

The A(H7N9) viruses circulating in China are of great public health concern. Here, we report a molecular and structural study of the major surface proteins from several recent A(H7N9) influenza viruses. Our results improve the understanding of these evolving viruses and provide important information on their receptor preference that is central to ongoing pandemic risk assessment.
KEYWORDS: A(H7N9), receptor binding, glycan array, A(H7N9), hemagglutinin
ABSTRACT
The avian influenza A(H7N9) virus continues to cause human infections in China and is a major ongoing public health concern. Five epidemic waves of A(H7N9) infection have occurred since 2013, and the recent fifth epidemic wave saw the emergence of two distinct lineages with elevated numbers of human infection cases and broader geographic distribution of viral diseases compared to the first four epidemic waves. Moreover, highly pathogenic avian influenza (HPAI) A(H7N9) viruses were also isolated during the fifth epidemic wave. Here, we present a detailed structural and biochemical analysis of the surface hemagglutinin (HA) antigen from viruses isolated during this recent epidemic wave. Results highlight that, compared to the 2013 virus HAs, the fifth-wave virus HAs remained a weak binder to human glycan receptor analogs. We also studied three mutations, V177K-K184T-G219S, that were recently reported to switch a 2013 A(H7N9) HA to human-type receptor specificity. Our results indicate that these mutations could also switch the H7 HA receptor preference to a predominantly human binding specificity for both fifth-wave H7 HAs analyzed in this study.
IMPORTANCE The A(H7N9) viruses circulating in China are of great public health concern. Here, we report a molecular and structural study of the major surface proteins from several recent A(H7N9) influenza viruses. Our results improve the understanding of these evolving viruses and provide important information on their receptor preference that is central to ongoing pandemic risk assessment.
INTRODUCTION
Since March 2013, when the first human infection with avian A(H7N9) influenza viruses was reported to the World Health Organization (WHO) (1), A(H7N9) viruses continue to be enzootic in poultry in China, and their reassortment with A(H9N2) viruses has continued to generate multiple genotypes (2). Human A(H7N9) infection is an ongoing epidemic in China, with sporadic spreading to Hong Kong, Taiwan, Macao, Malaysia, and Canada (3). The virus has infected humans in five distinct annual waves. During the fifth wave (1 October 2016 through 30 September 2017), 766 cases of A(H7N9) virus infection with 288 deaths (38% mortality rate) were reported in mainland China (4), numbering almost as many as the combined total for the previous four waves. During the first four epidemic waves, all human infections were solely caused by low-pathogenicity avian influenza (LPAI) virus. However, in addition to LPAI viruses, human infections with highly pathogenic avian influenza (HPAI) A(H7N9) virus were identified in the fifth wave (5, 6). In addition, the geographic distribution of A(H7N9) virus was widespread in China during the fifth epidemic wave. Although the human infections were mainly tied to exposures to infected poultry in live bird markets without reported human-to-human infection, the number of human infection clusters increased in the fifth-wave epidemic (7). Furthermore, two separate lineages (Pearl River Delta lineage and Yangtze River Delta lineage) have diverged among the fifth epidemic viruses (8, 9). All of these epidemiology data have indicated that A(H7N9) virus is actively evolving and continues to be considered the flu strain with the greatest potential to cause a pandemic (https://www.cdc.gov/flu/pandemic-resources/monitoring/irat-virus-summaries.htm) (10, 11).
The receptor binding specificity of the influenza virus coat protein hemagglutinin (HA) is considered one of the major barriers for transmission of avian influenza viruses in humans. Human seasonal influenza viruses bind to α2-6-linked sialic acids (SA), whereas avian influenza viruses predominantly bind to α2-3-linked SA (12, 13). In the past 100 years, only three influenza subtypes have successfully adapted to the human population, causing four pandemics, A(H1N1) in 1918 and 2009, A(H2N2) in 1957, and A(H3N2) in 1968 (14–17). Previous studies have shown that the switch of receptor binding specificity from avian-like to human-like only involved several key residues within the HA receptor binding site (RBS): E190D and G225D for A(H1N1) and Q226L and G228S for A(H2N2) and A(H3N2) (18, 19). While it is still unknown what changes will occur in A(H7N9) viruses should they adapt to humans, recent studies by de Vries et al. reported that three amino acid mutations, V177G/K-K184T-G219S (V186G/K-K193T-G228S in H3 numbering) on the A/Shanghai/2/2013 HA, were able to confer a switch in specificity for human-type receptors and promote binding to human trachea epithelial cells (20).
Here, as part of our ongoing risk assessment activities, we have expressed recombinant HAs (rHA) of A(H7N9) viruses from the fifth epidemic wave. Three-dimensional structures were solved for the HAs of both Yangtze River Delta and Pearl River Delta lineages. The glycan binding preferences of these recent 5th-wave LPAI and HPAI rHAs were also assessed. Finally, the V177G/K-K184T-G219S mutations that were reported to switch receptor specificity were introduced into the more contemporary Yangtze River Delta and Pearl River Delta lineage virus HAs in order to assess whether these changes would allow a switch to human receptor specificity in these circulating viruses.
RESULTS AND DISCUSSION
Overall structure.
In order to study the fifth-wave epidemic A(H7N9) HAs from a structural perspective, a number of rHAs were expressed in a baculovirus expression system, and trimeric rHAs without a foldon or His tag were produced for crystallization studies (Tables 1 and 2). The overall structures of GD3, HK61, and HK125 apo-HAs were found to be similar to that of the A(H7N9) A/Shanghai/2/2013 (SH2) strain (PDB entry 4LN6) (21), with superimposed monomer Cα atom root mean square deviations (RMSDs) of 0.56 Å, 0.87 Å, and 0.62 Å, respectively. The HA monomer is composed of a globular head containing the receptor binding site (RBS), a membrane-proximal domain that includes a central helical stalk, and the HA1/HA2 cleavage site (Fig. 1A). Like SH2 HA, five asparagine-linked glycosylation sites are predicted in GD3, HK61, and HK125 HA monomers. Due to the flexibility of glycans in glycoprotein crystals, our structural data only showed interpretable carbohydrate electron density at Asn28 and Asn231 in HA1 and Asn82 in HA2 for GD3 and only Asn231 in HA1 and Asn82 in HA2 for HK61 and HK125 HAs.
TABLE 1.
Recombinant HA proteins and mutations used in this study
| Influenza virus strain | Virus subtype | GISAID database no. | Mutation(s) | Name used here |
|---|---|---|---|---|
| A/Hong Kong/61/2016 LPAI (Pearl River Delta lineage) | A(H7N9) | EPI872958 | None (WTa) | HK61 |
| V177G-K184T-G219S | HK61-GTS | |||
| V177K-K184T-G219S | HK61-KTS | |||
| A/Hong Kong/125/2017 LPAI (Yangtze River Delta lineage) | A(H7N9) | EPI977395 | None (WT) | HK125 |
| V177G-K184T-G219S | HK125-GTS | |||
| V177K-K184T-G219S | HK125-KTS | |||
| A/Guangdong/17SF003/2016 HPAI (Yangtze River Delta lineage) | A(H7N9) | EPI919607 | None (WT) | GD3 |
| A/Taiwan/1/2017 HPAI (Yangtze River Delta lineage) | A(H7N9) | EPI917065 | None (WT) | TW1 |
| A/Switzerland/9715293/2013 | A(H3N2) | EPI814528 | None (WT) | HuH3 |
| A/northern pintail/Washington/40964/2014 | A(H5N8) | EPI690378 | None (WT) | avH5 |
WT, wild type.
TABLE 2.
Data collection and refinement statistics for the A(H7N9) HA crystal structures
| Parameter | Value(s)a for: |
|||
|---|---|---|---|---|
| GD3 Apo | HK61 Apo | HK125 Apo | HK125 LSTb | |
| Data collection | ||||
| Space group | C2 | C2 | C2 | C2 |
| Cell dimensions (Å; a, b, c) | 203.13, 117.05, 119.97 | 210.01, 97.53, 191.81 | 202.64, 116.89, 119.63 | 203.57, 117.87, 119.86 |
| Cell angle (°; a, b, c) | 90, 124.46, 90 | 90, 108.58, 90 | 90, 124.161, 90 | 90, 124.213, 90 |
| Resolution (Å) | 50–2.7 (2.75–2.70) | 50–2.95 (3.06–2.95) | 50–2.95 (3.06–2.95) | 50–3.55 (3.68–3.55) |
| Rsym | 0.053 (0.549) | 0.158 (0.674) | 0.112 (0.827) | 0.126 (0.789) |
| Rpim | 0.035 (0.367) | 0.108 (0.454) | 0.069 (0.500) | 0.076 (0.476) |
| I/σ | 22.3 (1.8) | 6.4 (1.4) | 18.4 (1.5) | 10.5 (1.5) |
| Completeness (%) | 98.7 (97.3) | 93.1 (71.4) | 98.9 (92.2) | 99.7 (98.7) |
| Redundancy | 3.2 (3.2) | 3.1 (2.9) | 3.7 (3.7) | 3.7 (3.6) |
| CC1/2b | 0.99 (0.888) | 0.998 (0.824) | 0.936 (0.723) | 0.914 (0.696) |
| Refinement | ||||
| Resolution (Å) | 50–2.7 (2.80–2.70) | 50–2.95 (3.06–2.95) | 50–2.95 (3.06–2.95) | 50–3.55 (3.68–3.55) |
| No. of reflections (total) | 62,809 | 68,692 | 48,207 | 26,155 |
| No. of reflections (test) | 3,265 | 3,659 | 2,675 | 1,297 |
| No. of atoms | 11,559 | 22,934 | 11,496 | 11,667 |
| Rwork/Rfree | 0.221/0.258 | 0.236/0.267 | 0.234/0.279 | 0.225/0.274 |
| B values (Å2) | ||||
| Protein | 75.3 | 42.7 | 88.4 | 65.1 |
| Ligand | 121.7 | |||
| Water | ||||
| Wilson B value (Å2) | 61.3 | 56.9 | 78.5 | 46.7 |
| RMSD bond length (Å) | 0.009 | 0.015 | 0.008 | 0.015 |
| RMSD bond angle (°) | 1.271 | 1.896 | 1.334 | 2.168 |
| MolProbity scoresc (%) | ||||
| Favored | 92.6 | 94.1 | 92.4 | 97.0 |
| Outliers | 0.4 | 0.4 | 1.0 | 2.7 |
| PDB entry | 6D7U | 6D7C | 6D8B | 6D8D |
Values in parentheses are for the outer shell.
CC1/2, Pearson correlation coefficient between two random half data sets.
Values are percentages of residues in the favored and outlier regions analyzed by MolProbity (51).
FIG 1.
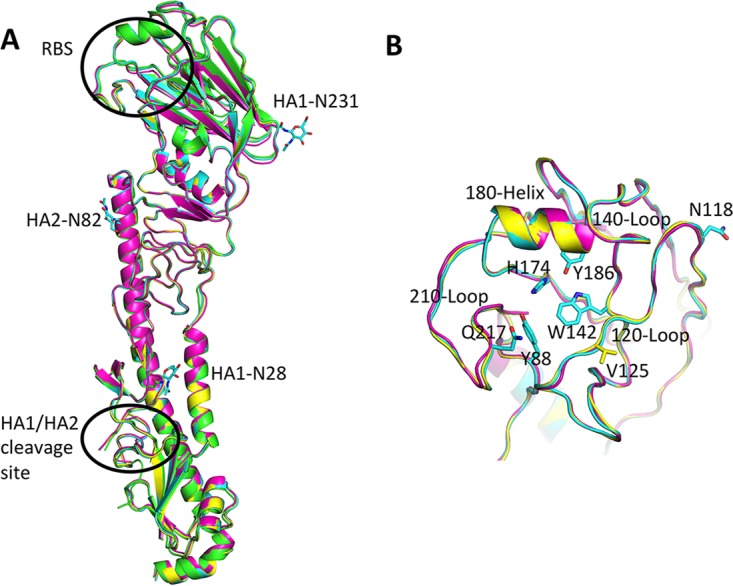
Overall structure of HA. (A) The superimposed HA monomers from SH2 (green), GD3 (cyan), HK61 (magenta), and HK125 (yellow) HAs. (B) A close view of the RBS. All discussed residues in the text are shown in sticks. All structural figures were generated with MacPyMOL (53).
The RBS of the fifth-epidemic-wave H7 HAs were structurally similar to all published influenza A virus HAs. They are composed of three structural elements: a 180-helix (positions 178 to 186), 210-loop (positions 211 to 219), and 120-loop (positions 123 to 128). At the base of the pocket, four highly conserved residues, Y88, W142, H174, and Y186 (equivalent to Y98, W152, H183, and Y195 in H3 numbering), are present and conserved in these recent HAs. As for other H7 HAs, the 140-loop has a unique feature that produces a 6-Å movement toward the RBS compared to group 2 H3 HAs (Fig. 1B) (21, 22). This elongated loop has also been observed for H10 HAs, which is in the same subclade as H7 in group 2 HA (23, 24), and previous data indicate that the 150-loop plays a role in restricting the host specificity of H10N8 viruses (24).
Sequence alignment of HA1s from the fifth-epidemic-wave HAs with that of SH2 highlighted several amino acid differences (Fig. 2). Significantly, a basic four-amino-acid insertion was found within the cleavage sites of TW1 and GD3 Yangtze River Delta lineage HAs, resulting in HPAI A(H7N9) viruses. Thus far, this insertion has only been identified in conjunction with an avian Q217 (226 in the H3 numbering) residue within the RBS. A Q217L substitution is believed to be one of the key residues necessary to switch from avian-like receptor binding to human-like receptor binding in A(H2N2) and A(H3N2) (12). For the other amino acid differences, only one A125V mutation in both HK125 and TW1 HAs was located on the 120-loop within the RBS region (Fig. 1B). While this change may not result in any significant conformational change on this loop, it may still affect loop stability and/or antigenicity. In addition, the TW1 HA possessed two substitutions, A118N and A120T, which introduce a glycosylation site at residue 118. While our attempt to obtain a TW1 HA structure was unsuccessful, the location of this site is away from the RBS (Fig. 1B) and thus is not expected to directly affect receptor specificity.
FIG 2.

Structural base sequence alignment of HA1 from A(H7N9) HAs. The receptor binding site (RBS), as well as seasonal H1 (1, Sa; 2, Sb; 3, Ca; 4, Cb) and H3 (A/B/C/D/E) antigenic sites, are labeled.
Antigenic properties of A(H7N9) viruses from two different lineages.
While human seasonal influenza virus A(H1N1) HAs have four distinct antigenic sites (Sa, Sb, Ca, and Cb) (25), five antigenic sites (A, B, C, D, and E) have been described for human seasonal influenza virus A(H3N2) HAs (26). Since the outbreak of avian A(H7N9) viruses in China in 2013, candidate vaccine viruses (CVVs) were developed and remained unchanged for the first four epidemic waves, as the circulating A(H7N9) viruses had been antigenically stable. However, during the fifth epidemic wave, A(H7N9) viruses showed much more genetic diversity and two genetic lineages were identified (9). Hemagglutination inhibition (HI) testing of the fifth-epidemic Yangtze River Delta lineage viruses, including the HPAI strains, highlighted significant antigenic differences compared with the CVVs developed from the A(H7N9) viruses of 2013 (27). As a result, the WHO has recommended the development of new CVVs to cover the fifth-epidemic-wave viruses. The HK125 virus selected as an LPAI vaccine strain has A112T, S118N, A125V, R130K, L168I, and M227I substitutions on HA1 compared to the sequence of SH2 HA, with four substitutions located on antigenic site A (Fig. 2 and 3A and B). The GD3 virus was selected as an HPAI CVV strain and has more substitutions, including I38T, A112P, S118N, K164E, L168I, L217Q, M227I, G261R, and I317V (Fig. 2), that are located on antigenic sites A, C, and D (Fig. 3C). These changes alter the HA surface properties, which could affect the antigenicity of the CVVs. The underlying mechanism that drove the divergence of the two H7 lineages during the fifth epidemic wave is unknown.
FIG 3.
Antigenicity of H7 HA. (A) The surface view of SH2 with H3 equivalent antigenic sites are shown in different colors. A, pale green; B, wheat; C, slate; D, pale cyan; E, pale yellow. The potential glycosylation sites are shown in orange. (B) Surface view of HK125 with amino acids that differ from SH2 are labeled and shown in red. (C) Surface view of GD3 with amino acids that differ from SH2 are labeled and shown in red.
Glycan binding analyses.
In order to assess these 5th-wave A(H7N9) RBS substitutions with respect to receptor specificity, glycan microarray analysis was performed using these rHAs. Previous work revealed that the 2013 A(H7N9) HAs possessed a weak human receptor binding preference by glycan array analysis, and this corresponded to the presence of a leucine at residue 217, a position equivalent to residue 226 in A(H3N2) viruses (21), and possibly a G177V substitution (28, 29). Since then, few HA sequence changes have been detected, and there has been no significant change in either the antigenic or receptor binding sites during the first four epidemic waves. Several fifth-epidemic-wave H7 HAs, rHAs for HK61, HK125, GD3, and TW1, were recombinantly expressed, and their binding profiles were compared to those of huH3 and avH5 rHAs (Fig. 4A to F). LPAI H7 HAs, HK61 from Pearl River Delta lineage and HK125 from Yangtze River Delta lineage, revealed binding profiles that were very similar to what was reported with the 2013 SH2 HA (21). Both had a more restricted binding profile to avian α2-3-linked sialosides than the avian H5 HA binding profile. They also revealed very weak binding to human α2-6 receptors (glycans 41 to 64; glycan structures are listed in Table 3), with only one strong binding signal to the branched glycan, Galβ1-3(Neu5Acα2-6)GlcNAcβ1-3Galβ1-4Glc (glycan 60, LS-tetrasaccharide b [LSTb]) (Fig. 4A and B). The HPAI H7 HAs GD3 and TW1, both from Yangtze River Delta lineage and with glutamine at position 217, revealed a strong avian receptor binding preference and no human-like receptor binding (Fig. 4C and D). The binding of LSTb had been reported in previous H7 HA studies (21, 30), although the significance of this glycan is not clear, since it has only been found in human milk (31). While attempts were made to determine the complex structure of LSTb with both HK61 and HK125 HAs, only an HK125/LSTb complex structure was successfully solved at 3.55 Å. Hydrogen bonds are formed between SIA and Y88 as well as T126 and S127. The branched GAL forms one hydrogen bond with K184 (Fig. 5A). The superimposition of this RBS/LSTb structure with a previous 2013 SH2/LSTb complex (21) highlighted the lack of significant differences between the two complexes (Fig. 5B).
FIG 4.

Glycan microarray and BLI binding analysis of 5th-wave H7 rHAs compared to human and avian rHAs. (A) HK61, (B) HK125, (C) GD3, (D) TW1, (E) huH3, and (F) avH5. Colored bars highlight glycans that contain α2-3 SA (blue) and α2-6 SA (red), α2-6/α2-3-mixed SA (purple), N-glycolyl SA (green), α2-8 SA (brown), β2-6 and 9-O-acetyl SA, and non-SA (gray). Error bars reflect the standard deviations from the signal for six independent replicates on the array. Structures of each of the numbered glycans are found in Table 3. (G and H) The binding kinetics of rHAs to specific biotinylated glycans (3-SLNLNb and 6-SLNLNb) immobilized onto biosensors were analyzed by BLI. Error bars reflect the standard errors from three independent experiments.
TABLE 3.
Glycan microarray for recHAs
| Glycan no.a | Glycan structureb | Bindingc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HK61 | HK61_GTS | HK61_KTS | HK125 | HK125_GTS | HK125_KTS | GD3 | TW1 | avH5 | huH3 | ||
| 1 | Neu5Acα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 2 | Neu5Acα | +++ | +++ | +++ | +++ | NB | +++ | NB | NB | NB | NB |
| 3 | Neu5Acβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 4 | Neu5Acα2–3(6-O-Su)Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 5 | Neu5Acα2-3Galβ1-3[6OSO3]GalNAcα | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 6 | Neu5Acα2-3Galβ1-4[6OSO3]GlcNAcβ | +++ | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 7 | Neu5Acα2-3Galβ1-4(Fucα1-3)[6OSO3]GlcNAcβ-propyl-NH2 | +++ | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 8 | Neu5Acα2-3Galβ1-3[6OSO3]GlcNAc-propyl-NH2 | +++ | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 9 | Neu5Acα2-3Galβ1-3(Neu5Acα2-3Galβ1-4)GlcNAcβ | + | NB | NB | ++ | NB | NB | +++ | +++ | +++ | NB |
| 10 | Neu5Acα2-3Galβ1-3(Neu5Acα2-3Galβ1-4GlcNAcβ1-6)GalNAcα | NB | NB | NB | +++ | NB | NB | NB | ++ | +++ | NB |
| 11 | Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-3(Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | NB |
| 12 | Neu5Acα(2-3)-Galβ(1-4)-GlcNAcβ(1-3)-Galβ(1-4)-GlcNAcβ(1-2)-Manα(1-3)-[Neu5Acα(2-3)-Galβ(1-4)-GlcNAcβ(1-3)-Galβ(1-4)-GlcNAcβ(1-2)-Manα(1-6)]-Manβ(1-4)-GlcNAcβ(1-4)-GlcNAcβ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | NB |
| 13 | Neu5Acα2-3Galβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 14 | Neu5Acα2-3Galβ1-3GalNAcα | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 15 | Neu5Acα2-3Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 16 | Neu5Acα2-3Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 17 | Neu5Acα2-3Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 18 | Neu5Acα2-3Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 19 | Neu5Acα2-3Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 20 | Neu5Acα2-3Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 21 | Neu5Acα2-3GalNAcβ1-4GlcNAcβ | NB | NB | NB | NB | NB | ++ | +++ | +++ | +++ | NB |
| 22 | Neu5Acα2-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ | +++ | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 23 | Neu5Aca2-3Galβ1-3GlcNAcβ1-3Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 24 | Neu5Acα2-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ | NB | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 25 | Neu5Acα2-3Galβ1-4GlcNAcβ1-3Galβ1-3GlcNAcβ | +++ | NB | NB | +++ | NB | NB | +++ | +++ | +++ | NB |
| 26 | Neu5Acα2-3Galβ1-3GalNAcα | NB | NB | NB | NB | NB | NB | ++ | +++ | +++ | NB |
| 27 | Galβ1-3(Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-6)GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | + | NB |
| 28 | Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | +++ | +++ | NB |
| 29 | Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 30 | Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ | NB | NB | NB | NB | NB | NB | +++ | +++ | +++ | NB |
| 31 | Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ | + | NB | NB | ++ | NB | NB | +++ | +++ | +++ | NB |
| 32 | Neu5Acα2-3Galβ1-3[Fucα1-4]GlcNAcβ1-3Galβ1-4[Fucα1-3]GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | +++ | +++ | NB |
| 33 | Neu5Acα2-3Galβ1-3[Fucα1-3]GlcNAcβ1-3Galβ1-4[Fucα1-3]GlcNAcβ | +++ | NB | NB | +++ | +++ | NB | +++ | +++ | +++ | NB |
| 34 | Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ | +++ | +++ | NB | +++ | +++ | +++ | +++ | +++ | +++ | NB |
| 35 | Neu5Acα2-3(GalNAcβ1-4)Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 36 | Neu5Acα2-3(GalNAcβ1-4)Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 37 | Neu5Acα2-3(GalNAcβ1-4)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 38 | Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 39 | Fucα1-2Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 40 | Fucα1-2Galβ1-3GalNAcβ1-4(Neu5Acα2-3)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 41 | Neu5Acα2-6Galβ1-4[6OSO3]GlcNAcβ | + | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 42 | Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-3(Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 43 | Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-3(Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | NB |
| 44 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-2Manα1-3[Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-2Manα1-6]Manβ1-4GlcNAcβ1-4GlcNAcβ | + | +++ | +++ | NB | + | +++ | NB | NB | NB | +++ |
| 45 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-2Manα1-3[Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-2Manα1-6]-Manβ1-4GlcNAcβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 46 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3[Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-6]GalNAca | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 47 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3[Neu5Acα2-6Galβ1-4GlcNAcβ1-6]GalNAca | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 48 | Neu5Acα2-6GalNAcα | NB | NB | ++ | NB | NB | + | NB | NB | NB | NB |
| 49 | Neu5Acα2-6Galβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 50 | Neu5Acα2-6Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 51 | Neu5Acα2-6Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 52 | Neu5Acα2-6Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | +++ |
| 53 | Neu5Acα2-6Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | +++ |
| 54 | Neu5Acα2-6GalNAcβ1-4GlcNAcβ | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 55 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 56 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 57 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 58 | Neu5Aca2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 59 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)GlcNAcβ | NB | NB | +++ | NB | NB | +++ | NB | NB | NB | +++ |
| 60 | Galβ1-3(Neu5Acα2-6)GlcNAcβ1-4Galβ1-4Glcβ-Sp10 | +++ | +++ | +++ | +++ | NB | +++ | NB | NB | NB | NB |
| 61 | Neu5Acα2-6[Galβ1-3]GalNAca | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 62 | Neu5Acα2-6Galβ1-4GlcNAcβ1-6[Galβ1-3]GalNAca | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 63 | Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-6[Galβ1-3]GalNAca | NB | NB | NB | NB | NB | + | NB | NB | NB | +++ |
| 64 | Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-3(Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ | ++ | NB | +++ | NB | NB | +++ | NB | NB | NB | NB |
| 65 | Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-3(Neu5Acα2-3Galβ1-4GlcNAcβ1-2Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ | +++ | + | +++ | +++ | +++ | +++ | +++ | +++ | +++ | NB |
| 66 | Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GalNAcα | +++ | NB | +++ | +++ | NB | +++ | +++ | +++ | +++ | NB |
| 67 | Neu5Acα2-3(Neu5Acα2-6)GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 68 | Neu5Gcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 69 | Neu5Gcα2-3Galβ1-3(Fucα1-4)GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 70 | Neu5Gcα2-3Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 71 | Neu5Gcα2-3Galβ1-4(Fucα1-3)GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | +++ | +++ | NB |
| 72 | Neu5Gcα2-3Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | ++ | NB |
| 73 | Neu5Gcα2-6GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 74 | Neu5Gcα2-6Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 75 | Neu5Acα2-8Neu5Acα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 76 | Neu5Acα2-8Neu5Acα2-8Neu5Acα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 77 | Neu5Acα2-8Neu5Acα2-3(GalNAcβ1-4)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 78 | Neu5Acα2-8Neu5Acα2-3Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 79 | Neu5Acα2-8Neu5Acα2-8Neu5Acα2-3(GalNAcβ1-4)Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 80 | Neu5Acα2-8Neu5Acα2-8Neu5Acα2-3Galβ1-4Glcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 81 | Neu5Acα2-8Neu5Acβ-Sp17 | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 82 | Neu5Acα2-8Neu5Acα2-8Neu5Acβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 83 | Neu5Acβ2-6GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 84 | Neu5Acβ2-6Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 85 | Neu5Gcβ2-6Galβ1-4GlcNAc | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 86 | Galβ1-3(Neu5Acβ2-6)GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 87 | [9NAc]Neu5Acα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 88 | [9NAc]Neu5Acα2-6Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 89 | Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 90 | Galβ1-3GlcNAcβ1-3Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | ++ | NB | +++ | NB |
| 91 | Galβ1-4GlcNAcβ1-2Manα1-3[Galβ1-4GlcNAcβ1-2Manα1-6]Manβ1-4GlcNAcβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 92 | GalNAcα1-3(Fucα1-2)Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 93 | GalNAcα1-3(Fucα1-2)Galβ1-4GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 94 | Galα1-3(Fucα1-2)Galβ1-3GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 95 | Galα1-3(Fucα1-2)Galβ1-4(Fucα1-3)GlcNAcβ | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
| 96 | Galβ1-3GalNAcα | NB | NB | NB | NB | NB | NB | NB | NB | NB | NB |
Different categories of glycans on the array are grouped together: glycans 1 to 3, sialic acid; glycans 4 to 40, α2-3 sialosides; glycans 41 to 63, α2-6 sialosides; glycans 64 to 67, mixed α2-3/α 2-6 biantennaries; glycans 68 to 74, N-glycolylneuraminic acid-containing glycans; glycans 75 to 82, α2-8-linked sialosides; glycans 83 to 88, 2-6-linked and 9-O-acetylated sialic acids; glycans 89 to 96, asialo glycans.
Neu5Ac, sialic acid; Neu5Gc, N-glycolylneuraminic acid; OSO3, sulfate; Gal, galactose; Fuc, fucose; Glc, d-glucose; GlcNAc, N-acetyl-d-glucosamine; GalNAc, N-acetyl-d-galactosamine; Man, d-mannose; 9NAc, 9-O-acetyl.
Significant binding of samples to glycans was qualitatively estimated based on the relative strength of the signal for the data shown. Symbols for fluorescence intensities: >10,000, +++; 5,000 to 10,000, ++; 1,250 to 4,999, +; <1,250, NB.
FIG 5.

Structure of HK125 in complex with LSTb. (A) Interaction of the LSTb glycan with the HK125 RBS. HKL125 is shown as a yellow cartoon, while LSTb (orange) and interacting HA residues are shown as sticks. Black dashed lines indicate hydrogen bonds. (B) Alignment of the RBS of HK125/LSTb complex (yellow/orange) with SH2 (green/teal) (PDB entry 4LN8).
The 2017 rHAs were also analyzed by biolayer interferometry (BLI) for their ability to bind to both 3SLNLNb and 6SLNLNb (Fig. 4G to H and Table 3). While there was 3SLNLNb binding to HK61, HK125, TW1, and SH2 rHAs, their binding signals were weaker than what was seen with the control avH5 HA (Fig. 4G). Overall, binding of these A(H7N9) rHAs to the human receptor analog, 6SLNLNb, was much reduced (≤50% compared to 3SLNLNb signal). However, 6SLNLNb binding at the higher rHA concentrations used (3 nM) was still significant for HK61, HK125, and GD3 (but not TW1) compared to that for the avH5 rHA (Fig. 4H). While this was expected for HK61 and HK125, both with L217, the GD3 rHA with avian Q217 was surprising, particularly as glycan array data for GD3 revealed a specific α2-3 binding profile and TW1 (also with avian Q217) showed no binding by BLI. Interestingly, the GD3 HA was previously reported to have dual α2-3- and α2-6-linked glycan receptor specificity (6, 32), and our data here are consistent with those reports. In addition, similar results were reported for the A/Shanghai/1/2013 (SH1) rHA (21). By BLI, the GD3 rHA binding signal to the α2-3 receptor analog was much stronger than that of the other H7 rHAs that were tested (Fig. 4G). The reason for the discrepancy between these assays is unclear. Previous studies with A(H5N1) viruses reported receptor binding changes that were detected by enzyme-linked immunosorbent assay (ELISA) (33) but not by glycan microarray (34). For virus experiments, these discrepancies may be due to differences in virus preparations or the antibodies used for the assays. It has also been suggested that BLI and ELISA type assays are more sensitive for detecting subtle changes in binding avidity to α2-6 sialoglycans and thus can detect weak binding to human-type receptors, whereas the glycan microarray is a more stringent test of binding specificity (34).
The biological relevance of each assay is also unknown and is complicated, because switching receptor binding is not considered the only major barrier for transmission of avian influenza viruses in humans. For example, A(H5N1) viruses bearing single-amino-acid substitutions that yielded receptor binding changes, detected by ELISA, were not sufficient for efficient infection and transmission in ferrets (35).
Adaptation to binding human receptors.
A recently published study by de Vries et al. used molecular modeling to identify three amino acid mutations, V177G/K-K184T-G219S (V186G/K-K193T-G228S in H3 numbering), on the 2013 SH2 HA that switched the HA RBS to bind human receptors (20). While the de Vries study used a 2013 HA (SH2), it is unknown if the same switch would be seen on A(H7N9) HAs from these two recent lineages. To address this, V177G-K184T-G219S (GTSmut) and V177K-K184T-G219S (KTSmut) triple mutations were introduced on both the HK61 and HK125 HA frameworks, and rHAs were expressed and analyzed by glycan microarray (Fig. 6A to D) and BLI (Fig. 6E and F). Compared to the wild-type (WT) rHA (Fig. 4A and B), the triple mutation GTSmut appeared to reduce the number of glycans binding to HK61 and HK125 on the array (Fig. 6A and B). Strong α2-3-linked signals were still apparent for biantennary glycans (glycans 11 and 12) as well as longer linear sialosides with α1-3 fucosylation (33 and 34). Interestingly, while binding to both α1-3 fucosylated glycans (33 and 34) was maintained by the HK125 GTSmut, the HK61 GTSmut lost binding to glycan 33, suggesting that while the core fucose helped binding to these rHAs, the β1-3 linkage between terminal galactose and N-acetylglucosamine (Galβ1-3GlcNAc) was detrimental to binding. Interestingly, in the de Vries et al. studies with the 2013 SH2 HA, there was little binding to the α2-6 linked sialosides with this H7 rHA on the array. Likewise, BLI results showed reduced binding to both glycans for both GTSmut proteins compared to that of their corresponding WT rHAs (Fig. 6E and F). The KTSmut variants, however, yielded a significant switch in HA binding preference from the α2-3-linked sialosides to α2-6-linked sialosides (Fig. 6C and D). The KTSmut on both HK61 and HK125 frameworks resulted in increased binding to sulfated (glycan 41), biantennary/branched (43, 44, and 46), and linear (54, 56, 58, and 59) α2-6-linked sialosides, and binding was maintained for the internal LSTb (60). Binding to α2-3-linked sialosides was also reduced, including binding to both α1-3 fucosylated glycans (33 and 34), although weak binding to glycan 34 was still apparent for the HK125 KTSmut. Strong α2-3-linked signals were still apparent for biantennary glycans (11 and 12), similar to what was observed for the GTSmut proteins. Similarly, BLI results showed that glycan binding increased for both HK61 and HK125 KTSmut rHAs compared to that of their WT counterparts, and binding to both 3SLNLNb and 6SLNLNb was similar for both KTSmut proteins (Fig. 6E and F).
FIG 6.

Glycan microarray and BLI binding analysis of 5th-wave H7 rHAs with RBS mutations. (A) HK61-GTSmut, (B) HK125-GTSmut, (C) HK61-KTSmut, and (D) HK125-KTSmut. Colored bars highlight glycans as listed in Fig. 4. Error bars reflect the standard deviations from the signal for six independent replicates on the array. Structures of each of the numbered glycans are found in Table 3. (E and F) The binding kinetics of mutated rHAs to specific biotinylated glycans (3-SLNLNb and 6-SLNLNb) immobilized onto biosensors were analyzed by BLI. Error bars reflect the standard errors from three independent experiments.
The three positions 177, 184, and 219, identified by de Vries et al. (20), have previously been shown to affect HA receptor binding. A valine at residue position 177 resulted in an overall increase in substrates bound on an A(H7N7) HA (21), and a G177V mutation was also reported as a potential position for adaptation of avian H7 to binding human receptors (28, 29). The positively charged lysine at position 184 is positioned on the alpha helix of the RBS and points toward the pocket (21). This position is important for receptor switching in other influenza A virus subtypes. K184T for A(H10N8) resulted in glycan binding to human-type receptors (24), and a K184S substitution was also reported to bind human receptors when introduced with three other substitutions on an H5 HA (36) and was reported to be essential for the A(H3N2) 1968 Hong Kong pandemic (37). Molecular modeling by de Vries et al. concluded that K184 could interfere with the receptor glycan that projects out of the RBS and predicted that a K184T substitution would permit binding of biantennary glycans across two monomers in the trimer. Thus far, the V177K and G219S substitutions have not been identified in any of the >1,800 available A(H7N9) HA sequences submitted to the GISAID database.
Conclusions.
The avian A(H7N9) virus epidemic is still active in China, with an increased number of human infections. A recent study of 40 human A(H7N9) clusters from the five waves of disease activity in China found a stable pattern in number and size. These results suggest that the human-to-human transmission risk has not changed since the virus emerged in 2013 (38), and our glycan binding analyses here agree with this view. The recent description of the V177K-K184T-G219S triple mutation and its effect on switching receptor specificity to human glycans is a cause for concern. However, while results from the de Vries et al. manuscript revealed a switch in receptor binding from avian to human specificity, results described here on the more recent HK61 (Pearl River Delta lineage) and HK125 (Yangtze River Delta lineage) HA frameworks showed a significant but not complete loss of binding to avian-like (α2-3-linked) receptors and increased binding to human-like (α2-6 linked) receptors on the glycan microarrays. Indeed, although the HA receptor binding preference is important, it is not the only requirement for efficient human-to-human transmission (39, 40). However, it is a critical factor that requires continued monitoring for changes that may lead to direct adaptation to humans or to a species that could be considered an intermediate mixing vessel (41–43) for adaptation. Previously, an incomplete switch was also observed in a study with A(H5N1) viruses, and the corresponding rescued mutant (D187G-E190D-K193S-Q226L-G228S) viruses showed no improvement in transmission efficiency compared to the parental viruses, indicating that additional molecular changes would be required for A(H5N1) viruses to fully adapt to humans (36). With the recent lifting of the moratorium on gain-of-function (GoF) experiments that had prevented these animal experiments, future studies with a V177K-K184T-G219S triple mutation reverse genetics virus would need to be approved and performed in order to complete a full risk assessment analysis.
MATERIALS AND METHODS
Recombinant HA cloning and expression.
cDNA encoding residues 1 to 321 of the LPAI HA1 chain (1 to 325 for HPAI) and 1 to 174 of the HA2 chain of the mature ectodomain from each HA listed in Table 1 were synthesized as codon-optimized constructs (Genscript Inc.) and subcloned into the baculovirus transfer vector pAcGP67-B (BD Biosciences) in frame with an N-terminal baculovirus GP67 signal peptide and a C-terminal thrombin cleavage site, a T4 fibritin sequence for generating functional trimers, and a His tag to aid purification (44). HA mutations were introduced by mutagenesis of the wild-type constructs using a QuikChange lightning site-directed mutagenesis kit (Agilent Technologies). Transfection and virus amplification were carried out by utilizing AB Vector's baculovirus transfer vectors and their suggested transfection protocol (AB Vector, San Diego, CA).
Protein expression and purification.
Secreted soluble rHA protein was recovered from the cell culture supernatant by tangential flow filtration through a 30-kDa-molecular-weight-cutoff membrane, metal affinity chromatography, and gel filtration chromatography. For structural studies, rHA proteins were subjected to trypsin digestion (1:1,000, wt/wt, ratio of trypsin to protein). HAs were buffer exchanged into 10 mM Tris-HCl, 50 mM NaCl, pH 8.0, and concentrated to 11 to 14 mg/ml for crystallization trials.
Crystallization, ligand soaking, and data collection.
Initial crystallization trials were set up using a Formulatrix NT8 (Formulatrix, Inc., Bedford, MA). Conditions in which crystals were observed were optimized at 20°C using a modified method for microbatch under oil (45). The GD HA was crystallized with 0.2 M ammonium formate, pH 6.6, 20% polyethylene glycol (PEG) 3350, whereas the HK61 HA was crystallized with 0.2 M magnesium chloride, 0.1 M HEPES-NaOH, pH 7.5, 25% PEG 3350. The HK125 HA was crystallized with 0.2 M magnesium acetate, 20% PEG 3350. Attempts to produce receptor analog complex crystals were performed by soaking crystals for 3 h in the crystallization buffer containing 10 mM either 3′-sialyl-N-acetyllactosamine (3-SLN), 6′-sialyl-N-acetyllactosamine (6-SLN), or LS-tetrasaccharide b (LSTb) (Dextra Laboratories, UK). Only the HK125 HA/LSTb complex yielded diffraction-quality crystals. All crystals were flash-cooled at 100 K, and data sets were collected at the Argonne National Laboratory Advanced Photon Source (APS), beamline 22_ID, and processed with the DENZO-SACLEPACK suite (46). More specific information for each HA is included in Table 2.
Structure determination and refinement.
All H7 HA structures were determined by molecular replacement with Phaser (47) using the HA structure from A/Shanghai/2/2013 (SH2) (PDB entry 4LN6). Models were then mutated to their correct sequences, rebuilt by Coot (48), and refined with Phenix (49) and REFMAC using TLS refinement (50). The final models were assessed using MolProbity (51). Statistics on data processing and refinement are presented in Table 2. Unless specified otherwise, all residue numbering is according to that of the mature H7 protein.
Glycan binding analyses.
Glycan microarray slides and biotinylated glycans used in this study were produced under contract from the Centers for Disease Control and Prevention by James Paulson at The Scripps Research Institute (La Jolla, CA). Recombinant HA glycan microarray analyses were performed as described previously (21, 52). Briefly, recombinant HA-antibody complexes were prepared by mixing rHA (15 μg), mouse anti-penta-His-Alexa Fluor 488 (Qiagen), and anti-mouse-IgG-Alexa Fluor 488 (Thermo Fisher Scientific) in a molar ratio of 4:2:1, respectively. These prepared mixtures of complexes were incubated for 1 h on ice, diluted to 0.5 ml with a phosphate-buffered saline buffer containing 2% (wt/vol) bovine serum albumin (PBS-BSA), and incubated on the microarray slide in a 4°C humidified chamber for 1.5 h. Slides were subsequently washed by successive rinses in PBS with 0.05% Tween 20 (PBS-T), PBS, and deionized water and then immediately subjected to imaging. Fluorescence intensities were detected using an Innoscan 1100AL scanner (Innopsys, USA), and image analyses were carried out using ImaGene 9 image analysis software (BioDiscovery Inc., USA). Table 3 lists the glycans used in these experiments as well as a tabulated qualitative assessment of binding for each protein analyzed.
For kinetic studies, biotinylated receptor analogs Neu5Ac(α2-3)Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)GlcNAcb-biotin (3SLNLNb) and Neu5Ac(α2-6)Gal(β1-4)GlcNAc(β1-3)Gal(β1-4)GlcNAcb-biotin (6SLNLNb) were precoupled to streptavidin-coated biosensors (Pall Fortebio LLC). Binding of recombinant HA, prepared from 6 μM trimer in kinetics buffer (PBS containing 0.02% [vol/vol] Tween 20, 0.005% [wt/vol] sodium azide, and 100 μg/ml bovine serum albumin) in serial 2-fold dilutions, was analyzed by BLI using an Octet Red384 system (Pall Fortebio LLC) according to the manufacturer's instructions. Data were analyzed using the system software and fitted to a 1:1 binding model.
Accession number(s).
The atomic coordinates and structure factors of each HA model described here are available from the RCSB PDB database (www.pdb.org) under the accession codes 6D7U, 6D7C, 6D8B, and 6D8D.
ACKNOWLEDGMENTS
This work was funded by the Centers for Disease Control and Prevention. We thank the staff of SER-CAT sector 22 at the Advanced Photon Source (APS) for their help in data collection. The U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357, supports use of the Advanced Photon Source at Argonne National Laboratory. The Consortium for Functional Glycomics (CFG), funded by National Institute of General Medical Sciences grant GM62116, produced glycan microarrays under contract for the Centers for Disease Control and Prevention.
We thank the WHO Global Influenza Surveillance and Response System (GISRS) and, in particular, the Chinese Center for Disease Control and Prevention for depositing their A(H7N9) HA sequences in the GISAID database.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention or the Agency for Toxic Substances and Disease Registry.
REFERENCES
- 1.WHO. 2013. Avian influenza A (H7N9) virus. World Health Organization, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/influenza_h7n9/en/index.html. [Google Scholar]
- 2.Yang L, Zhu W, Li X, Chen M, Wu J, Yu P, Qi S, Huang Y, Shi W, Dong J, Zhao X, Huang W, Li Z, Zeng X, Bo H, Chen T, Chen W, Liu J, Zhang Y, Liang Z, Shi W, Shu Y, Wang D. 2017. Genesis and spread of newly emerged highly pathogenic H7N9 avian viruses in mainland China. J Virol 91:e01277-17. doi: 10.1128/JVI.01277-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin Q, Lin Z, Chiu AP, He D. 2016. Seasonality of influenza A(H7N9) virus in China-fitting simple epidemic models to human cases. PLoS One 11:e0151333. doi: 10.1371/journal.pone.0151333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO. 2017. Human infection with avian influenza A(H7N9) virus–China. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/don/13-september-2017-ah7n9-china/en/. [Google Scholar]
- 5.Zhang F, Bi Y, Wang J, Wong G, Shi W, Hu F, Yang Y, Yang L, Deng X, Jiang S, He X, Liu Y, Yin C, Zhong N, Gao GF. 2017. Human infections with recently-emerging highly pathogenic H7N9 avian influenza virus in China. J Infect 75:71–75. doi: 10.1016/j.jinf.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhu W, Zhou J, Li Z, Yang L, Li X, Huang W, Zou S, Chen W, Wei H, Tang J, Liu L, Dong J, Wang D, Shu Y. 2017. Biological characterisation of the emerged highly pathogenic avian influenza (HPAI) A(H7N9) viruses in humans, in mainland China, 2016 to 2017. Euro Surveill 22:30533. doi: 10.2807/1560-7917.ES.2017.22.19.30533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kile JC, Ren R, Liu L, Greene CM, Roguski K, Iuliano AD, Jang Y, Jones J, Thor S, Song Y, Zhou S, Trock SC, Dugan V, Wentworth DE, Levine MZ, Uyeki TM, Katz JM, Jernigan DB, Olsen SJ, Fry AM, Azziz-Baumgartner E, Davis CT. 2017. Update: increase in human infections with novel Asian lineage avian influenza A(H7N9) viruses during the fifth epidemic–China, October 1, 2016–August 7, 2017. MMWR Morb Mortal Wkly Rep 66:928–932. doi: 10.15585/mmwr.mm6635a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su S, Gu M, Liu D, Cui J, Gao GF, Zhou J, Liu X. 2017. Epidemiology, evolution, and pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China. Trends Microbiol 25:713–728. doi: 10.1016/j.tim.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Yang L, Zhu W, Zhang Y, Zou S, Bo H, Gao R, Dong J, Huang W, Guo J, Li Z, Zhao X, Li X, Xin L, Zhou J, Chen T, Dong L, Wei H, Li X, Liu L, Tang J, Lan Y, Yang J, Shu Y. 2016. Two outbreak sources of influenza A (H7N9) viruses have been established in China. J Virol 90:5561–5573. doi: 10.1128/JVI.03173-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trock SC, Burke SA, Cox NJ. 2015. Development of framework for assessing influenza virus pandemic risk. Emerg Infect Dis 21:1372–1378. doi: 10.3201/eid2108.141086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke SA, Trock SC. 2018. Use of influenza risk assessment tool for prepandemic preparedness. Emerg Infect Dis 24:471–477. doi: 10.3201/eid2403.171852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connor RJ, Kawaoka Y, Webster RG, Paulson JC. 1994. Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology 205:17–23. doi: 10.1006/viro.1994.1615. [DOI] [PubMed] [Google Scholar]
- 13.Matrosovich M, Tuzikov A, Bovin N, Gambaryan A, Klimov A, Castrucci MR, Donatelli I, Kawaoka Y. 2000. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J Virol 74:8502–8512. doi: 10.1128/JVI.74.18.8502-8512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholtissek C, Rohde W, Von Hoyningen V, Rott R. 1978. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 87:13–20. doi: 10.1016/0042-6822(78)90153-8. [DOI] [PubMed] [Google Scholar]
- 15.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bean WJ, Schell M, Katz J, Kawaoka Y, Naeve C, Gorman O, Webster RG. 1992. Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J Virol 66:1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reid AH, Taubenberger JK. 2003. The origin of the 1918 pandemic influenza virus: a continuing enigma. J Gen Virol 84:2285–2292. doi: 10.1099/vir.0.19302-0. [DOI] [PubMed] [Google Scholar]
- 18.Rogers GN, D'Souza BL. 1989. Receptor binding properties of human and animal H1 influenza virus isolates. Virology 173:317–322. doi: 10.1016/0042-6822(89)90249-3. [DOI] [PubMed] [Google Scholar]
- 19.Rogers GN, Daniels RS, Skehel JJ, Wiley DC, Wang XF, Higa HH, Paulson JC. 1985. Host-mediated selection of influenza virus receptor variants. Sialic acid-alpha 2,6Gal-specific clones of A/duck/Ukraine/1/63 revert to sialic acid-alpha 2,3Gal-specific wild type in ovo. J Biol Chem 260:7362–7367. [PubMed] [Google Scholar]
- 20.de Vries RP, Peng W, Grant OC, Thompson AJ, Zhu X, Bouwman KM, de la Pena ATT, van Breemen MJ, Ambepitiya Wickramasinghe IN, de Haan CAM, Yu W, McBride R, Sanders RW, Woods RJ, Verheije MH, Wilson IA, Paulson JC. 2017. Three mutations switch H7N9 influenza to human-type receptor specificity. PLoS Pathog 13:e1006390. doi: 10.1371/journal.ppat.1006390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H, Carney PJ, Chang JC, Villanueva JM, Stevens J. 2013. Structural analysis of the hemagglutinin from the recent 2013 H7N9 influenza virus. J Virol 87:12433–12446. doi: 10.1128/JVI.01854-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H, Carney PJ, Donis RO, Stevens J. 2012. Structure and receptor complexes of the hemagglutinin from a highly pathogenic H7N7 influenza virus. J Virol 86:8645–8652. doi: 10.1128/JVI.00281-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Carney PJ, Chang JC, Villanueva JM, Stevens J. 2015. Structure and receptor binding preferences of recombinant hemagglutinins from avian and human H6 and H10 influenza A virus subtypes. J Virol 89:4612–4623. doi: 10.1128/JVI.03456-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzarum N, de Vries RP, Peng W, Thompson AJ, Bouwman KM, McBride R, Yu W, Zhu X, Verheije MH, Paulson JC, Wilson IA. 2017. The 150-loop restricts the host specificity of human H10N8 influenza virus. Cell Rep 19:235–245. doi: 10.1016/j.celrep.2017.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brownlee GG, Fodor E. 2001. The predicted antigenicity of the haemagglutinin of the 1918 Spanish influenza pandemic suggests an avian origin. Philos Trans R Soc Lond B Biol Sci 356:1871–1876. doi: 10.1098/rstb.2001.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiley DC, Wilson IA, Skehel JJ. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- 27.WHO. 2017. Zoonotic influenza viruses: antigenic and genetic characteristics and development of candidate vaccine viruses for pandemic preparedness. Wkly Epidemiol Rec 92:129–144. [PubMed] [Google Scholar]
- 28.Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, McCauley JW, Collins PJ, Walker PA, Skehel JJ, Gamblin SJ. 2013. Receptor binding by an H7N9 influenza virus from humans. Nature 499:496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 29.Dortmans JC, Dekkers J, Wickramasinghe IN, Verheije MH, Rottier PJ, van Kuppeveld FJ, de Vries E, de Haan CA. 2013. Adaptation of novel H7N9 influenza A virus to human receptors. Sci Rep 3:3058. doi: 10.1038/srep03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H, Chen LM, Carney PJ, Donis RO, Stevens J. 2010. Structures of receptor complexes of a North American H7N2 influenza hemagglutinin with a loop deletion in the receptor binding site. PLoS Pathog 6:e1001081. doi: 10.1371/journal.ppat.1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinstein J, de Souza-e Silva U, Paulson JC. 1982. Sialylation of glycoprotein oligosaccharides N-linked to asparagine. Enzymatic characterization of a Gal beta 1 to 3(4)GlcNAc alpha 2 to 3 sialyltransferase and a Gal beta 1 to 4GlcNAc alpha 2 to 6 sialyltransferase from rat liver. J Biol Chem 257:13845–13853. [PubMed] [Google Scholar]
- 32.Imai M, Watanabe T, Kiso M, Nakajima N, Yamayoshi S, Iwatsuki-Horimoto K, Hatta M, Yamada S, Ito M, Sakai-Tagawa Y, Shirakura M, Takashita E, Fujisaki S, McBride R, Thompson AJ, Takahashi K, Maemura T, Mitake H, Chiba S, Zhong G, Fan S, Oishi K, Yasuhara A, Takada K, Nakao T, Fukuyama S, Yamashita M, Lopes TJS, Neumann G, Odagiri T, Watanabe S, Shu Y, Paulson JC, Hasegawa H, Kawaoka Y. 2017. A highly pathogenic avian H7N9 influenza virus isolated from a human is lethal in some ferrets infected via respiratory droplets. Cell Host Microbe 22:615–626. doi: 10.1016/j.chom.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Kiso M, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 34.Chen LM, Blixt O, Stevens J, Lipatov AS, Davis CT, Collins BE, Cox NJ, Paulson JC, Donis RO. 2012. In vitro evolution of H5N1 avian influenza virus toward human-type receptor specificity. Virology 422:105–113. doi: 10.1016/j.virol.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yen HL, Lipatov AS, Ilyushina NA, Govorkova EA, Franks J, Yilmaz N, Douglas A, Hay A, Krauss S, Rehg JE, Hoffmann E, Webster RG. 2007. Inefficient transmission of H5N1 influenza viruses in a ferret contact model. J Virol 81:6890–6898. doi: 10.1128/JVI.00170-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maines TR, Chen LM, Van Hoeven N, Tumpey TM, Blixt O, Belser JA, Gustin KM, Pearce MB, Pappas C, Stevens J, Cox NJ, Paulson JC, Raman R, Sasisekharan R, Katz JM, Donis RO. 2011. Effect of receptor binding domain mutations on receptor binding and transmissibility of avian influenza H5N1 viruses. Virology 413:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Poucke S, Doedt J, Baumann J, Qiu Y, Matrosovich T, Klenk HD, Van Reeth K, Matrosovich M. 2015. Role of substitutions in the hemagglutinin in the emergence of the 1968 pandemic influenza virus. J Virol 89:12211–12216. doi: 10.1128/JVI.01292-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L, Chen E, Bao C, Xiang N, Wu J, Wu S, Shi J, Wang X, Zheng Y, Zhang Y, Ren R, Greene CM, Havers F, Iuliano AD, Song Y, Li C, Chen T, Wang Y, Li D, Ni D, Zhang Y, Feng Z, Uyeki TM, Li Q. 17 February 2018. Clusters of human infection and human-to-human transmission of avian influenza A(H7N9) virus, 2013–2017. Emerg Infect Dis doi: 10.3201/eid2402.171565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrauwen EJ, Fouchier RA. 2014. Host adaptation and transmission of influenza A viruses in mammals. Emerg Microbes Infect 3:e9. doi: 10.1038/emi.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann G, Kawaoka Y. 2015. Transmission of influenza A viruses. Virology 479–480:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L, Bao L, Deng W, Zhu H, Li F, Chen T, Lv Q, Yuan J, Xu Y, Li Y, Yao Y, Gu S, Yu P, Chen H, Qin C. 2014. Rapid adaptation of avian H7N9 virus in pigs. Virology 452–453:231–236. [DOI] [PubMed] [Google Scholar]
- 42.Kida H, Ito T, Yasuda J, Shimizu Y, Itakura C, Shortridge KF, Kawaoka Y, Webster RG. 1994. Potential for transmission of avian influenza viruses to pigs. J Gen Virol 75(Part 9):2183–2188. [DOI] [PubMed] [Google Scholar]
- 43.De Vleeschauwer A, Van Poucke S, Braeckmans D, Van Doorsselaere J, Van Reeth K. 2009. Efficient transmission of swine-adapted but not wholly avian influenza viruses among pigs and from pigs to ferrets. J Infect Dis 200:1884–1892. doi: 10.1086/648475. [DOI] [PubMed] [Google Scholar]
- 44.Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 45.Chayen NE, Shaw-Steward PD, Blow DM. 1992. Microbatch crystallization under oil–a new technique allowing many small volume crystallization experiments. J Cryst Growth 122:176–180. [Google Scholar]
- 46.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winn MD, Isupov MN, Murshudov GN. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57:122–133. doi: 10.1107/S0907444900014736. [DOI] [PubMed] [Google Scholar]
- 51.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H, Carney PJ, Mishin VP, Guo Z, Chang JC, Wentworth DE, Gubareva LV, Stevens J. 2016. Molecular characterizations of surface proteins hemagglutinin and neuraminidase from recent H5Nx avian influenza viruses. J Virol 90:5770–5784. doi: 10.1128/JVI.00180-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeLano WL. 2002. The PyMol molecular graphics systems. DeLano Scientific, San Carlos, CA, USA. [Google Scholar]