

MicroRNAs are expressed by diverse viruses, but for only a few is there any understanding of their in vivo function. PyVs can cause serious disease in immunocompromised hosts. Therefore, increased knowledge of how these viruses interact with the immune response is of clinical relevance. Here we show a novel activity for a viral miRNA locus in promoting virus shedding. This work indicates that in addition to any role for the PyV miRNA locus in long-term persistence, it also has biological activity during the acute phase. As this mutant phenotype is alleviated by infection of mice lacking an adaptive immune response, our work also connects the in vivo activity of the PyV miRNA locus to the immune response. Given that PyV-associated disease is associated with alterations in the immune response, our findings help to better understand how the balance between PyVs and the immune response becomes altered in pathogenic states.
KEYWORDS: polyomavirus, miRNA, viral shedding, viral persistence, viruria
ABSTRACT
Polyomaviruses (PyVs) can cause serious disease in immunosuppressed hosts. Several pathogenic PyVs encode microRNAs (miRNAs), small RNAs that regulate gene expression via RNA silencing. Despite recent advances in understanding the activities of PyV miRNAs, the biological functions of PyV miRNAs during in vivo infections are mostly unknown. The studies presented here used murine polyomavirus (MuPyV) as a model to assess the roles of the PyV miRNAs in a natural host. This analysis revealed that a MuPyV mutant that is unable to express miRNAs has enhanced viral DNA loads in select tissues at late times after infection. This is consistent with the PyV miRNAs functioning to reduce viral replication during the persistent phase of infection in a natural host. Additionally, the MuPyV miRNA locus promotes viruria during the acute phase of infection as evidenced by a defect in shedding during infection with the miRNA mutant virus. The viruria defect of the miRNA mutant virus could be rescued by infecting Rag2−/− mice. These findings implicate the miRNA locus as functioning in both the persistent and acute phases of infection and suggest a role for MuPyV miRNA in evading the adaptive immune response.
IMPORTANCE MicroRNAs are expressed by diverse viruses, but for only a few is there any understanding of their in vivo function. PyVs can cause serious disease in immunocompromised hosts. Therefore, increased knowledge of how these viruses interact with the immune response is of clinical relevance. Here we show a novel activity for a viral miRNA locus in promoting virus shedding. This work indicates that in addition to any role for the PyV miRNA locus in long-term persistence, it also has biological activity during the acute phase. As this mutant phenotype is alleviated by infection of mice lacking an adaptive immune response, our work also connects the in vivo activity of the PyV miRNA locus to the immune response. Given that PyV-associated disease is associated with alterations in the immune response, our findings help to better understand how the balance between PyVs and the immune response becomes altered in pathogenic states.
INTRODUCTION
The polyomavirus (PyV) family is comprised of a large number of viruses that predominantly infect vertebrates (1). The founding member of the PyVs, murine polyomavirus (MuPyV), has been the most tractable in vivo model for studying the PyV life cycle and has played an important role in understanding PyV-mediated transformation and the antiviral response (2, 3, 4). MuPyV is thought to be transmitted to newborn mice via a respiratory route, possibly via inhalation of urine containing virions (5, 6, 7). Primary replication, which occurs at 1 to 6 days postinfection (p.i.) in nonciliated epithelial clara cells of the lungs (7), is followed by dissemination and replication in secondary tissues at 7 to 12 days postinfection (6, 8). Viral replication has been observed in various secondary organs, including kidneys, salivary glands, spleen, lymph nodes, heart, liver, skin, lungs, and mammary glands (8, 9, 10). Virus replication peaks at around 12 days after infection, concurrent with a rise in viral shedding in the urine and saliva (6). A rise in anti-MuPyV antibody titers occurs at 7 to 15 days p.i. and precedes clearance of detectable virus in most organs by 22 to 30 days (8).
A reduction in viral loads marks the transition from the acute phase to the persistent phase, which is generally defined by the continued presence of viral DNA (vDNA) in tissues (3). Viral DNA is maintained in select tissues, such as bone marrow, spleen, kidney, salivary glands, and mammary glands (8, 9). The viral DNA in these tissues is thought to represent a persistent reservoir of “smoldering” viral replication in semipermissive cells (4), although a latent state has yet to be ruled out. A change in the microenvironment (e.g., tissue damage), immune status (e.g., pregnancy or immunosuppression), or hormone levels (e.g., pregnancy or stress), can “reactivate” or increase replication of MuPyV from persistent reservoirs (11, 12). This results in episodic shedding of MuPyV in urine and saliva, which contaminates the surrounding environment with infectious virus (6). Similar observations have been made for human PyVs, whereby increased viruria during pregnancy coincides with a rise in anti-PyV neutralizing antibodies (13, 14, 15).
Many polyomaviruses, including MuPyV, raccoon polyomavirus (RacPyV), simian virus 40 (SV40), JC polyomavirus (JCPyV), Merkel cell polyomavirus (MCPyV), and BK polyomavirus (BKPyV), encode microRNAs (miRNAs) (16, 17, 18, 19, 20). miRNAs are small regulatory RNAs (∼22 nucleotides [nt]) that repress gene expression via RNA silencing (21). The PyV miRNAs are encoded in the late orientation, opposite to the T antigen mRNAs. Consequently, miRNAs are perfectly complementary to the T antigen transcripts, permitting Argonaute 2 (Ago2)-mediated cleavage of these mRNAs. This results in a reduction in T antigen protein levels late in infection, a conserved function of PyV miRNAs (16, 17, 18, 19, 20, 22). Despite established roles of T antigen in promoting viral replication, miRNA-null laboratory strains of SV40 and MuPyV replicate at rates similar to those of their wild-type counterparts under standard cell culture conditions (16, 17). However, miRNAs expressed by nonrearranged BKPyV strains, which more closely resemble circulating BKPyV in humans and express lower levels of the early transcripts, are able to downregulate T antigen levels to sufficiently reduce viral replication in primary renal proximal tubule epithelial cells (23). Consistent with these results, MCPyV miRNA promotes long-term persistence by inhibiting DNA replication in a cell culture model (24), and the SV40 miRNAs reduce persistent SV40 DNA loads in Syrian golden hamster tissues (25). These studies suggest that the PyV miRNAs reduce virus replication by downregulating T antigen expression. However, the effects of miRNA expression on virus replication in a natural host have yet to be established.
In addition to regulating viral replication, independent studies indicate a role for the PyV miRNAs in inhibiting the host immune response. Downregulation of T antigen by the SV40 miRNAs decreases CD8 T-cell-mediated lysis in cell culture (16). miRNA-mediated downregulation of the host ULBP3 stress-induced ligand has been suggested to reduce killing of BKPyV- and JCPyV-infected cells by natural killer cells (26). MuPyV miRNAs reduce Smad2-mediated apoptosis during infection (27). These studies support a model whereby the PyV miRNAs modulate viral and host transcripts in order to extend the life of infected cells by reducing innate and adaptive immune responses.
Despite advances in understanding the expression and activities of PyV miRNAs, their functions during the viral life cycle in a natural host are poorly understood. In this study, we used MuPyV as a model to investigate the role of the PyV miRNA locus in viral dissemination, replication, and shedding during the acute and persistent phases of infection in mice. This work reveals activities of the MuPyV miRNA locus in both the persistent and acute phases of infection and suggests a link between miRNA function and the adaptive immune response.
RESULTS
Sequencing of the MuPyV miRNAs.
MuPyV miRNAs were initially identified via secondary-structure predictions and confirmed indirectly (17). This approach could only estimate the 5′ and 3′ termini of the derivative miRNAs. It also did not resolve whether MuPyV contains additional miRNA loci. To better characterize the MuPyV miRNAs, we conducted high-throughput sequencing of small RNA from NIH 3T3 cells infected with the MuPyV PTA strain (Fig. 1). These results mapped with precision the termini of the MuPyV miRNAs and confirmed that only the single, previously identified pre-miRNA locus gives rise to MuPyV-encoded miRNA derivatives. We next used high-throughput sequencing to determine if the MuPyV miRNA is detectable during in vivo infection. Six mice were infected with MuPyV PTA, and kidneys were harvested from each mouse at 7 days p.i. during the acute phase and at 4 weeks p.i. during the persistent phase. This analysis revealed that all of the mice from the acute phase and one of the mice from the persistent phase were positive for detectable MuPyV miRNA (Table 1). The miRNA detected was the same dominant derivative detected in infected cultured cells (Fig. 1). Because all of these in vivo results were near the limit of detection of our assay, these results do not rule out expression in any samples, but they do conclusively demonstrate that the miRNA is detectable in the kidney at different times postinfection. We conclude that the MuPyV miRNA is specifically processed and detectable in vivo in at least some samples during the acute and persistent phases of infection.
FIG 1.

Identification of MuPyV miRNAs by small RNA sequencing. NIH 3T3 cells were infected with MuPyV PTA at an MOI of 0.1. A small RNA library was prepared at 7 days p.i. Circles on the graph represent the read counts of the 5′ ends of small RNA reads mapping to the MuPyV PTA genome (NCBI accession number U27812), positioned on the x axis. The arrow below the graph represents the pre-miRNA hairpin position and direction. The MuPyV PTA reference sequence encoding the miRNAs is indicated. Sequences of the three most abundant small RNAs mapping to the 5′ (bold/red) and 3′ (bold/blue) arms of the pre-miRNA hairpin are shown aligned to the pre-miRNA. Numbers represent the read counts.
TABLE 1.
Small RNA-seq results for detection of MuPyV miRNA in kidney
| ID | Wk p.i. | Detection |
|---|---|---|
| Ex2-4 | 1 | + |
| Ex2-5 | 1 | + |
| Ex2-6 | 1 | + |
| Ex2-14 | 4 | + |
| Ex2-15 | 4 | − |
| Ex2-16 | 4 | − |
MuPyV miRNAs correlate with reduced vDNA loads during the persistent phase of infection.
PTA-dl1013 is a deletion mutant of the MuPyV PTA strain that does not express miRNAs (17). Importantly, although PTA-dl1013 contains a small deletion mutation that eliminates miRNA expression, it has no observable effects on early gene function as assayed by virus yield or transformation activity (17). Previous studies reported that adult C57BL/6 mice infected with PTA-dl1013 maintained vDNA loads comparable to or slightly (up to 5-fold) higher than those of PTA in spleen and kidney during the acute phase of infection (up to 34 days p.i.) (17). To confirm these results and determine whether the miRNAs function at later times postinfection, we examined MuPyV DNA loads by quantitative PCR (qPCR) in spleens, kidneys, and livers of adult C57BL/6 mice infected with 105 IU of PTA or PTA-dl1013 via intraperitoneal (i.p.) injection during the acute (1 and 4 weeks p.i.) and persistent (10, 16, and 28 weeks p.i.) phases of infection (Fig. 2).
FIG 2.

Quantitation of PTA and PTA-dl1013 during the acute and persistent phases of infection. Adult C57BL/6 female mice were inoculated with 1 × 105 IU of either PTA (white circles) or PTA-dl1013 (gray circles) via i.p. injection, and genome levels in spleen, kidney, and liver were quantified at different times postinfection by using a TaqMan probe and primers that recognize both genomes. The limit of detection for the qPCRs was 10 copies. The input DNA amounts were 20 ng (at 1, 4, and 10 weeks p.i.), 40 ng (at 16 weeks), or 200 ng (at 28 weeks). Genome copy numbers were normalized per 1 μg. Circles represent individual animals. The black bars represent the average genome copy number. The gray bars represent the limit of detection (LOD) of the assay after normalization. Samples that were below the limit of detection are graphed at the limit of detection. P values were calculated using the Mann-Whitney U test. n.t., not tested.
High virus yields (2 × 105 to 5 × 106 genome equivalents/μg DNA) were observed in spleen and kidney after 1 and 4 weeks of infection with PTA, consistent with robust virus replication during the acute phase of replication (Fig. 2). Lower levels were observed in liver during these times (Fig. 2). Virus loads diminished to ≤103 genome equivalents/μg DNA at later times after infection (16 and 28 weeks p.i.), consistent with a low level of virus replication during the persistent phase of replication. These levels were only slightly above the limit of detection of our PCR assay, even when increased input DNA was assayed.
In comparison to PTA levels, PTA-dl1013 levels were 4.1-fold lower in spleen, but not kidney, at 1 week p.i. (Fig. 2, closed symbols). No significant differences in PTA and PTA-dl1013 DNA levels were detected at 4 weeks or 10 weeks after infection. However, at 16 weeks, PTA-dl1013 genome levels were approximately 10-fold higher than PTA levels in spleen and kidney. By 28 weeks, only low levels of both PTA and PTA-dl1013 genomes were detected, and no significant differences were observed. These results confirm that the miRNAs are not required for infection and replication in tissues during the acute phase of infection and indicate that miRNA expression correlates with reduced genomic DNA in certain tissues during the persistent phase of infection.
The results summarized in Fig. 2 reveal a substantial variation in viral copies between different mice. To minimize variations due to interhost differences, we compared levels of PTA and PTA-dl1013 in individual animals. C57BL/6 mice were coinfected with equivalent amounts of PTA and PTA-dl1013 by i.p. injection. Tissues were then harvested during the acute (2 and 4 weeks) and persistent (10 and 16 weeks) phases of infection, and genome levels were determined by strain-specific qPCR (Fig. 3A to C). Similar to the case for individually infected mice, we observed high levels of both PTA and PTA-dl1013 in tissues during the acute phase of infection, with MuPyV levels decreasing during the persistent phase of infection (Fig. 3D). We note that PTA was not detected in the spleen or kidney by 16 weeks p.i., while PTA-dl1013 was present at 103 to 104 copies/μg DNA (Fig. 3D).
FIG 3.

Quantitation of PTA and PTA-dl1013 in tissues of coinfected mice. (A) Schematic diagram of the qPCR strategy to specifically recognize and quantitate the PTA and PTA-dl1013 genomes. The PTA-specific probe was designed to recognize the 21-bp sequence deleted in PTA-dl1013. The PTA-dl1013-specific probe was designed to recognize the sequences upstream and downstream of the 21-bp deletion in PTA-dl1013. The primers recognize the sequences in both PTA and PTA-dl1013 flanking the probe-binding location. (B) Genome copy number standards of PTA and PTA-dl1013 were made by serial dilution of purified plasmids containing the PTA or PTA-dl1013 genome. qPCR analysis of PTA and PTA-dl1013 standards using the universal MuPyV primers/probe confirmed that the difference between the standards was less than one threshold cycle (CT) value at each dilution. (C) qPCR analysis of the PTA and PTA-dl1013 106 copy number standards using the PTA- and PTA-dl1013-specific probes demonstrated that PTA-dl1013 was undetected with the PTA probe, whereas PTA was largely undetected, or far less efficiently detected, with the PTA-dl1013 probe. This confirms that the PTA and PTA-dl1013 probes specifically recognize their respective genomes. n.d., samples in which CT values were undetermined. The CT values are the average from reactions run in triplicate, which were used to calculate the CT value and standard deviation (SD). (D) qPCR analysis of PTA and PTA-dl1013 genome levels in the spleens, kidneys, livers, and bladders of adult C57BL/6 mice inoculated with 5 × 104 IU of PTA and 5 × 104 IU of PTA-dl1013 via i.p. injection. Tissues were harvested at indicated time points. DNA was recovered and subjected to qPCR analysis using PTA- and PTA-dl1013-specific probes. The limit of detection (LOD) was 10 copies. Samples in which PTA or PTA-dl1013 were below the limit of detection were graphed at the limit of detection. The quantities were normalized to 1 μg of total DNA. P values were determined using the Mann-Whitney U test.
To compensate for variation in different animals, we calculated the ratio of PTA to PTA-dl1013 genomes in individual mice. This analysis revealed that during the acute phase of infection (2 weeks p.i.), the levels of PTA and PTA-dl1013 were comparable in spleen and kidney (Fig. 4). At later times, PTA-dl1013 copy numbers were 10-fold higher than PTA copy numbers in the spleen and kidney at weeks 10 and 16 and 100-fold higher in the kidney at week 16 (Fig. 4). We did not observe substantial differences in the liver but did observe 7-fold-higher PTA-dl1013 levels in the bladder at week 4 (Fig. 4). These results are consistent with the elevated levels of PTA-dl1013 detected in the spleen and kidney at 16 weeks following infections with the individual virus strains (Fig. 2) and suggest that MuPyV miRNA expression correlates with reduced viral loads in select tissues during the persistent phase of infection.
FIG 4.

Ratio of PTA and PTA-dl1013 in tissues of coinfected mice. The ratios of PTA and PTA-dl1013 genome copies from Fig. 3 in the indicated organs of individual mice coinfected with PTA and PTA-dl1013 are shown. Values below the limit of detection were set at the limit of detection. The circles represent log10(PTA/PTA-dl1013). P values were calculated using one-sample t test.
MuPyV miRNAs promote viruria during the acute phase of infection.
Early epidemiological studies of MuPyV revealed that high titers of infectious virus are excreted in the urine of mice during the acute phase of infection and then are episodically shed during the persistent phase of infection (6). The periodic shedding of MuPyV and many other PyVs in the urine is considered a primary route of transmission (6, 28). Therefore, we investigated whether the MuPyV miRNAs affects the magnitude and/or frequency of MuPyV shedding in the urine during the acute and persistent phases of infection. To test this, we collected urine specimens periodically postinfection and quantitated viral genomes. To assess whether PTA-dl1013 could maintain long-term persistence, we treated mice with cyclosporine at day 474 postinfection in order to immunosuppress the mice, which is correlated with increased PyV replication (4).
In the majority of mice individually infected with either PTA or PTA-dl1013, we observed vDNA shedding during the acute phase of infection (13 to 28 days p.i.) (Fig. 5), indicating that the MuPyV miRNAs are not required for establishing viruria. Both strains were episodically shed throughout the persistent phase (28 to 87 day p.i.), which demonstrates that the MuPyV miRNAs are not required for persistent viruria (Fig. 5). The magnitude and timing of virus shedding by individual mice infected varied substantially (Fig. 5), and thus no statistical differences between PTA and PTA-dl1013 levels could be determined.
FIG 5.

Quantitation of viruria of mice infected with either PTA or PTA-dl1013. Four C57BL/6 female mice were infected with 105 IU of either PTA or PTA-dl1013, and DNA genome copies in 2 μl urine were quantified at various times p.i. qPCR analysis was performed using the universal primer/probes set that recognize both the PTA and PTA-dl1013 genomes. (A and B) Genome equivalents in individual mice infected with wild-type (A) or PTA-dl1013 (B). Samples in which copy numbers were below the limit of detection (10 copies) were graphed at the limit of detection. (C) Average number of genome copies (± standard error [SE]) of PTA (circles) and PTA-dl1013 (triangles).
To circumvent the differences in shedding due to variations between individual mice, we tested whether the presence of the miRNA locus could correlate with viral shedding in mice coinfected with both PTA and PTA-dl1013. Adult C57BL/6 female mice were coinfected with equal amounts of the PTA and PTA-dl1013, and PTA and PTA-dl1013 DNAs were quantitated during the acute and persistent phases of infection (Fig. 6A and Supplemental Data File S1 in the supplemental material). We did not detect substantial quantities of either PTA or PTA-dl1013 genomes in the urine of ∼40% of the mice at any time point during the acute phase of infection (Supplemental Data File S1). The remaining “high-shedder” mice expressed readily detectable MuPyV DNA in the urine (Fig. 6A). MuPyV in these samples was detected at high levels beginning by 10 days p.i. and peaked between 13 to 27 days, after which the number of shedding mice and the magnitude of shedding began to decrease. Thereafter, we observed episodic shedding events throughout the persistent phase of infection (28 to 87 days p.i.). These findings reveal that while some mice rarely or never shed detectable virus under our assay conditions, ∼60% are competent for acute and persistent viruria.
FIG 6.

Quantitation of PTA and PTA-dl1013 DNA shedding in the urine. Mice were coinfected with 5 × 104 IU of PTA and 5 × 104 IU of PTA-dl1013 via i.p. injection, urine samples were collected at the indicated times, and strain-specific qPCR was used to quantify wild-type and mutant genomes. The numbers of mice tested at each time were 18 to 30 mice during days 1 to 27 p.i., 10 to 20 mice during days 28 to 55 p.i., and 4 mice during days 58 to 492 p.i. (A) Dot plot of genome equivalents of PTA (blue circles) and PTA-dl1013 (yellow triangles) in 2 μl of urine from mice that shed viral genome copies equal to or greater that the limit of detection of the qPCR assay. Mice that did not shed above the limit of detection or time points in which genomes were not detected were not included in this analysis. Lines represent the median values of PTA (solid blue) and PTA-dl1013) (dashed yellow). Significant differences (P ≤ 0.05) between PTA and PTA-dl1013 genome levels are marked by asterisks. (B) Comparison of the ratio of wild-type and mutant genomes in individual mice coinfected with PTA and PTA-dl1013. When only one genome (either PTA or PTA-dl1013) could be detected, the other genome was set at the limit of detection in order to calculate the PTA/PTA-dl1013 ratio. The line represents the average log10(PTA/PTA-dl1013) ± standard error of the mean (SEM). P values were calculated by using a one-sample t test (*, P ≤ 0.05).
The median PTA levels were significantly higher (5- to 100-fold) than the median PTA-dl1013 levels during the majority of the acute phase of infection (10, 13, 15, 17, 20, and 27 days p.i.) in coinfected mice (Fig. 6A). To minimize the variance in individual mice, we determined the ratio of PTA DNA to PTA-dl1013 DNA in each individual mouse. These data confirmed that PTA was shed at significantly higher levels on average (∼10-fold) than PTA-dl1013 during days 10 to 17 of the acute phase of infection in individual coinfected mice (Fig. 6B). We did not observe statistically significant differences in the magnitude of viral shedding at the majority of times tested during the persistent phase of infection (Fig. 6A and B and Supplemental Data File S1). Although this may be due to high variance in the timing and the magnitude of shedding, as well as the lower number of mice tested, this suggests that the MuPyV miRNAs do not promote viruria during the persistent phase of infection. In fact, we observed 84 PTA-dl1013 shedding events that were above the limit of detection from days 34 through 87, compared to 33 for PTA (Fig. 6A). This implies that the MuPyV miRNAs may limit detectable shedding during the persistent phase of infection, consistent with the reduced PTA levels in tissues (Fig. 2 to 4). At very late times postinfection (days 467 to 492), we observed PTA and PTA-dl1013 shedding events in a small number of mice. Thus, the MuPyV miRNAs are not required to maintain long-term persistent infections. These data further indicate that the MuPyV miRNA locus promotes viral shedding during the acute phase of infection but limits the number of MuPyV shedding events during the persistent phase of infection.
The shedding defect of miRNA-null MuPyV is mitigated in Rag2−/− mice.
The host immune response plays an important role in controlling MuPyV infection (2–4), and two lines of evidence suggest that PyV miRNAs may function to reduce the adaptive immune response. First, miRNA-mediated downregulation of T-antigens decreases susceptibility to cytotoxic T-cell-mediated lysis to SV40-infected cultured cells (16). Second, miRNA-null strains of SV40 and JCPyV have been isolated only from severely immunocompromised hosts, suggesting that an active immune response provides selective pressure to maintain miRNA expression (29). Since the absence of the MuPyV miRNA results in decreased virus shedding during the acute phase of infection, we tested whether the defect in acute viral shedding of PTA-dl1013 is mitigated in C57BL/6 Rag2−/− mice, which lack mature T and B cells. C57BL/6 Rag2−/− mice were coinfected with PTA and PTA-dl1013, and the shedding of DNA in urine was monitored. Unlike wild-type mice, where ∼40% of animals did not shed detectable vDNA during the acute phase of infection, all infected C57BL/6 Rag2−/− mice shed detectable levels of MuPyV by 10 days postinfection (Fig. 7 and Supplemental Data File S1). Levels of both PTA and PTA-dl1013 DNAs in urine were found to continually increase in the coinfected C57BL/6 Rag2−/− mice up to 20 days p.i., whereas MuPyV levels leveled off and began to decrease by 20 days p.i. (Fig. 7). These observations are consistent with the host adaptive immune response being a major factor that controls MuPyV shedding. Importantly, unlike in wild-type C57BL/6 mice, C57BL/6 Rag2−/− mice shed PTA-dl1013 DNA at levels similar to those for PTA during the acute phase of infection (10 to 13 days p.i.) (Fig. 7). These data indicate that the MuPyV miRNA locus promotes viruria during the acute phase of infection in a manner that is dependent on the host having an intact adaptive immune response.
FIG 7.
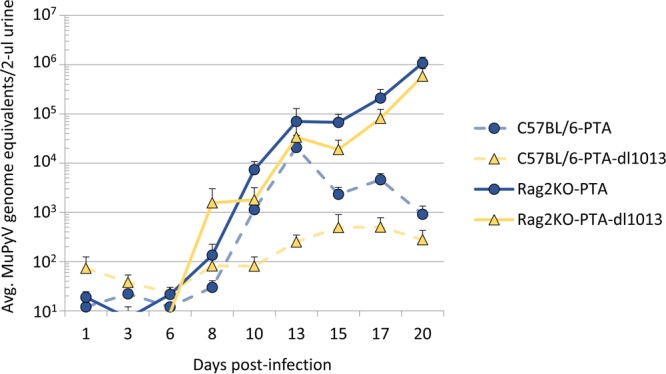
Analysis of PTA and PTA-dl1013 shedding in C57BLC57bl/6 Rag2−/− mice. Nine female C57BL/6-RAG2−/− mice were coinfected with 5 ×104 IU of PTA and 5 ×104 IU of PTA-dl1013 via i.p. injection. Urine was collected during the acute phase of infection, and genomic DNA was quantified by strain-specific qPCR analysis. Values for wild-type C57BL/6 mice coinfected with PTA (blue circles/dashed lines) and PTA-dl1013 (yellow triangles/dashed line) viruses are from Fig. 6. PTA genomes (blue circles/solid line) and PTA-dl1013 virus (yellow triangles/solid line) from C57BL/6-Rag2−/− mice are presented. Plots represent the average (±SEM; n = 8 or 9) at the indicated times.
DISCUSSION
The natural in vivo functions of PyV miRNAs are poorly understood. The results presented here reveal that, consistent with previous studies in cell culture models and a nonnatural host (23–25), the PyV miRNA locus functions to reduce viral loads during persistent infection of a natural host. In addition, we uncovered novel effects of the MuPyV miRNA locus on viruria during the persistent and acute phases of infection that provide new insights into PyV miRNA biology.
Similar to the results of previous studies (17), we did not observe large differences in the gross levels of the PTA (wild-type) and PTA-dl1013 (miRNA-null) strains of MuPyV in tissues during the acute phase of infection (weeks 1, 2, and 4 p.i.). This further confirms that the MuPyV miRNAs are not required for acute viral infection or replication in vivo under laboratory conditions. However, at weeks 10 and 16 p.i., we observed higher levels of PTA-dl1013 DNA, relative to PTA DNA, in the kidneys and spleens of mice following single infection or coinfection with PTA and PTA-dl1013 (Fig. 2 to 4). Consistent with the PyV literature (8, 25), our results (Table 1) suggest that an extreme minority of cells per organ are being infected. Our data show that the miRNA is made in vivo in all of the acute infected samples assayed and in at least one of the three persistently infected mice assayed (Table 1). Because the small transcriptome sequencing (RNA-seq) miRNA detection assay we employed is near the limit of detection, we expect that our inability to detect the miRNA in 2 of the 3 persistently infected samples is likely a technical limitation as opposed to the miRNA not being expressed/functioning at these times postinfection. In support of this interpretation, Zhang et al. (25) have previously shown that Syrian golden hamsters infected with SV40 miRNA mutant viruses also display higher virus loads during the persistent phase of infection. These results are consistent with the notion that miRNA-mediated downregulation of T antigen reduces virus replication in specific tissues/cell types at late times after infection to promote viral persistence (23, 24).
Our assessment of MuPyV viruria revealed that the MuPyV miRNAs are not required to establish or maintain long-term viral persistence (Fig. 5 and 6). However, we did observe more PTA-dl1013 than PTA shedding events during the persistent phase of infection (34 to 87 days p.i.) (Fig. 6A and Supplemental Data File S1 in the supplemental material). This is consistent with increased PTA-dl1013 levels in tissues and further indicates that the MuPyV miRNAs may limit viral replication events during the persistent phase of infection. Though the function of this activity remains unclear, it may be important to prevent priming of the host immune response in order to transmit to naive hosts more efficiently. Alternatively, this activity could prevent virus-associated pathologies in the host, which would presumably decrease viral fitness. Future experiments will address the relevant role of miRNA-mediated reduction of viral loads in urine and tissues during the persistent phase of infection.
During the acute phase of infection (days 10 to 20) in wild-type C57BL/6 mice, only ∼60% of mice shed substantial levels of MuPyV DNA (Fig. 6 and 7 and Supplemental Data File S1). This is in contrast to infections in immunodeficient C57BL/6 Rag2−/− mice, in which 100% of mice shed substantial levels of MuPyV DNA (Fig. 7 and Supplemental Data File S1). This suggests that the adaptive immune response is a major factor controlling viruria. Analysis of wild-type C57BL/6 mice coinfected with PTA and PTA-dl1013 revealed that PTA-dl1013 DNA was shed at significantly lower levels than PTA (Fig. 6), which indicates that the MuPyV miRNAs promote viral shedding at early times postinfection. Importantly, the defect in PTA-dl1013 was largely mitigated during infection of immunodeficient C57BL/6 Rag2−/− mice (Fig. 7), suggesting that the MuPyV miRNAs help evade a component of the host adaptive immune response to promote viruria. We note that PTA-dl1013 shedding was not fully rescued by infecting C57BL/6 Rag2−/− mice, possibly indicating that the MuPyV miRNAs have additional activities (for example, reduction of smad2-mediated apoptosis [27]) in promoting viruria independent of the adaptive immune response. These combined data demonstrate that the MuPyV miRNA locus promotes viral shedding by at least indirectly interacting with the adaptive immune response. This activity could be important in the wild as a means to increase transmission of the virus to susceptible hosts during the acute phase of infection.
The exact mechanism(s) by which MuPyV miRNA expression enhances virus shedding in immunocompetent mice remains unclear. Because we observed miRNA locus-dependent promotion of shedding in coinfected immunocompetent mice, this indicates that MuPyV miRNAs may function via a cell autonomous mechanism to repress the effects of the adaptive immune response. This is consistent with previous findings that the SV40 miRNAs can reduce CD8 T-cell-mediated lysis. However, it is unclear why the miRNAs would promote viruria only during a narrow window of time postinfection. One possibility is that the MuPyV miRNAs increase dissemination to the target cells required for subsequent viruria, possibly by reducing the cytotoxic T lymphocyte (CTL) response to promote infection/replication in lymphocytes early during the acute phase of infection. In support of this, at 1 week p.i., but not at 2 and 4 weeks p.i., we observed ∼4-fold-higher PTA DNA levels specifically in the spleen (Fig. 2 to 4), indicating that the MuPyV miRNAs may promote infection of lymphocytes early during infection. Alternatively, the cell-mediated adaptive immune response to MuPyV may change over time, making the miRNAs less effective at promoting viruria. While the CD8+ T cell response to MuPyV is predominately to T antigen, the CD8+ T cell response has also been shown to be directed toward a VP2-derived nonamer peptide (30). Thus, as the infection progresses, miRNA-mediated downregulation of T antigens may become less effective at muting the CTL response as the host T cell response develops more so against the late VP proteins. Future studies are required to more precisely define the relevant components of the immune response important for hindering the miRNA mutant virus.
In summary, this study demonstrates functionality of the MuPyV miRNA locus during both the persistent and acute phases of infection and at least indirectly implicates miRNA function in altering some component of the adaptive immune response. Given the clinical relevance of understanding the interplay between polyomaviruses and the immune response, this work provides a useful, quantitative, and noninvasive system for future efforts at understanding PyV miRNA effects on adaptive immunity.
MATERIALS AND METHODS
Cell cultures and virus strains.
Primary baby mouse kidney (BMK) cell cultures were prepared from 2- to 4-week-old female C57BL/6 mice (Jackson Laboratories). Kidneys were excised, washed in phosphate-buffered saline (PBS), and incubated in Dulbecco modified Eagle medium (DMEM) containing 0.25% trypsin overnight at 4°C. Kidneys were homogenized, incubated in trypsin at 37°C with agitation, and suspended in DMEM plus 10% fetal bovine serum (FBS) (Cellgro). Cells were washed by centrifugation and plated at a density of 107 cells per 100-mm dish. NIH 3T3 cells were maintained in DMEM plus 10% FBS.
The PTA and PTA-dl1013 virus strains have been described previously (17). Virus was propagated by infecting primary BMK cells (80% confluent) with PTA or PTA-dl1013 at a multiplicity of infection (MOI) of 0.05. Virus stocks were prepared after 10 days by scraping the cells into a portion of the culture medium. The cell extracts were then subjected to three freeze/thaw cycles, clarified by centrifugation, and stored at −80°C.
Virus titers were determined by immunofluorescence. NIH 3T3 cells in 12- or 24-well plates at 80 to 90% confluence were infected with dilutions of the PTA and PTA-dl1013 virus stocks for 1 h. Infected cells were maintained in DMEM plus 2% FBS for 40 h and then fixed and permeabilized with 4% paraformaldehyde and 1% NP-40. After blocking with 10% goat serum overnight, cells were incubated with rabbit anti-PyV antibody (a gift from Richard Consigi) for 30 min. After three PBS washes, cells were stained with Cy3-goat anti-rabbit IgG (Abcam) for 10 min and washed. The numbers of total and stained cells per field were quantified by using an inverted fluorescence microscope (Leica), and infectious titers (IU/ml) were determined.
Animal infections and tissue analysis.
Female C57BL/6 (Jackson Laboratories) and C57BL/6 Rag2−/− mice were inoculated with 1 × 105 IU of either PTA or PTA-dl1013 via intraperitoneal (i.p.) injection between 4 and 5 weeks of age. For coinfections, mice were inoculated (i.p.) with 5 × 104 IU of PTA and 5 × 104 IU of PTA-dl1013. Cyclosporine was administered as described in reference 31. Tissues were harvested at the indicated times and flash frozen in liquid nitrogen. Homogenates were prepared by grinding with a mortar and pestle chilled with liquid nitrogen. DNA was extracted from 25 mg of tissue by using the DNeasy blood and tissue kit (Qiagen). Recovered DNA was quantitated by the NanoDrop procedure and diluted to 100 ng/μl or 10 ng/μl for qPCR analysis. Animals were cared for in accordance with protocols approved by the Institutional Animal Care and Use Committee (AUP-2017-00037) and consistent with National Institutes of Health guidelines for the care and use of animals.
Urine analysis.
To collect urine, mice were placed on wire-bottomed cages for 2 to 4 h. Urine was collected on Parafilm, and 50-μl aliquots (or the total if less than 50 μl) were used for DNA extraction with the QIAamp viral RNA minikit (Qiagen) following the manufacturer's protocol to remove PCR inhibitors. DNA was eluted in 60 μl of Tris-EDTA (TE) buffer. MuPyV copy numbers were normalized to that in 2 μl of urine.
qPCR.
Concentrated pBluescript-sk+PTA and pBluescript-sk+PTA-dl1013 vectors were obtained via Maxi Prep (Invitrogen). Plasmids were prepared at 85.5 ng/μl (1010 copies of PTA or PTA-dl1013 per μl) and then serially diluted. For analysis of tissues from individually infected mice, 20 ng of DNA (for samples taken 1, 4, and 10 weeks p.i.) or 200 ng of DNA (10 and 28 weeks p.i.) in 2 μl of TE buffer was added to an 8-μl reaction mixture containing 5 μl of 2× gene expression master mix (Applied Biosystems), 0.75 μM MuPyV sense primer (CGCACATACTGCTGGAAGAAGA), 1.0 μM MuPyV antisense primer (TCTTGGTCGCTTTCTGGATACAG), and 100 nM MuPyV TaqMan MGB (Applied Biosystems) probe (6-carboxyfluorescein [FAM]-ATCCTTGTGTTGCTGAGCCCGATGA-NFQ) as described previously (32). For specific detection of PTA and PTA-dl1013 in urine, 2 μl of DNA (of the 60 μl extracted unless otherwise indicated in Supplemental Data File S1 in the supplemental material) was added to an 8-μl reaction mixture containing 5 μl 2× gene expression master mix (Applied Biosystems), 0.75 μM PTA/PTA-dl1013 sense primer (GATGAGCTGGGGTACTTGT), 0.75 μM PTA/PTA-dl1013 antisense primer (TGTATCCAGAAAGCGACCAAG), and 100 nM PTA-specific TaqMan MGB (Applied Biosystems) probe (FAM-TAGGATGTCCAAATACAGATCCTC-NFQ) or PTA-dl1013-specific TaqMan MGB (Applied Biosystems) probe (FAM-CTCCGGTTCCATTGGCATGT-NFQ). The PTA and PTAdl1013 standards (107/μl to 101/μl) were confirmed by using the universal MuPyV primers/probe, which detect both PTA and PTA-dl1013, to ensure that the copy number of each standard was equal. Assays were performed using 384-well format plates in a ViiA 7 real-time PCR system (Applied Biosystems). The limit of detection was 10 copies.
Small RNA-seq.
For small RNA-seq of MuPyV miRNA in NIH 3T3 cells, NIH 3T3 cells were infected with MuPyV PTA at an MOI of 0.1. At 1 week p.i., small RNAs (<70 nt) were isolated using TRIzol reagent (Thermo Fisher Scientific) as described in reference 33. Small RNA libraries were prepared for Illumina small RNA-seq using the multiplex small RNA library preparation set (New England BioLabs) and sequenced on the Illumina HiSeq 2500 platform (Genomic Sequencing and Analysis Facility, University of Texas at Austin). Adapter sequences were removed from reads using FastX clipper from the FastX toolkit software (http://hannonlab.cshl.edu/fastx_toolkit). The preprocessed reads were then mapped to the reference MuPyV PTA reference genome (GenBank accession no. U27812.1) using the SHRiMP2 software package (34). Small RNA reads for the MuPyV miRNAs were quantitated using custom scripts. For small RNA-seq analysis of MuPyV-infected kidney tissue, total RNA was extracted with TRIzol from frozen mouse kidney tissue samples harvested at either 1 week or 4 weeks postinfection as indicated. Small RNA-seq libraries were prepared with the NEBNext Multiplex Small RNA Library Prep Set for Illumina (E7300) according to the manufacturer's instructions. The resulting libraries were sequenced on an Illumina NextSeq 500 single-read 75-nt-length run. The 3′ adapter sequences were trimmed from the fastq files with cutadapt (35). Trimmed reads were then mapped to the MuPyV strain PTA reference genomic sequence (GenBank accession no. U27812.1) using Bowtie 2 (36) with the options “–local –score-min G,16,1.” Only reads mapping perfectly to the MuPyV 3p miRNA were counted in the analysis.
Statistical analyses.
The one-sample t test (two tailed) and Mann-Whitney U test (two tailed) were performed using the Real Statistics resource package for Excel (http://www.real-statistics.com/).
Accession number(s).
All RNA-seq data can be accessed through NCBI BioProject PRJNA472371.
Supplementary Material
ACKNOWLEDGMENTS
The work described in this article was supported by a Burroughs Wellcome Investigators in Pathogenesis Award to C.S.S. and a grant from the Cancer Prevention and Research Institute of Texas (RP140842).
We thank Lauren Ehrlich and Edward Marcotte for reagents, Luis Villarreal for insights regarding viral persistence, and Aaron Stark for technical support.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02131-17.
REFERENCES
- 1.Johne R, Buck CB, Allander T, Atwood WJ, Garcea RL, Imperiale MJ, Major EO, Ramqvist T, Norkin LC. 2011. Taxonomical developments in the family Polyomaviridae. Arch Virol 156:1627–1634. doi: 10.1007/s00705-011-1008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin TL. 2001. Polyoma virus: old findings and new challenges. Virology 289:167–173. doi: 10.1006/viro.2001.1124. [DOI] [PubMed] [Google Scholar]
- 3.Ramqvist T, Dalianis T. 2009. Murine polyomavirus tumour specific transplantation antigens and viral persistence in relation to the immune response, and tumour development. Semin Cancer Biol 19:236–243. doi: 10.1016/j.semcancer.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Swanson PA, Lukacher AE, Szomolanyi-Tsuda E. 2009. Immunity to polyomavirus infection: the polyomavirus-mouse model. Semin Cancer Biol 19:244–251. doi: 10.1016/j.semcancer.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe WP, Hartley JW, Brodsky I, Huebner RJ, Law LW. 1958. Observations on the spread of mouse polyoma virus infection. Nature 182:1617–16179. [DOI] [PubMed] [Google Scholar]
- 6.Rowe WP. 1961. The epidemiology of mouse polyoma virus infection. Bacteriol Rev 25:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gottlieb K, Villarreal LP. 2000. The distribution and kinetics of polyomavirus in lungs of intranasally infected newborn mice. Virology 266:52–65. doi: 10.1006/viro.1999.0030. [DOI] [PubMed] [Google Scholar]
- 8.Dubensky TW, Villarreal LP. 1984. The primary site of replication alters the eventual site of persistent infection by polyomavirus in mice. J Virol 50:541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wirth JJ, Amalfitano A, Gross R, Oldstone MB, Fluck MM. 1992. Organ- and age-specific replication of polyomavirus in mice. J Virol 66:3278–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berke Z, Dalianis T. 1993. Persistence of polyomavirus in mice infected as adults differs from that observed in mice infected as newborns. J Virol 67:4369–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atencio IA, Shadan FF, Zhou XJ, Vaziri ND, Villarreal LP. 1993. Adult mouse kidneys become permissive to acute polyomavirus infection and reactivate persistent infections in response to cellular damage and regeneration. J Virol 67:1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCance DJ, Mims CA. 1979. Reactivation of polyoma virus in kidneys of persistently infected mice during pregnancy. Infect Immun 25:998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coleman DV, Gardner SD, Mulholland C, Fridiksdottir V, Porter AA, Lilford R, Valdimarsson H. 1983. Human polyomavirus in pregnancy. A model for the study of defense mechanisms to virus reactivation. Clin Exp Immunol 53:289–296. [PMC free article] [PubMed] [Google Scholar]
- 14.Eash S, Manley K, Gasparovic M, Querbes W, Atwood WJ. 2006. The human polyomaviruses. Cell Mol Life Sci 63:865–876. doi: 10.1007/s00018-005-5454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson PE, Field AM, Gardner SD, Coleman DV. 1981. Occurrence of IgM antibodies against BK and JC polyomaviruses during pregnancy. J Clin Pathol 34:674–679. doi: 10.1136/jcp.34.6.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435:682–686. doi: 10.1038/nature03576. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan CS, Sung CK, Pack CD, Grundhoff AT, Lukacher AE, Benjamin T, Ganem D. 2009. Murine polyomavirus encodes a microRNA that cleaves early RNA transcripts but is not essential for experimental infection. Virology 387:157–167. doi: 10.1016/j.virol.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seo GJ, Chen CJ, Sullivan CS. 2009. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 383:183–187. doi: 10.1016/j.virol.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Seo GJ, Fink LH, O'Hara B, Atwood WJ, Sullivan CS. 2008. Evolutionarily conserved function of a viral microRNA. J Virol 82:9823–9828. doi: 10.1128/JVI.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CJ, Cox JE, Azarm K, Wylie KN, Woolard KD, Pesavento PA, Sullivan CS. 2015. Identification of a polyomavirus microRNA highly expressed in tumors. Virology 476:43–53. doi: 10.1016/j.virol.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 22.Chen CJ, Cox JE, Kincaid RP, Martinez A, Sullivan CS. 2013. Divergent microRNA targetomes of closely related circulating strains of a polyomavirus. J Virol 87:11135–11147. doi: 10.1128/JVI.01711-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broekema NM, Imperiale MJ. 2013. miRNA regulation of BK polyomavirus replication during early infection. Proc Natl Acad Sci U S A 110:8200–8205. doi: 10.1073/pnas.1301907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Theiss JM, Günther T, Alawi M, Neumann F, Tessmer U, Fischer N, Grundhoff A. 2015. A comprehensive analysis of replicating Merkel cell polyomavirus genomes delineates the viral transcription program and suggests a role for mcv-miR-M1 in episomal persistence. PLoS Pathog 11:e1004974. doi: 10.1371/journal.ppat.1004974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Sroller V, Zanwar P, Chen CJ, Halvorson SJ, Ajami NJ, Hecksel CW, Swain JL, Wong C, Sullivan CS, Butel JS. 2014. Viral microRNA Effects on pathogenesis of polyomavirus SV40 infections in Syrian golden hamsters. PLoS Pathog 10:e1003912. doi: 10.1371/journal.ppat.1003912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauman Y, Nachmani D, Vitenshtein A, Tsukerman P, Drayman N, Stern-Ginossar N, Lankry D, Gruda R, Mandelboim O. 2011. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe 9:93–102. doi: 10.1016/j.chom.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Sung SK, Yim H, Andrews E, Benjamin TL. 2014. A mouse polyomavirus-encoded microRNA targets the cellular apoptosis pathway through Smad2 inhibition. Virology 468-470:57–62. doi: 10.1016/j.virol.2014.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gosert R, Rinaldo CH, Funk GA, Egli A, Ramos E, Drachenberg CB, Hirsch HH. 2008. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J Exp Med 205:841–852. doi: 10.1084/jem.20072097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CJ, Burke JM, Kincaid RP, Azarm KD, Mireles N, Butel JS, Sullivan CS. 2014. Naturally arising strains of polyomaviruses with severely attenuated microRNA expression. J Virol 88:12683–12693. doi: 10.1128/JVI.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swanson PA, Pack CD, Hadley A, Wang C-R, Stroynowski I, Jensen PE, Lukacher AE. 2008. An MHC class Ib–restricted CD8 T cell response confers antiviral immunity. J Exp Med 205:1647–1657. doi: 10.1084/jem.20080570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gattazzo F, Molon S, Morbidoni V, Braghetta P, Blaauw B, Urciuolo A, Bonaldo P. 2014. Cyclosporin A promotes in vivo myogenic response in collagen VI-deficient myopathic mice. Front Aging Neurosci 6:244. doi: 10.3389/fnagi.2014.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kemball CC, Lee ED, Vezys V, Pearson TC, Larsen CP, Lukacher AE. 2005. Late priming and variability of epitope-specific CD8+ T cell responses during a persistent virus infection. J Immunol 174:7950–7960. doi: 10.4049/jimmunol.174.12.7950. [DOI] [PubMed] [Google Scholar]
- 33.Burke JM, Kuny CV, Kincaid RP, Sullivan CS. 2015. Identification, validation, and characterization of noncanonical miRNAs. Methods 91:57–68. doi: 10.1016/j.ymeth.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol 5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 36.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.