 hpi. (A) ISRE activity and percent GFP-positive THP-1 and THP-1KO-18 cells. (B) ISRE and viral luciferase activities in THP-1 and THP-1KO-18 cells. (C) ISRE activity and percent GFP-positive THP-1 and THP-1KO-11 cells. (D) ISRE and viral luciferase activities in THP-1 and THP-1KO-11 cells. (E) THP-1 and THP-1KO-18 cells were infected with HIV-1GFP (MOI of 1). Viral DNA (gag) at 48 hpi was quantified by quantitative PCR and normalized to GAPDH. Data shown are means and standard deviations of triplicate measurements. (F) THP-1 and THP-1KO-18 cells were infected with HIV-1GFP (MOI of 1), and 2-LTR circles were quantified by quantitative PCR and normalized to the level of GAPDH. Data are presented as means and standard deviations of technical triplicates from one of three independent experiments. ns, not significant. (G) THP-1 and THP-1KO-18 cells were infected with HIV-1GFP (MOI of 1). Proviral integrants at 24 hpi were quantified by Alu-PCR and normalized to the level of GAPDH. Data are presented as means and standard deviations of technical triplicates from one of three independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Taken together, the data show that the intracellular amount of TREX1 profoundly influences innate immune induction by HIV-1 infection across a wide dynamic range of protein levels. The strikingly deficient ISG induction in TREX1-overexpressing cells that are otherwise competent for cytosolic DNA sensing, and which have equal to greater levels of infection and proviral integrants, indicates that complete integration-competent genomes are not the viral nucleic acids targeted by cellular sensors. Rather, the results imply that the targets of TREX1 exonuclease activity are partial HIV-1 genomes that do not contribute to productive infection.
Pharmacological destabilization of cytosolic viral cores prior to completion of reverse transcription exposes viral PAMPs that trigger innate immunity.
We investigated whether partially reverse transcribed viral cDNAs are the active trigger for ISG production with a second experimental approach. We hypothesized that if incomplete reverse transcription intermediates are the key PAMPs, exposing them to cytosolic sensors prior to completion of reverse transcription would trigger an innate immune response. We first determined the time course of completed reverse transcription in THP-1 cells with a nevirapine time-of-addition assay (30). Nevirapine was added at sequential time points after infection with HIV-1 Lailuc, cells were harvested 2 days later, and virally encoded luciferase expression was measured, which is indicative of virions that have completed reverse transcription. As shown in Fig. 7A, no luciferase activity was detected in THP-1 cells if reverse transcription was interrupted with nevirapine at time points up to 6 h postinfection (hpi), indicating that no particles had completed reverse transcription by this time. By 11 h, some particles had completed reverse transcription, as indicated by the appearance of resistance to inhibition by nevirapine.
FIG 7.

Pharmacological disruption of cytosolic viral cores prior to completion of reverse transcription exposes reverse transcription-dependent viral PAMPs that trigger innate immunity. PF74 treatment of HIV-infected THP-1 cells at time points prior to completion of reverse transcription inhibits productive infection but not ISRE induction. (A and B) THP-1 cells were infected with HIV-1luc (MOI of 1), and nevirapine (5 μM) or PF74 (10 μM) was added at the indicated times postinfection. Viral luciferase activities were assayed for all samples at 48 hpi. ISRE luciferase activity was measured at the same time as viral luciferase. This very close correlation with reverse transcription was observed in more than eight experiments. (C and D) THP-1 cells were infected with HIV-1GFP (MOI of 1) and nevirapine (5 μM) or PF74 (10 μM) was added at the time of infection (t0). Percent GFP-positive cells was measured at 48 hpi. ISRE activity was measured at 48 hpi. ns, not significant. (E and F) PAMPs generated in cells treated with PF74 require reverse transcription of viral RNA. Viral luciferase activity and ISRE luciferase activity, as indicated, were determined at 48 hpi in THP-1 cells treated with a dimethyl sulfoxide (DMSO) control, nevirapine (5 μM), PF74 (10 μM), or both drugs together, with or without infection with HIV-1luc or empty particles (EP). ns, not significant. (G and H) THP-1 cells were infected with HIV-1GFP (MOI of 1), and PF74 (10 μM or 1 μM) or dimethyl sulfoxide was added at the time of infection (t0). Percent positive GFP cells was measured at 48 h pi (***, P < 0.001). ISRE activity was measured at 48 hpi. Data presented in all panels in this figure are from one representative experiment of three experiments, with error bars representing standard deviations of triplicate measurements.
We then employed PF74, a small molecule that binds to the HIV-1 capsid and mediates its rapid disassembly (31, 32). In order to expose reverse transcription intermediates at intervals prior to completed reverse transcription. The PF74-induced block to infection had a virtually identical time course as the nevirapine block (Fig. 7A). The drug produced nearly complete inhibition of infection when added up to 6 h after infection, with viral luciferase activity at 6% of the no-drug control. Drug addition at 11 h resulted in viral luciferase activity of 24% of the no-drug control.
However, nevirapine and PF74 effects on ISRE induction were markedly different. PF74 triggered ISRE induction when added within the initial 4-h window. Figure 7A and B show a PF74 time-of-addition experiment in which reporter virus (HIV-1luc) infection and ISRE activity over that of the no-virus control were tracked simultaneously in THP-1 cells, with both measurements made at 48 h after infection. Note that at the earliest time points of addition (0 and 3 h after infection), PF74 resulted in 6.3-fold and 7.5-fold increases in ISRE activity, respectively, despite significant inhibition of infection. ISRE induction above the level in uninfected cells was observed even when PF74 was added simultaneously with virus (time 0, or t0). In contrast to PF74, in the nevirapine time-of-addition experiment, there was no significant increase in ISRE induction when drug was added during the first 3 h (Fig. 7B). These effects were reproduced with experiments performed with HIV-1GFP infections. PF74 addition at t0 inhibited infectivity by 9-fold (Fig. 7C), while resulting in ISRE induction of 5-fold above that uninfected cells (Fig. 7D).
The observed induction of ISRE activity in the presence of 10 μM PF74 added at t0 were reverse transcription dependent. Experiments with the D185E catalytic reverse transcriptase mutant and empty (genome-less) particles did not lead to measurable infection or ISG induction (Fig. 7E and F). The possibility that the reverse transcriptase templates on a nonspecific cellular RNA or RNAs were excluded by PF74 experiments with vesicular stomatitis virus G protein (VSV-G) pseudotyped empty (genome-less) particles (Fig. 7F). PF74 has also been reported to inhibit HIV infection at lower concentrations (∼1 μM) in HeLa cells (32). Addition of 1 μM PF74 at the time of infection did not block HIV-1 in our experiments (Fig. 7G) or affect ISRE activity (Fig. 7H). Both cyclophilin A and CPSF6 (cleavage and polyadenylation specificity factor subunit 6) have been shown to modulate the antiviral effects of PF74. We speculate that these dose effects may be cell type specific and cofactor expression level dependent.
Numerous laboratories have demonstrated that ≥10 μM PF74 concentrations inhibit reverse transcription (31, 32). In light of our results that demonstrate induction of ISRE by reverse transcription-dependent PAMPs that accumulate in the presence of 10 μM PF74, we next determined and compared the effects of 10 μM PF74 and nevirapine addition at t0 on levels of reverse transcription products generated at different times after infection with HIV-1.
Consistent with previous reports, 10 μM PF74 inhibited reverse transcription, but the inhibition was less than that caused by nevirapine. The accumulation of gag DNA at 24 hpi was reduced to 37% of the control level by PF74 in contrast to 3.6% of the control level by nevirapine at an MOI of 0.5 (Fig. 8A). This pattern was also observed with early DNA levels at 24 hpi (Fig. 8B). Interestingly, 10 μM PF74 treatment at t0 resulted in no inhibition of gag DNA accumulation at 6 hpi (Fig. 8C). Again, PF74 addition at t0 resulted in no inhibition of early viral DNA accumulation at 6 hpi (Fig. 8D), whereas 2- to 4-fold inhibitions were observed at this time point with nevirapine treatment at t0.
FIG 8.
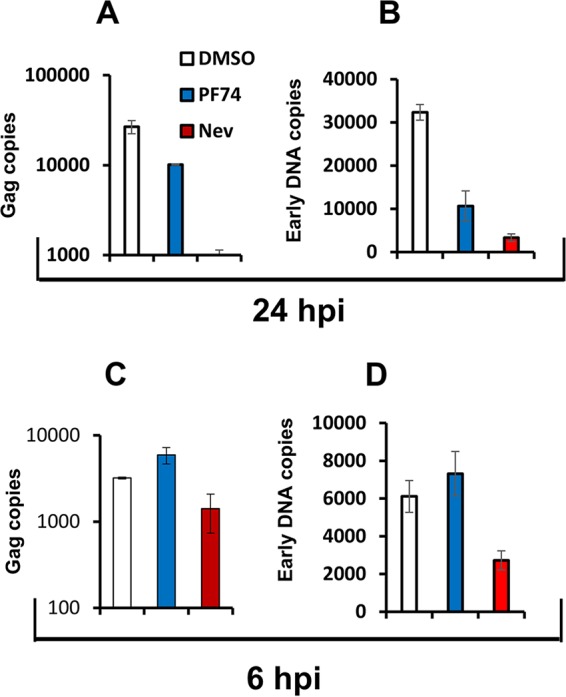
Inhibition of reverse transcription by 10 μM PF74 is delayed relative to inhibition by nevirapine. THP-1 cells were infected with HIV-1GFP (MOI of 0.5) and treated with dimethyl sulfoxide (DMSO), PF74 (10 μM), or nevirapine (5 μM) added at the time of infection. DNA extracted at 6 and 24 hpi was analyzed for first strand transfer (U3) and gag reverse transcription products. Data shown are means and standard deviations of technical triplicates from one of three representative experiments. (A and C) Copies of gag DNA quantified at 24 and 6 hpi. (B and D) Copy numbers of early reverse transcription products (U3) quantified 24 and 6 hpi.
These results indicate that PF74 inhibits reverse transcription at a later stage than nevirapine. This difference likely contributes to the differential effects of the two drugs on ISRE induction. PF74-induced destabilization of viral capsid leads to a later block in reverse transcription that is permissive of accumulation of viral DNA, in contrast to the effect of direct RT inhibition by nevirapine.
Another potential explanation could be differences in how the two treatments expose core-contained genomes to the cytosol. The Fig. 7 data suggest that PF74 may induce uncoating and release of viral DNA PAMPs from most cores in a given cell more or less simultaneously (synchronized uncoating) as opposed to a more gradual (asynchronous) release from cores during the course of natural uncoating. Asynchronous release may allow TREX1 to more efficiently digest viral DNA, as opposed to sudden DNA elevations that may saturate the enzyme. This concept is supported by comparing the PF74 and nevirapine time courses on ISG induction. Both treatments inhibit infection by inhibiting reverse transcription. However, inhibition of reverse transcriptase activity increases stability of the HIV-1 core (33) and delays uncoating (21), so that cytosolic exposure of reverse transcription intermediates may be delayed and asynchronous when nevirapine is added at 2 and 4 h after infection.
cGAS knockout led to no increase in single-cycle infections, demonstrating that HIV-1 DNA-triggered signaling is not rapid enough to impair completion of the initial ISG-triggering infection cycle.
We used the CRISPR/Cas9 system to generate cGAS gene (MB21D1)-knockout THP-1 cells (Fig. 9A and details in the legend). As show in Fig. 9B, definitive knockout was achieved. Gene-inactivating deletions in the MB21D1 locus were confirmed by sequencing (data not shown). Expression of cGAS protein (Fig. 9C) and mRNA (Fig. 9D) was eliminated. The cGAS−/− THP-1 cells were transfected with dsDNA and poly(I·C). The knockout abrogated ISG induction by dsDNA (Fig. 9E), whereas sensing of poly(I·C) was equivalent in cGAS+/+ and cGAS−/− cells (Fig. 9F), indicating that RIG-I-like receptor signaling pathways were intact in the cGAS−/− THP-1 cells. The cGAS knockout THP-1 cells were completely impaired for sensing of reverse-transcribed HIV-1 DNA (Fig. 9G). Treatments with cGAMP (Fig. 9H) and IFN-α (Fig. 9I) indicated that STING (stimulator of interferon genes)-mediated and type I IFN receptor (IFNAR)-mediated signaling was intact in cGAS KO cells.
FIG 9.

Generation of cGAS gene knockout THP-1 cells. (A) A pair of guide RNAs was designed to target sites 558 bp apart in the first exon of the MB21D1 locus on chromosome 6, downstream of the initiator ATG codon. Single-cell knockout clones were derived by limiting dilution 3 days after electroporation of CRISPR components. The clones were screened for genomic DNA editing by PCR amplifications spanning the cut sites. Binding sites of the CRISPR guide RNAs used (A and B) are shown by lightning bolt symbols. Numbered horizontal arrows show PCR primers 1 to 4 that span the cut sites and were used in various combinations to identify gene editing. (B) PCR on genomic DNA. Sequencing of genomic DNA derived from this single cell clone identified the gene edits to be a 568-bp deletion in one allele and a 557-bp inversion with a 1-bp deletion in the other allele. Primers 1 and 4 yield a 1,145-bp product in parental THP-1 cells. In the KO clone, there was a 587-bp PCR product, indicating a fully deleted allele. A second band was indistinguishable on the gel from the 1,145-bp product. Sequencing of these PCR products confirmed the deletion between sites A and B in one allele and revealed that the larger product represents an inversion as well as a 1-bp deletion in the other allele. (C) Immunoblot of THP-1 and cGAS KO THP-1 cells with anti-cGAS antibody (bsm-51379M MB21D1 [7E3]; Biomass Antibodies). (D) cGAS mRNA was quantified by qRT-PCR and normalized to the level of GAPDH. (E and F) cGAS KO THP-1 cells were transfected with dsDNA and poly(I·C), and ISRE activity was measured 24 h later. (G) ISRE activity at 48 hpi in THP-1 cells and cGAS KO THP-1 cells infected with increasing MOIs of HIV-1 (LAI) luciferase reporter virus. (H and I) cGAS KO THP-1 cells were treated with cGAMP and IFN-α, and ISRE activity was measured 24 h later.
To investigate whether incoming viral particles could induce ISGs rapidly enough to restrict the first round of infection, we infected THP-1 cells and cGAS KO THP-1 cells with HIV-1 Lailuc (Fig. 10A) and HIV-1GFP (Fig. 10C) reporter viruses at a range of MOIs, including viral inputs that induced ISRE in parental cells. However, cGAS knockout produced no increase in single-cycle infection (Fig. 10B and D), establishing that at normal endogenous levels of TREX1, cGAS-mediated signaling pathways are not rapid enough to impair the initial ISG-triggering infection cycle. To additionally test this finding and to study whether TREX1 deficiency might trigger cGAS-independent ISGs that may have a more rapid time course of induction, CRISPR/Cas9-mediated TREX1 gene knockouts were carried out in cGAS−/− cells. Two clones with sequence-confirmed deletions, designated double-knockout (DKO) clone 15 and clone 26, were used for these studies. Again, no decrease in infection in DKO cells was observed. Interestingly, cells with both TREX1 and cGAS knocked out did not demonstrate elevated ISRE activity at the baseline uninfected state or an enhanced ISRE induction post-HIV-1 infection, and this was associated with complete rescue of infectivity to wt levels. This confirmed that ISGs triggered in TREX1 KO are cGAS dependent in cells of monocyte-macrophage lineage.
FIG 10.

HIV-1 DNA-triggered signaling is not rapid enough to impair the initial ISG-triggering infection cycle. The THP-1 cells, THP-1 cGAS KO, and THP-1 DKO cells were infected with luciferase- or GFP-encoding single-cycle viruses at a range of MOIs. Samples were assayed for ISRE and viral luciferase activities or analyzed for percent positive GFP by fluorescence-activated cell sorting analysis at 48 hpi. (A and B) ISRE activity and viral luciferase activity were determined at 48 hpi in THP-1, THP-1 cGAS KO, and THP-1 DKO clone 15, and THP-1 DKO clone 26 cells infected with HIV (LAI) luciferase reporter virus. (C and D) ISRE activity and percent positive GFP cells were determined at 48 hpi in THP-1, THP-1 cGAS KO, THP-1 DKO clone 15, and THP-1 DKO clone 26, and THP-1 TREX1KO-11 cells infected with HIV-1GFP reporter virus. The results represent means and standard deviations of biological triplicates from two independent experiments.
Viral PAMPs exposed by PF74 treatment are sensed by cGAS.
Finally, we examined whether viral PAMPs sensed in PF74-treated cells are sensed by cGAS. As observed above, infection levels were equivalent in the +/+ and −/− cells, and PF74 treatment reduced infection of both (Fig. 11A). However, HIV-1 ISRE induction was abrogated in cGAS KO cells (Fig. 11B). Collectively, these data show that the viral PAMPs sensed in PF74-treated cells are partially reverse transcribed viral DNA genomes that require a catalytically active reverse transcriptase and are sensed by the cGAS-STING pathway.
FIG 11.
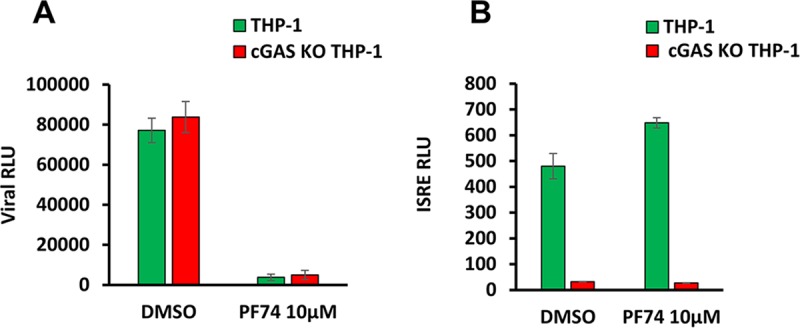
Viral PAMPs exposed by PF74 treatment are sensed by cGAS. Viral luciferase activity (A) and ISRE activity (B) was determined at 48 hpi in THP-1 cells and cGAS KO THP-1 cells treated with dimethyl sulfoxide (DMSO) or 10 μM PF74. The results represent means and standard deviations of technical triplicates from one of three independent experiments.
DISCUSSION
In this study, we add to the evidence that TREX1 is a pivotal determinant of HIV-1's innate immunogenicity. We show that the intracellular level of this protein profoundly affects HIV-1 early events over a large dynamic expression range. Incomplete products of reverse transcription are the PAMPs that are sensed by cells and that trigger ISG induction during the early events of the HIV-1 life cycle. The results support the concept that manipulation of cellular TREX1 levels could represent a means of enhancing innate immune detection and control of the virus.
In one approach, we experimentally increased and decreased TREX1 levels in an informative cell model with intact nucleic acid sensing and ISG signaling repertoires, the THP-1 human monocytic cell line. Stably elevating TREX1 levels suppressed ISG induction while simultaneously supporting equal to increased levels of HIV-1 infectivity and integrated proviruses. In contrast, stably reducing TREX1 levels caused log-fold increases in ISG induction while significantly reducing viral infectivity. The strikingly deficient innate immune induction in THP-1HI cells, despite equal or higher integrated provirus numbers, provides evidence that the relevant HIV-1 DNA PAMPs are not complete, integration-competent genomes. THP-1HI cells had undiminished capacity to detect cytosolic DNA, as demonstrated by exogenous DNA transfection experiments. The experiments indicate that full-length genomes are not targets of TREX1 exonuclease activity. The targets are partial cDNAs that are generated by reverse transcription and are of sufficient length to be sensed.
In another approach, pharmacological disruption of the capsid at early postentry time points exposed reverse transcription products that stimulated sensors that led to ISG induction. To our knowledge, this is the first study to experimentally dissect the roles of partial reverse-transcribed and full-length integration-competent DNA genomes in innate sensing of HIV-1. Collectively, these studies demonstrate that complete genomes are not required for innate sensing of this virus and that partial DNA genomes are sufficient.
Lack of degradation of full-length 3′-processed genomes by TREX1, which are otherwise absolutely ideal target substrates for this exonuclease, suggests that full-length integration-competent DNA remains shielded from cytosolic antiviral effectors during transit to the nucleus. Further studies to specifically examine the role of DNA binding by integrase may further our understanding of TREX1 interactions with viral DNA. Although we previously observed that dominantly interfering GFP fusions to the LEDGF/p75 integrase binding domain (IBD) retain their antiviral activity when confined to the cytoplasm (34), which suggests that integrase within the preintegration complex (PIC) is accessible prior to nuclear entry, the present data suggest that complete genomes are neither sensed nor degraded. It may be that the ends of the fully double-stranded genomes are protected from TREX1 exonucleolytic activity by being bound or sterically shielded by proteins contained in the preintegration complex.
The viral capsid is an important target for small-molecule development because of its central role in multiple aspects of the viral life cycle. Our results suggest that small molecules that facilitate capsid disassembly may enhance innate immunity to HIV-1. A key finding is the differential effect on ISRE induction by nevirapine and PF74. While both drugs inhibited productive infection effectively, PF74 treatment at the time of infection or at early postentry time points enabled ISRE induction by incomplete DNA transcripts whereas similarly timed exposure to nevirapine did not. A delayed time course of inhibition of reverse transcription by 10 μM PF74 may be permissive of generation of viral DNA PAMPs to levels above the cytosolic threshold levels required for sending. Differences in how DNA intermediate PAMPs are exposed to the cellular milieu may be involved, and the dynamics of uncoating may be a determinant in their immunogenicity.
A previous study proposed a model in which HIV-1 has evolved to optimally utilize host cofactor cleavage and polyadenylation specificity factor subunit 6 (CPSF6) and cyclophilins to cloak its reverse-transcribed immune stimulatory genomes (35). Manipulation of capsid interactions with cyclophilins by pharmacological and nonpharmacological methods resulted in sensing of viral PAMPs in macrophages. Consistent with those findings, our studies demonstrate that facilitated exposure of encapsidated viral PAMPs can result in activation of innate immunity by HIV-1. Although studies described in the present manuscript were limited to HIV-1, a previous study showed that the gammaretrovirus glyco-Gag protein that enhances the stability of viral cores' reduced sensing of murine leukemia virus (MLV) infection (23), suggesting that a link between stability of cores and innate sensing may be conserved across retroviruses. Additional studies with PF74 as well as other small molecules that target the viral capsid would be useful for further understanding these effects.
This work extends previous studies that demonstrated the dependence of type I interferon induction by HIV-1 on reverse transcription and defined the lentiviral PAMP to consist of reverse-transcribed cDNAs. Careful understanding of which reverse transcription products can induce ISGs is of importance to strategies to harness innate immune inhibition of the virus. If complete genomes do not stimulate innate immunity, antivirals that enhance production of partial DNA viral PAMPs would optimally combine direct inhibition of productive infection with facilitating timely innate immune control.
MATERIALS AND METHODS
Cells, antibodies, drugs, and reagents.
Human embryonic kidney 293T (HEK293T) cells were obtained from the American Type Culture Collection (ATCC). THP-1 Lucia ISG reporter cells were obtained from InvivoGen. HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% inactivated fetal calf serum, penicillin-streptomycin, and l-glutamine, and THP-1 Lucia cells and all THP-1 Lucia-derived cells were maintained in RPMI medium containing 10% fetal bovine serum, 1% nonessential amino acids, and penicillin-streptomycin, Normocin (InvivoGen), and Zeocin (InvivoGen) antibiotics. Rabbit anti-TREX1 was purchased from Sigma (HPA035437), and mouse anti-cGAS was purchased from Bioss antibodies (MB21D1, 7E3). Nevirapine and AZT were obtained from NIH AIDs Reagents Program. Poly(I·C), herring testis (HT) DNA, and PF74 were purchased from Sigma. Poly(I·C) and HT-DNA were transfected into cells using Lipofectamine 2000 (Invitrogen).
Plasmid constructions and generation of stable cell lines.
TREX1 cDNA was cloned into a lentiviral transfer construct with a deletion of U3 and containing a spleen focus-forming virus (SFFV) promoter and puromycin selection marker. Briefly, TSIN-TREX-HA-IRES-puro was constructed by PCR amplifying TREX1 cDNA with primers containing BamH1/PmeI sites and reverse primer containing the coding sequence for HA that was inserted in frame immediately after amino acid 369 of human TREX1. The PCR product was cloned into the BamH1/PmeI/calf intestinal phosphatase (CIP)-digested lentiviral transfer construct. Lentiviral vectors were made in 293T cells with a transfer vector, pCMVΔR8.91, and a VSV-G expression plasmid and purified by ultracentrifugation, as described previously (36). TREX1 overexpression stable cell lines were generated by transducing THP-1 Lucia cells and selection in puromycin (2.5 μg/ml). A short hairpin RNA (shRNA) targeting cGAS (TRCN0000128310) was purchased from Sigma-Aldrich.
CRISPR/Cas9-mediated gene targeting.
TREX1 and cGAS knockout cell lines were generated by the CRISPR/Cas9 system. Two to three gRNAs targeting the coding sequence were designed for each gene. For MB21D1, target sequences were 5′-GGCTTCCGCACGGAATGCCA-3′ and 5′-CACGTGCTCATAGTAGCTCC-3′. For TREX1, target sequences were 5′-TCCCCTTCGGATCTTAACAC-3′, 5′-AGTTCCTCCACCACCGCGTG-3′, and 5′-CCAGACTCGCACACGGCTGA-3′. Guide RNAs were designed using an online CRISPR design tool (http://tools.genome-engineering.org). A pair of oligonucleotides carrying the 20-nucleotide guide sequence were annealed, ligated into a Cas9 nuclease and single-guide RNA (sgRNA) expression plasmid PX458, and verified by sequencing. pSpCas9 (BB)-2A-GFP (PX458), a gift from Feng Zhang (plasmid 48138; Addgene) is described by Ran et al. (37). CRISPR plasmids were transfected into THP-1 Lucia cells using electroporation (Neon system; Invitrogen) following optimization performed per the manufacturer's instruction. At 72 h posttransfection, single-cell cloning was carried out by limiting dilution in 96-well plates. Clones were chosen from plates having <30 colonies per plate, expanded, and screened by PCR amplification of the targeted gDNA region. PCR products spanning the CRISPR target sites were blunt cloned into a PCR blunt cloning vector (Stratagene). Up to 10 clones for each single-cell clone were sequenced.
Immunoblotting.
Cell lysates were collected in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 1% NP-40, 150 mM Tris-HCl, pH 8.0) plus protease inhibitors (Complete-Mini; Boehringer); samples were sonicated and centrifuged at 14,000 rpm for 10 min at 4°C to precipitate and remove cell debris. Protein optical densities (ODs) were measured at 595 nm to quantify proteins, and equivalent amounts of proteins (20 μg) were immunoblotted for TREX1 using rabbit anti-TREX1 (Sigma) or rat anti-HA antibody (Roche) as indicated in the figures. Briefly, proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, blocked in 10% milk, and incubated overnight at 4°C with primary antibodies diluted in Tris-buffered saline plus Tween (TBST)–5% milk plus 0.1% Tween 20. Membranes were washed three times in TBS–0.1% Tween 20 for 10 min each and incubated for 1 h with secondary antibodies at room temperature diluted 1/4,000 in TBS–5% milk–0.1% Tween 20. Bound antibodies were detected by enhanced chemiluminescence (ECL) and imaged with Bio-Rad software. Membranes were reblotted with anti-tubulin monoclonal antibody (MAb; 1/2,000) and anti-mouse secondary antibody to assess loading.
Viruses.
HIV-1 Lai-based reporter viruses were used in this study. pNL4.3R-E-Δ426 and pNL4.3R-E-GFP have been previously described (36). pLailuc R-E-Δ426 (referred to as HIVluc in this study) was constructed by exchanging an SalI/XhoI fragment from pNL4.3R-E-Δ426 into the corresponding fragment in pHIV-Lai. This virus has a truncated envelope with 426 amino acids deleted, luciferase in the nef slot, and a frameshift in vpr. The pHIVluc D185E reverse transcription mutant (7) was created by site-directed mutagenesis (QuikChange Lightning; Agilent). Single-round VSV-G pseudotyped luciferase and GFP reporter virus stocks were generated using previously described methods (36). Briefly, 293T cells were transfected with reporter virus plasmid and pMD.G (VSV-G). Viral stocks were collected 48 h posttransfection, filtered (0.45-μm pore size), and sucrose purified. All experiments were performed with viral stocks treated with 1 μl/ml Turbo DNase (Thermo Fisher) for 30 min at 37°C. Titers of viral stocks were determined on GHOST cells by FACScan (BD Biosciences). Empty (genome-less) particles were prepared by cotransfection of 293T cells with pVSV-G and the packaging construct pCMV-dR8.91, and the viral stocks were collected, concentrated over sucrose, and treated with DNase as described for reporter viruses.
Interferon-stimulated gene induction and HIV-1 infection in target cells.
THP-1 Lucia ISG cells were seeded at a density of 100,000/well in 96-well plates and transduced with viral stocks at a range of MOIs, as indicated in the figures. Cells were transduced by spinoculation at 2,000 rpm for 2 h. For time-of-addition assays, drugs were added at the noted concentrations at the time of transduction (time zero p.i.) and at various time points postinfection. Nevirapine, AZT, and raltegravir were used at 5 μM, and PF74 was used at 10 μM. At 48 hpi, supernatants were quantified for Lucia luciferase with QuantiLuc detection reagent (InvivoGen). Briefly, 20 μl of supernatant was mixed with 100 μl of detection reagent, and luciferase counts were scored on a luminescence counter (Hidex). Cells were subsequently analyzed for firefly luciferase reporter expression with SteadyGlo luciferase detection reagent (Promega) or for GFP expression by FACScan (BD Biosciences).
Real-time PCR quantification of TREX1 mRNA, cGAS mRNA, and mRNA from representative interferon-stimulated genes.
Total RNA was isolated using a Qiagen RNeasy Plus minikit. cDNA was generated from total RNA using Transcriptor First Strand cDNA Synthesis (Roche) for RT-PCR. Two microliters of cDNA was quantified using target gene-specific primers as well as human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers using a serially diluted plasmid standard and a Bio-Rad analyzer. PCR amplification programs were performed with initial denaturation at 95°C for 10 min followed by 35 cycles of 95°C for 10 s, 56°C for 10 s, and 72°C for 20 s and a melting step after amplification (65°C to 95°C, temperature transition rate of 0.11°C/s). Samples were quantified in triplicate. For oligoadenylate synthase (OAS) and interferon beta genes, target gene mRNA abundance in THP-1HI and THP-1KO cells was first normalized to GAPDH mRNA abundance and then normalized to THP-1 cells, and fold induction was calculated using the ΔΔCT (where CT is threshold cycle) method.
Real-time PCR quantification of viral reverse transcription products and proviral integrants.
THP-1 Lucia cells were transduced with reporter viruses, and cells were harvested at various times after infection. Real-time PCR was performed as previously described with primers for early and late reverse transcription products and proviral integrants as described previously (36). Strong-stop DNA was measured according to Dismuke and Aiken (38).
ACKNOWLEDGMENTS
This work was supported by NIH grant AI077344 (E.P.) and a gift from the Tietze Foundation.
We thank members of the Poeschla laboratory for comments and suggestions.
Experiments were conceived and designed by S.K., J.M., E.P.; the majority of the experiments were performed by S.K., with help from J.M. and technical advice from J.M. and E.P. S.K., J.M., D.D., and E.P. analyzed the data. S.K. and E.P. wrote the paper, with review and corrections by J.M. and D.D.
REFERENCES
- 1.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silvin A, Manel N. 2015. Innate immune sensing of HIV infection. Curr Opin Immunol 32:54–60. doi: 10.1016/j.coi.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazur DJ, Perrino FW. 2001. Structure and expression of the TREX1 and TREX2 3′ → 5′ exonuclease genes. J Biol Chem 276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 7.Mazur DJ, Perrino FW. 1999. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′ → 5′ exonucleases. J Biol Chem 274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 8.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. 2008. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem 283:31649–31656. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stetson DB, Ko JS, Heidmann T, Medzhitov R. 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- 11.Gray EE, Treuting PM, Woodward JJ, Stetson DB. 2015. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J Immunol 195:1939–1943. doi: 10.4049/jimmunol.1500969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. 2006. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakaya Y, Lilue J, Stavrou S, Moran EA, Ross SR. 2017. AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio 8:e00944-17. doi: 10.1128/mBio.00944-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. 2007. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet 80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasan M, Yan N. 2014. Safeguard against DNA sensing: the role of TREX1 in HIV-1 infection and autoimmune diseases. Front Microbiol 5:193. doi: 10.3389/fmicb.2014.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. 2007. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 18.Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. 2007. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 19.Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. 2013. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol 14:61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC 3rd, Lehrman MA, Yan N. 2015. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 43:463–474. doi: 10.1016/j.immuni.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu WS, Hughes SH. 2012. HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2:a006882. doi: 10.1101/cshperspect.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler LA, Trifonova RT, Vrbanac V, Barteneva NS, Liu X, Bollman B, Onofrey L, Mulik S, Ranjbar S, Luster AD, Tager AM, Lieberman J. 2016. TREX1 knockdown induces an interferon response to HIV that delays viral infection in humanized mice. Cell Rep 15:1715–1727. doi: 10.1016/j.celrep.2016.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stavrou S, Nitta T, Kotla S, Ha D, Nagashima K, Rein AR, Fan H, Ross SR. 2013. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc Natl Acad Sci U S A 110:9078–9083. doi: 10.1073/pnas.1217399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stavrou S, Blouch K, Kotla S, Bass A, Ross SR. 2015. Nucleic acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe 17:478–488. doi: 10.1016/j.chom.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goujon C, Arfi V, Pertel T, Luban J, Lienard J, Rigal D, Darlix JL, Cimarelli A. 2008. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol 82:12335–12345. doi: 10.1128/JVI.01181-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fye JM, Coffin SR, Orebaugh CD, Hollis T, Perrino FW. 2014. The Arg-62 residues of the TREX1 exonuclease act across the dimer interface contributing to catalysis in the opposing protomers. J Biol Chem 289:11556–11565. doi: 10.1074/jbc.M114.559252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fye JM, Orebaugh CD, Coffin SR, Hollis T, Perrino FW. 2011. Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutieres syndrome. J Biol Chem 286:32373–32382. doi: 10.1074/jbc.M111.276287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Iaco A, Luban J. 2011. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8:98. doi: 10.1186/1742-4690-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito A, Ferhadian D, Sowd GA, Serrao E, Shi J, Halambage UD, Teng S, Soto J, Siddiqui MA, Engelman AN, Aiken C, Yamashita M. 2016. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J Virol 90:5808–5823. doi: 10.1128/JVI.03116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Fricke T, Diaz-Griffero F. 2013. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J Virol 87:683–687. doi: 10.1128/JVI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meehan A, Saenz D, Morrison J, Peretz M, Poeschla E. 2011. LEDGF dominant interference proteins demonstrate pre-nuclear exposure of HIV-1 integrase and synergize with LEDGF depletion to destroy viral infectivity. J Virol 85:3570–3583. doi: 10.1128/JVI.01295-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 37.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dismuke DJ, Aiken C. 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J Virol 80:3712–3720. doi: 10.1128/JVI.80.8.3712-3720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]