

T-20 remains the only membrane fusion inhibitor available for the treatment of viral infection, but its relatively low anti-HIV activity and genetic barrier for drug resistance have significantly limited its clinical application. Here we report two new lipopeptide-based fusion inhibitors (LP-50 and LP-51) showing extremely potent inhibitory activities against diverse HIV-1, HIV-2, SIV, and T-20-resistant variants. Promisingly, both inhibitors exhibited potent and long-lasting ex vivo anti-HIV activity and could efficiently suppress viral loads to undetectable levels in SHIV-infected monkey models. We believe that LP-50 and LP-51 are the most potent and broad-spectrum fusion inhibitors known to date and thus have high potential for clinical development.
KEYWORDS: HIV, membrane fusion, fusion inhibitor, T-20, lipopeptide
ABSTRACT
T-20 (enfuvirtide) is the only approved viral fusion inhibitor that is used for the treatment of human immunodeficiency virus type 1 (HIV-1) infection; however, it has relatively low antiviral activity and easily induces drug resistance. We recently reported a T-20-based lipopeptide fusion inhibitor (LP-40) showing improved anti-HIV activity (X. Ding et al., J Virol 91:e00831-17, 2017, https://doi.org/10.1128/JVI.00831-17). In this study, we designed LP-50 and LP-51 by refining the structure and function of LP-40. The two new lipopeptides showed dramatically enhanced secondary structure and binding stability and were exceptionally potent inhibitors of HIV-1, HIV-2, simian immunodeficiency virus (SIV), and chimeric simian-human immunodeficiency virus (SHIV), with mean 50% inhibitory concentrations (IC50s) in the very low picomolar range. They also exhibited dramatically increased potencies in inhibiting a panel of T-20- and LP-40-resistant mutant viruses. In line with their in vitro data, LP-50 and LP-51 exhibited extremely potent and long-lasting ex vivo anti-HIV activities in rhesus monkeys: serum dilution peaks that inhibited 50% of virus infection were >15,200-fold higher than those for T-20 and LP-40. Low-dose, short-term monotherapy of LP-51 could sharply reduce viral loads to undetectable levels in acutely and chronically SHIV infected monkey models. To our knowledge, LP-50 and LP-51 are the most potent and broad HIV-1/2 and SIV fusion inhibitors, which can be developed for clinical use and can serve as tools for exploration of the mechanisms of viral entry and inhibition.
IMPORTANCE T-20 remains the only membrane fusion inhibitor available for the treatment of viral infection, but its relatively low anti-HIV activity and genetic barrier for drug resistance have significantly limited its clinical application. Here we report two new lipopeptide-based fusion inhibitors (LP-50 and LP-51) showing extremely potent inhibitory activities against diverse HIV-1, HIV-2, SIV, and T-20-resistant variants. Promisingly, both inhibitors exhibited potent and long-lasting ex vivo anti-HIV activity and could efficiently suppress viral loads to undetectable levels in SHIV-infected monkey models. We believe that LP-50 and LP-51 are the most potent and broad-spectrum fusion inhibitors known to date and thus have high potential for clinical development.
INTRODUCTION
Highly active antiretroviral therapy (HAART) has been very successful in treating human immunodeficiency virus type 1 (HIV-1) infection; it can suppress the virus to below the limit of detection, thus leading to profound reductions in morbidity and mortality from AIDS. However, HAART doses not eradicate the virus; rather, lifelong treatment is required, which ultimately causes severe adverse effects and drug resistance. Therefore, there is a pressing need to develop new drugs that target different steps of the viral life cycle, including cell entry, reverse transcription, integration, and virion maturation.
An early entry step of HIV is mediated by its trimeric envelope (Env) glycoproteins, consisting of the surface subunit gp120 and the noncovalently associated transmembrane subunit gp41 (1, 2). Sequential binding of gp120 to the cellular receptor CD4 and a coreceptor (CCR5 or CXCR4) triggers a cascade of conformational changes in the Env complex and further activates the fusogenic activity of gp41. In the current model, the fusion peptide (FP) at the N terminus of gp41 is exposed and is inserted into the cell membrane, resulting in a prehairpin intermediate (PHI) that bridges the viral and cellular membranes. Then the C-terminal heptad repeats (CHR) of gp41 fold into the hydrophobic grooves created by the trimeric N-terminal heptad repeat (NHR) coiled-coil in an antiparallel manner, generating a stable six-helix bundle (6-HB) structure that drives the two membranes into the close apposition required for fusion (3–5). Among the six FDA-approved classes of anti-HIV drugs, only two inhibitors target the entry process. Maraviroc binds the coreceptor CCR5 and blocks its interaction with gp120, and thus, it is used to treat infections by CCR5-tropic HIV-1 isolates (6, 7). Enfuvirtide (T-20), a 36-mer peptide derived from the CHR of gp41, binds to the NHR to prevent the formation of the viral 6-HB structure (8–10). T-20 is effective in combination therapy of HIV-1 infection, but it has relatively low antiviral activity and a short half-life, thus requiring high-dose injections (90 mg twice daily). More disappointingly, T-20 inherits a low genetic barrier to inducing drug resistance (11, 12). In succession to T-20, T1249 was designed as a second-generation fusion inhibitor with significantly improved pharmaceutical profiles (13, 14); however, its clinical development was stopped due to formulation difficulties.
Earlier studies have demonstrated that genetically anchoring fusion inhibitor peptides to the cell membrane is a viable strategy for increasing antiviral activity (15, 16). Emerging studies suggest that lipid conjugation is a more efficient way to design peptides that block the viral fusion step (17–22). The resulting lipopeptides are considered to interact preferentially with the cell membranes, thus raising the local concentrations of the inhibitors at the target site (17, 21). For example, C34-Chol, which was developed by conjugating a cholesterol group to the C terminus of the CHR-derived peptide C34, showed sharply increased anti-HIV potency and an extended half-life in vivo (17). By conjugating different lipids (fatty acid, cholesterol, sphingolipids) to short peptides that mainly target the deep pocket on the NHR helices, we previously generated a panel of lipopeptides, such as LP-11 and LP-19, that also rendered dramatically increased in vitro, ex vivo, and in vivo antiviral activities and stability (19, 20). In short-term monotherapy, LP-19 could reduce viral loads to undetectable levels in both acutely and chronically simian-human immunodeficiency virus (SHIV) infected rhesus monkeys (20). Very recently, we developed LP-40 and LP-46 by replacing the C-terminal membrane-binding sequences of T-20 and T1249 with a fatty acid group, which sharply improved anti-HIV activity (23, 24). The crystal structures of LP-40 and LP-46 in complexes with target mimic peptides revealed the key binding motifs underlying the mechanisms of action of T-20, T1249, and their lipid derivatives (23, 24). In this study, we rationally designed two new lipopeptides, LP-50 and LP-51, by refining the structure and function of LP-40. Amazingly, both inhibitors had dramatically increased activities in inhibiting HIV-1, HIV-2, simian immunodeficiency virus (SIV), and T-20-resistant mutants; they exhibited extremely potent and long-lasting antiviral activities in rhesus monkeys. In short-term and low-dose monotherapies, LP-50 and LP-51 could sharply reduce viral loads to undetectable levels in SHIV-infected monkey models, highlighting their potential use for the treatment of HIV-1/2 infection.
RESULTS
Structure-based design of lipopeptide inhibitors with greatly increased α-helicity and binding affinity.
Guided by the crystal structure of LP-40 in 6-HBs, we designed two lipopeptides with dramatically improved pharmaceutical profiles. As shown in Fig. 1, LP-50 was generated by introducing multiple pairs of glutamic acid and lysine residues (EK motif) in the solvent-accessible sites (b, c and f, g) of LP-40 to promote the formation of intrahelical salt bridges per helix turn. LP-50 was further modified by introducing amino acids from the sequences of HIV-2 and SIV into its NHR-binding positions (sites a, d, and e), resulting in LP-51, with a peptide sequence in which >60% of amino acids were mutated. First, we used circular dichroism (CD) spectroscopy to determine the secondary structures of the inhibitors themselves. While T-20 and LP-40 were unstructured in solution, the two newly designed inhibitors displayed a typical α-helical conformation and greatly enhanced thermostability (Fig. 2A and B). Then we analyzed their interactions with an NHR-derived target mimic peptide (N39). Promisingly, all the inhibitors interacted with N39 to form α-helical structures (Fig. 2C); however, the binding stability of the inhibitors was markedly improved upon sequence optimization (Fig. 2D). While T-20 and LP-40 had melting temperature (Tm) values of 44°C and 51°C, respectively, LP-50 and LP-51 exhibited Tm values of 63°C and 72°C, respectively. These results indicated that the introduction of the EK motifs and the HIV-2 and SIV residues greatly enhanced the α-helical stability and binding affinity of T-20-based lipopeptide inhibitors.
FIG 1.
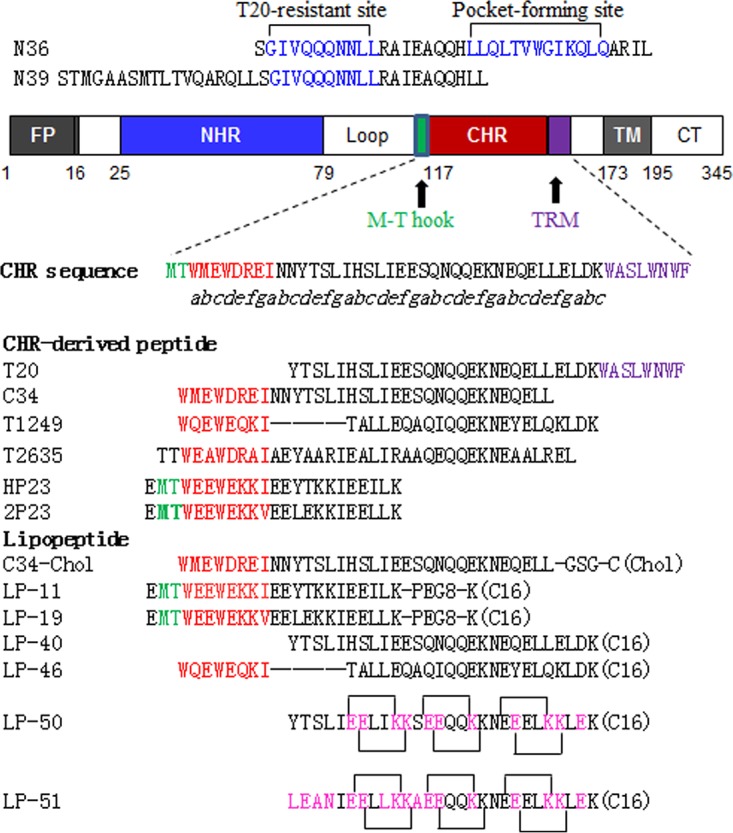
Schematic illustration of HIV gp41 and its NHR- and CHR-derived peptides. The gp41 numbering of HIV-1HXB2 is used. FP, fusion peptide; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; TRM, tryptophan-rich motif; TM, transmembrane domain; CT, cytoplasmic tail. The positions and sequences corresponding to the T20-resistant site and pocket-forming site in the NHR are shown in blue. The italic “abcdefg” corresponds to the positions of the amino acids per heptad repeat sequence. The positions and sequences corresponding to the M-T hook structure, pocket-binding domain, and TRM in the CHR are shown in green, red, and purple, respectively. (Chol) and (C16) represent cholesterol and palmitic acid, respectively; PEG8, 8-unit polyethylene glycol. Engineered residues in LP-50 and LP-51 are shown in pink, and the potential salt bridges are indicated by solid black lines.
FIG 2.

Biophysical properties of the new lipopeptide inhibitors determined by CD spectroscopy. (A and B) The secondary structures (A) and thermostability (B) of the isolated inhibitors at 10 μM were measured in PBS. (C and D) The secondary structures (C) and thermostability (D) of the inhibitors in the presence of the NHR-derived target peptide N39 were determined with 10 μM (final concentration) each peptide in PBS. The α-helical contents and Tm values are shown in parentheses. The experiments were repeated at least twice, and representative data are shown. NA, not applicable for calculation.
LP-50 and LP-51 have dramatically increased activities against divergent HIV-1 isolates.
The antiviral activities of LP-50 and LP-51 were first examined by use of a panel of replication-competent HIV-1 strains with different phenotypes (Table 1). Strikingly, LP-50 and LP-51 inhibited six representative viruses with 50% inhibitory concentrations (IC50s) of 35 and 25 pM, respectively, 396- and 555-fold lower than that of T-20 (13,868 pM) and 193- and 270-fold lower than that of LP-40 (6,759 pM), respectively. Then a large panel of HIV-1 pseudoviruses with their Envs representing the worldwide AIDS epidemic was constructed, and the inhibitory activities of LP-50 and LP-51 were determined by a single-cycle infection assay. As shown in Table 2, LP-50 and LP-51 inhibited divergent HIV-1 subtypes with mean IC50s of 22 and 26 pM, respectively, 1,637- and 1,385-fold lower than that of T-20 (36,020 pM) and 272- and 231-fold lower than that of LP-40 (5,994 pM), respectively. We found that both new inhibitors, like LP-40, had a 50% cytotoxic concentration (CC50) of >140 μM, suggesting an extremely high therapeutic selectivity index (CC50/IC50 ratio).
TABLE 1.
Inhibitory activities of T-20-derived lipopeptide fusion inhibitors against replication-competent HIV-1 isolatesa
| HIV-1 strain | Subtype | Tropism | Mean IC50 (pM) ± SD |
|||
|---|---|---|---|---|---|---|
| T-20 | LP-40 | LP-50 | LP-51 | |||
| NL4-3 | B | X4 | 53,097 ± 4,720 | 26,054 ± 983 | 71 ± 18 | 23 ± 6 |
| LAI | B | X4 | 2,437 ± 346 | 286 ± 74 | 25 ± 7 | 17 ± 3 |
| SG3 | B | X4 | 1,919 ± 321 | 425 ± 55 | 9 ± 2 | 9 ± 2 |
| JR-CSF | B | R5 | 6,601 ± 1,401 | 1,304 ± 137 | 19 ± 6 | 28 ± 9 |
| 89.6 | B | R5X4 | 11,549 ± 3,431 | 11,986 ± 6,072 | 57 ± 32 | 56 ± 11 |
| R3A | B | R5X4 | 7,605 ± 723 | 499 ± 251 | 26 ± 25 | 17 ± 13 |
| Mean | 13,868 | 6,759 | 35 | 25 | ||
The assay was performed in triplicate and was repeated three times.
TABLE 2.
Inhibitory activities of T-20-derived lipopeptide fusion inhibitors against divergent HIV-1 subtypesa
| Primary Env | Subtype | Mean IC50 (pM) ± SD |
|||
|---|---|---|---|---|---|
| T-20 | LP-40 | LP-50 | LP-51 | ||
| 92UG037.8 | A | 7,581 ± 2,117 | 3,667 ± 1,040 | 19 ± 6 | 17 ± 4 |
| 92RW020 | A | 3,582 ± 2,682 | 1,279 ± 71 | 37 ± 4 | 56 ± 8 |
| 398-F1_F6_20 | A | 16,913 ± 1,702 | 4,853 ± 1,099 | 12 ± 2 | 15 ± 4 |
| PVO | B | 71,948 ± 3,160 | 2,832 ± 179 | 24 ± 3 | 33 ± 5 |
| pREJO4541 | B | 57,733 ± 3,229 | 10,129 ± 1,086 | 9 ± 4 | 8 ± 1 |
| SF162 | B | 26,503 ± 3,030 | 5,138 ± 613 | 27 ± 4 | 26 ± 9 |
| JRFL | B | 8,998 ± 855 | 1,756 ± 96 | 39 ± 5 | 74 ± 21 |
| SC422661.8 | B | 16,473 ± 3,788 | 17,481 ± 2,084 | 29 ± 5 | 7 ± 1 |
| AC10.0.29 | B | 3,496 ± 970 | 1,092 ± 170 | 6 ± 2 | 9 ± 2 |
| TRO.11 | B | 6,175 ± 620 | 3,095 ± 476 | 26 ± 3 | 27 ± 6 |
| X2278_C2_B6 | B | 5,986 ± 447 | 1,665 ± 275 | 13 ± 1 | 11 ± 3 |
| B01 | B′ | 89,185 ± 6,922 | 8,630 ± 1,053 | 76 ± 7 | 93 ± 6 |
| B02 | B′ | 9,283 ± 2,120 | 975 ± 60 | 23 ± 2 | 29 ± 5 |
| B04 | B′ | 5,355 ± 568 | 1,272 ± 117 | 23 ± 6 | 44 ± 3 |
| 43-22 | B′ | 30,648 ± 2,054 | 1,354 ± 662 | 5 ± 1 | 8 ± 2 |
| Du156 | C | 14,433 ± 2,125 | 10,538 ± 724 | 6 ± 0 | 8 ± 1 |
| ZM53 M.PB12 | C | 25,809 ± 8,710 | 4,221 ± 715 | 21 ± 2 | 19 ± 3 |
| CAP210.2.00.E8 | C | 140,667 ± 16,565 | 29,190 ± 2,347 | 35 ± 2 | 62 ± 7 |
| CAP45.2.00.G3 | C | 141,963 ± 25,069 | 19,025 ± 4,703 | 9 ± 1 | 12 ± 3 |
| CE703010217_B6 | C | 41,847 ± 1,094 | 3,618 ± 83 | 27 ± 2 | 20 ± 2 |
| HIV_25710-2.43 | C | 13,974 ± 1,841 | 4,662 ± 271 | 26 ± 3 | 27 ± 3 |
| CE1176_A3 | C | 8,608 ± 5,859 | 1,422 ± 275 | 10 ± 2 | 15 ± 1 |
| X1632-S2-B10 | G | 12,240 ± 2,860 | 2,034 ± 404 | 19 ± 2 | 32 ± 7 |
| 246_F3_C10_2 | A/C | 39,126 ± 4,633 | 6,620 ± 2,366 | 20 ± 5 | 26 ± 2 |
| AE03 | A/E | 9,671 ± 2,403 | 3,367 ± 429 | 16 ± 3 | 17 ± 4 |
| GX11.13 | A/E | 22,219 ± 11,067 | 1,892 ± 933 | 10 ± 1 | 14 ± 1 |
| SHX335.24 | A/E | 48,420 ± 7,568 | 25,968 ± 4,444 | 11 ± 1 | 16 ± 3 |
| CNE8 | A/E | 29,069 ± 7,541 | 3,612 ± 133 | 40 ± 3 | 31 ± 1 |
| CNE55 | A/E | 21,888 ± 3,389 | 2,425 ± 449 | 14 ± 1 | 8 ± 1 |
| CH64.20 | B/C | 31,270 ± 20,276 | 4,358 ± 1,080 | 27 ± 2 | 29 ± 3 |
| CH070.1 | B/C | 163,316 ± 6,269 | 6,269 ± 2,286 | 37 ± 5 | 38 ± 6 |
| CH110 | B/C | 33,790 ± 2,139 | 3,631 ± 964 | 6 ± 1 | 8 ± 1 |
| CH119.10 | B/C | 17,993 ± 5,279 | 2,210 ± 21 | 19 ± 2 | 11 ± 2 |
| CH120.6 | B/C | 51,541 ± 27,076 | 4,591 ± 628 | 18 ± 2 | 19 ± 2 |
| BJOX002000.03.2 | B/C | 33,007 ± 18,876 | 4,923 ± 1,363 | 36 ± 3 | 31 ± 4 |
| Mean | 36,020 | 5,994 | 22 | 26 | |
The assay was performed in triplicate and was repeated three times.
LP-50 and LP-51 are highly potent inhibitors of T-20-resistant HIV-1 mutants as well as HIV-2 and SIV isolates.
We were interested in determining the inhibitory activities of LP-50 and LP-51 on T-20-resistant HIV-1 mutants, because our previous studies suggested that the template, LP-40, had no or minor improvement over T-20. As shown in Table 3, LP-50 and LP-51 displayed dramatically increased inhibitory activities against a panel of NL4-3 Env-based pseudoviruses carrying single or combined T-20-induced resistance mutations. In comparison, LP-51 showed much better inhibition than LP-50, as exemplified by their potencies on the V38A, N43K, I37T N43K, and V38A N42T mutants. Nonetheless, both the new lipopeptides also exhibited significantly higher IC50s against these mutants than against the wild-type (WT) and T-20-sensitive (D36G) viruses, highlighting the fact that the T-20-resistant mutations in the target site could still confer resistance, although LP-50 and LP-51 sustained their inhibitory activities at low nanomolar or subnanomolar concentrations.
TABLE 3.
Inhibitory activities of lipopeptide fusion inhibitors against T-20-resistant HIV-1 mutants, HIV-2 isolates, and SIV isolatesa
| Env | Mean IC50 (nM) ± SD |
|||
|---|---|---|---|---|
| T-20 | LP-40 | LP-50 | LP-51 | |
| T-20 sensitive: NL4-3D36G | 13.65 ± 0.48 | 0.61 ± 0.07 | <0.01 | <0.01 |
| T-20 resistant | ||||
| NL4-3WT | 152.23 ± 15.28 | 43.94 ± 12.29 | 0.08 ± 0.02 | 0.03 ± 0 |
| NL4-3I37T | 1,001.19 ± 55.94 | 349.57 ± 95.56 | 1.07 ± 0.16 | 0.59 ± 0.04 |
| NL4-3V38A | 2,780 ± 176.55 | 1,812.67 ± 496.51 | 6.53 ± 0.37 | 1.77 ± 0.24 |
| NL4-3V38M | 1,319.73 ± 33.1 | 2,747 ± 758.5 | 1.45 ± 0.47 | 0.4 ± 0.19 |
| NL4-3Q40H | 2,147.37 ± 447.3 | 1,293.5 ± 364.83 | 3.76 ± 0.66 | 0.58 ± 0.05 |
| NL4-3N43K | 729.12 ± 79.37 | 534.77 ± 169.81 | 5.87 ± 0.73 | 0.99 ± 0.14 |
| NL4-3G36S/V38M | 565.17 ± 57.28 | 604.52 ± 142.66 | 1.76 ± 0.08 | 0.38 ± 0.06 |
| NL4-3I37T/N43K | >4,200 | 3,296 ± 260.62 | 142.03 ± 166.5 | 13.11 ± 1.08 |
| NL4-3V38A/N42T | 2,756 ± 831.37 | 1,745.13 ± 84.18 | 60.32 ± 7.89 | 8.3 ± 0.88 |
| NL4-3E49K/N126K | 470.97 ± 34.19 | 648.93 ± 21.12 | 0.16 ± 0.03 | 0.1 ± 0.03 |
| HIV-2 or SIV | ||||
| HIV-2ROD | 496.73 ± 14.53 | 1,582.33 ± 271.65 | 0.09 ± 0.01 | 0.08 ± 0.02 |
| HIV-2ST | 1,395.44 ± 429.91 | >5,000 | 7.01 ± 0.75 | 1.7 ± 0.13 |
| SIV239 | 353.13 ± 18.6 | 435.33 ± 19.6 | 0.05 ± 0.01 | 0.04 ± 0.01 |
| SIVPBJ | 939.83 ± 125.77 | 1,695.33 ± 265.44 | 0.06 ± 0.01 | 0.07 ± 0.01 |
| SHIV | ||||
| SHIVSF162P3 | 4.94 ± 0.65 | 1.23 ± 0.15 | 0.07 ± 0 | 0.09 ± 0 |
| SHIV1157 | 11.48 ± 1.31 | 23.68 ± 4.7 | 0.04 ± 0 | 0.04 ± 0.01 |
| Control Env: VSV | >2,000 | >2,000 | >2,000 | >2,000 |
The assay was performed in triplicate and was repeated three times.
Next, we determined the inhibitory activities of LP-50 and LP-51 against HIV-2, SIV, and SHIV isolates. As shown in Table 3, LP-50 and LP-51 inhibited HIV-2 isolate ROD at IC50s of 0.09 and 0.08 nM, respectively, indicating 5,519- and 6,209-fold higher potency than T-20 (IC50, 496.73 nM) and 17,581- and 19,779-fold higher potency than LP-40 (IC50, 1,582.33 nM), respectively. While LP-50 and LP-51 inhibited HIV-2 isolate ST with IC50s of 7.01 and 1.7 nM, respectively, T-20 and LP-40 had IC50s of 1,395.44 and >5,000 nM, respectively. For the two SIV isolates tested, T-20, LP-40, LP-50, and LP-51 inhibited SIV239 with IC50s of 353.13, 435.33, 0.05, and 0.04 nM, respectively, and inhibited SIVPBJ with IC50s of 939.83, 1695.33, 0.06, and 0.07 nM, respectively. For the two SHIV isolates tested, T-20, LP-40, LP-50, and LP-51 inhibited SHIVSF162P3 with IC50s of 4.94, 1.23, 0.07, and 0.09 nM, respectively, while they inhibited SHIV1157ipd3N4 with IC50s of 11.48, 23.68, 0.04, and 0.04 nM, respectively. As a control, all the inhibitors had no inhibitory activities against a vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped virus at 2,000 nM. Therefore, we were amazed by the antiviral potencies of LP-50 and LP-51, with IC50s approaching very low picomolar concentrations, and by their broad-spectrum activities against diverse subtypes of HIV-1, HIV-2, SIV, and SHIV, as well as T-20 resistant mutants.
LP-50 and LP-51 exhibit exceptionally potent and long-lasting ex vivo antiviral activities.
We previously reported the potent and long-acting antiviral activities of LP-11 in a rat model and of LP-19 in a nonhuman primate model (19, 20). Here we used the same group of rhesus monkeys that we had used for LP-19 to determine the in vivo stabilities and antiviral activities of LP-40, LP-50, and LP-51. Each inhibitor was subcutaneously injected into six healthy rhesus monkeys, and their pharmacological kinetics in sera were measured pre- or postadministration, which could reflect the in vivo half-lives and antiviral potencies of inhibitors. Surprisingly, LP-40 reached a low serum peak level at 4 h after administration (Fig. 3A), and the dilution that inhibited 50% of virus infection was 40-fold, which was similar to that of the prodrug T-20 (46-fold), determined previously (20). In sharp contrast, LP-50 and LP-51 exhibited extremely high serum peak levels at 4 h after administration (Fig. 3B and C), with serum dilutions of 711,644- and 700,482-fold, respectively, showing that they were >15,200-fold more active than T-20 and LP-40. Compared to LP-19, which had a peak serum dilution of 4,329-fold determined previously (20), LP-50 and LP-51 were >160-fold more active (Fig. 3D). More importantly, while the serum antiviral activity of T-20 and LP-40 could not be detected after 6 h, the inhibitory activities of LP-50 and LP-51 persisted until 72 h postinjection. Specifically, LP-50 showed serum dilutions of 10,549-fold after 48 h and 182-fold after 72 h; LP-51 showed serum dilutions of 34,481-fold after 48 h and 1,122-fold after 72 h. In contrast, LP-19 had serum dilutions of 200-fold after 48 h and 40-fold after 72 h. Therefore, these results suggested that LP-50 and LP-51 have extremely potent and long-lasting in vivo antiviral activities.
FIG 3.

Ex vivo anti-HIV activities of the new lipopeptide inhibitors. (A through C) Each of the inhibitors (LP-40, LP-50, LP-51) was subcutaneously injected into six rhesus monkeys at 3 mg/kg, and monkey sera were harvested at different time points before or after injection. The inhibitory activities of sera from monkeys administered LP-40 (A), LP-50 (B), and LP-51 (C) against HIV-1NL4-3 were tested by a single-cycle infection assay, and serum dilutions required for 50% inhibition of virus infection were calculated. (D) Comparison of HIV-inhibitory activities of monkey sera. Since the same group of monkeys was used, the data for T-20 and LP-19 were obtained from a previous study (20). A Mann-Whitney U test was performed to judge the significance of the differences between treatment groups (P, 0.0005 for LP-50 versus LP-40; P, 0.0003 for LP-51 versus LP-40; P, 0.4458 for LP-40 versus T-20; P, 0.0003 for LP-50 versus T-20; P, 0.0003 for LP-51 versus T-20; P, 0.0042 for LP-50 versus LP-19; P, 0.0004 for LP-51 versus LP-19; P, 0.9459 for LP-50 versus LP-51).
High therapeutic efficacies of LP-50 and LP-51 in acute SHIV infection.
We were highly intrigued and eager to evaluate the in vivo therapeutic efficacies of LP-50 and LP-51 in nonhuman primate models. First, we determined their therapeutic efficacies in rhesus monkeys acutely infected with simian-human immunodeficiency virus (SHIVSF162P3), in which treatment was initiated on day 11 after virus inoculation, when acute infection had been established with the peak of virus replication. Fifteen monkeys were randomly assigned to three groups and were subcutaneously injected with normal saline (group F), LP-50 (group G), or LP-51 (group H) once daily for 4 weeks. As shown in Fig. 4A to C, the plasma viral loads of monkeys reached the highest levels (average, 7.61 log10 RNA copies/ml) and then declined naturally; however, LP-50 or LP-51 treatment sharply accelerated the decline rate relative to that for the normal-saline group. In the LP-50-treated group, the plasma viral loads in monkeys G1, G2, G3, G4, and G5 declined to undetectable levels (100 copies/ml) by days 28, 30, 24, 38, and 24, respectively, after the initiation of treatment, but temporary viral rebounds were observed at day 32 in monkey G3 and from day 28 to day 35 in monkey G5. In the LP-51-treated group, monkeys H1, H2, H3, H4, and H5 achieved undetectable plasma viral loads by days 30, 28, 24, 21, and 21, respectively. As expected, the viral loads rebounded in all of the treated monkeys from 10 to 21 days after the drugs were withdrawn, with averages of 11 days for the LP-50 group and 15 days for the LP-51 group.
FIG 4.

Therapeutic efficacies of LP-50 and LP-51 in acute SHIV infection. (A to C) Starting from day 11 after virus inoculation, 15 SHIVSF162P3-infected rhesus monkeys were subcutaneously treated with normal saline (A), LP-50 (B), or LP-51 (C) once daily for 4 weeks. The plasma viral loads of monkeys were measured at different time points by quantitative PCR. (D) Comparison of the viral load kinetics in monkeys treated with LP-50, LP-51, and normal saline control. A Mann-Whitney U test was performed to judge the significance of the differences during treatment (P, 0.0009 for LP-50 versus saline; P, 0.0006 for LP-51 versus saline; P, 0.3796 for LP-50 versus LP-51).
High therapeutic efficacy of LP-51 in chronic SHIV infection.
We next focused on evaluating the therapeutic efficacy of LP-51 in chronic SHIV infection. Since one rhesus monkey (designated J0) chronically infected by SHIVSF162P3 with a high viral load (6.47 log10 RNA copies/ml) was available, we started a pilot experiment by subcutaneously treating monkey J0 with LP-51 at 3 mg/kg of body weight once daily for 4 weeks. Astonishingly, the plasma viral load precipitated below the detection limit at day 7 after the initiation of treatment, which was the first blood sampling time (Fig. 5A). The viral load retained undetectable during the treatment but rebounded 7 days after LP-51 was stopped. After a 30-day break, a second-round of treatment was provided for monkey J0 with the same dosage of LP-51 (3 mg/kg once daily for 4 weeks). As shown in Fig. 5A, the viral load at the starting point was as high as 7.51 log10 RNA copies/ml, but it rapidly declined to an undetectable level after treatment for 21 days. Similarly, the viral load rebounded again after treatment cessation. After a 90-day break, we gave a third round of LP-51 treatment to monkey J0 with a reduced dosage (1 mg/kg once daily for 4 weeks). Again, the viral replication was fully controlled, declining to an undetectable level after treatment for 14 days, and efficacy was retained until 14 days after LP-51 was withdrawn.
FIG 5.
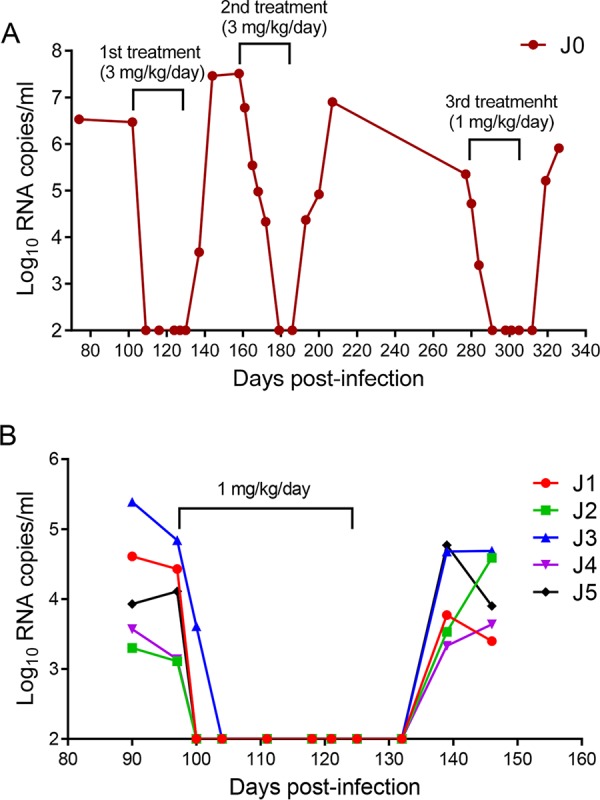
Therapeutic efficacy of LP-51 in chronic SHIV infection. (A) Pilot structurally interrupted treatment of a rhesus monkey chronically infected with SHIVSF162P3. LP-51 was subcutaneously administered at 3 mg/kg for the first and second rounds of treatment and at 1 mg/kg for the third round of treatment. (B) Treatment of rhesus monkeys chronically infected with SHIVSF162P3 (J1 to J5). After viral inoculation for 97 days, LP-51 was subcutaneously administered once daily for 4 weeks. The plasma viral loads of the monkeys were measured at different time points by quantitative PCR.
To validate the efficacy of LP-51 for the treatment of chronic SHIV infection, five rhesus monkeys (J1 to J5) were intravenously infected with SHIVSF162P3 over 3 months, by which time chronic infection had been established with set point viral loads of 3.11 to 4.84 log10 RNA copies/ml. Then the monkeys were subcutaneously treated with LP-51 at 1 mg/kg once daily for 4 weeks. Consistently, we observed rapid and precipitous declines in plasma viral loads to undetectable levels by day 3 in 4 of 5 monkeys and by day 7 in the remaining monkey with the highest baseline viral load (J3). Similarly, the viral loads rebounded in all of the treated monkeys from 10 to 21 days after LP-51 was stopped.
DISCUSSION
Membrane fusion is an essential step utilized by enveloped viruses and has been considered an ideal target for developing antiviral agents (1, 2); however, T-20 remains the only membrane fusion inhibitor available for the treatment of viral infection, and its clinical use has been compromised by its relatively low anti-HIV capacity and genetic barrier to inducing drug resistance. Thus, new drugs that block the viral fusion process are called for. In the past decade, we have dedicated our efforts to exploring the mechanism of HIV-1 Env-mediated membrane fusion and to developing novel viral fusion inhibitors with pharmaceutical profiles. In this study, we successfully generated two novel fusion inhibitor lipopeptides (LP-50 and LP-51) with greatly enhanced α-helicity and binding stability. Both LP-50 and LP-51 showed dramatically increased activities at inhibiting different subtypes and phenotypes of HIV-1 strains as well as HIV-2 and SIV isolates. More amazingly, LP-50 and LP-51 also exhibited dramatic improvements in in vivo antiviral activity and half-life and showed high therapeutic efficacy in rhesus monkey models. Especially, LP-51 could sharply reduce the viral loads to undetectable levels in both acutely and chronically SHIV infected monkeys. Therefore, our studies not only offer promising candidates for clinical development but also provide critical tools for elucidating the mechanisms of HIV-1/2 and SIV fusion/entry and their inhibition.
To overcome the disadvantages of T-20, several strategies have been applied in peptide engineering. Prominently, C34, a gp41 CHR-derived overlapping peptide, has been widely applied as an inhibitor-designing template because it is considered a core CHR sequence in the gp41 structure (6-HB), and its N-terminal segment contains a pocket-binding domain (PBD) that is critical for the interaction of the NHR and CHR helices (3, 5, 25). Compared to T-20, C34 and its derivatives, such as SC34EK, sifuvirtide, CP32M, and T2635, did exhibit improved activities in inhibiting both wild-type and T-20-resistant viruses (26–29). While the pocket-binding sequence has been used for inhibitor design, little attention has been paid to the C-terminal tryptophan-rich motif (TRM) of T-20, which is thought as a lipid binding domain (LBD) that can interact with the cell membrane to confer antiviral activity (30–32). Indeed, the mechanism of action of T-20 and its structural properties remain elusive, as exemplified by the fact that its target sites have been suggested to be on the NHR helices (8, 11), the CHR helices (33, 34), the fusion peptide (35, 36), or the transmembrane domain (32) of gp41 and the coreceptor binding site of gp120 (32, 37–40). We are interested in revisiting the mode of action of T-20 in an attempt to develop fusion inhibitors that differ from those specifically targeting the gp41 pocket site, such as LP-11 and LP-19 (19, 20). The design of the lipopeptide LP-40 and its structural characterization revealed the importance of both the N- and C-terminal amino acid sequences in T-20 and shed new light on the mechanisms of viral membrane fusion and inhibition, which have guided our current design of new inhibitors. Here, the successful creation of LP-50 and LP-51 has verified the two design strategies: lipid conjugation and the introduction of “salt bridge”-prone amino acids into the inhibitors, which might be simultaneously required to ensure that the lipopeptides have markedly enhanced α-helicity, thermostability, and binding affinity and dramatically increased antiviral potency. As evidenced by LP-51, integrating the HIV-2- or SIV-derived amino acids into the target-binding sites of the peptide sequence is also a viable approach to further enhancing the binding of an inhibitor to the target site and improving its inhibition of HIV-2, SIV, and T-20-resistant viruses. Actually, we are still excited by the fact that the antiviral activity of T-20 could be enhanced >1,600-fold by lipid conjugation and sequence optimization (Table 2), and we hope to characterize the underlying mechanism in detail.
The cholesteroylated peptide C34-Chol, with highly potent anti-HIV activity and an extended half-life, has been reported previously (17). Recently, Quinn et al. reported its initial clinical trial, which showed that in three HIV-positive subjects, a single subcutaneous low dose of C34-Chol resulted in a mean change in the HIV-1 RNA load of −0.9 log10 copies/ml (41). Although the trial was suspended due to serious injection site reactions (ISRs), the human pharmacodynamic and pharmacokinetic data suggested a continuing evaluation of C34-Chol as a long-acting antiretroviral drug (LA-ARV) after optimization of the drug formulation and administration route. We would like to address several advantages of LP-50 and LP-51 over C34-Chol. First, fatty acid has been proved to be a safe molecule for conjugating peptide drugs, such as liraglutide and semaglutide for type 2 diabetes, while one may have concerns about the safety of a cholesterol-based drug for long-term in vivo use. Indeed, LP-50 and LP-51 showed an extremely high therapeutic selectivity index, and no ISRs were observed in the repeatedly injected rhesus monkeys. Second, both the new inhibitors have shorter amino acid sequences (28-mer) than C34-Chol (38-mer) and require a relatively convenient conjugation protocol, which certainly benefits their production and cost-effectiveness. Third, we have data (not shown) revealing that the antiviral activity of C34-Chol is much lower than those of LP-50 and LP-51, especially in the inhibition of HIV-2 and SIV isolates.
By applying the M-T hook structure in peptide sequence and lipid conjugation, we previously developed a panel of lipopeptide fusion inhibitors, such as LP-11 and LP-19, that mainly target the gp41 pocket site and possess highly potent anti-HIV activity (19, 20, 42–44). LP-19 was also evaluated in nonhuman primate models and demonstrated its potent therapeutic efficacies in both acute and chronic infections (20). Compared to LP-19, LP-50 and LP-51 were about 20-fold more active in their in vitro potencies and 160-fold more active in their in vivo activities, and consistently, they were more effective in the treatment of SHIV-infected monkeys. Nonetheless, it is expected that LP-50 or LP-51 and LP-19 would be used in combination therapy, since our previous studies demonstrate a synergistic effect between their parental inhibitors (LP-11 and LP-40), which possess different target sites on the NHR of gp41 and different drug resistance profiles. Furthermore, LP-50 and LP-51 can be used for treating HIV-2, which has already caused 1 to 2 million infections worldwide and continues to spread in different regions. As virus entry inhibitors, they can also serve as prophylactics for the prevention of HIV transmission. The membrane-anchored lipopeptides might not only elevate the local concentrations of inhibitors at the site where viral fusion occurs, thus improving antiviral activity, but also bind to serum proteins, such as human serum albumin (HSA), thus greatly extending their in vivo half-lives. Therefore, we believe that the lipopeptide-based strategy is also applicable for the development of membrane fusion inhibitors against other enveloped viruses, such as influenza virus, respiratory syncytial virus (RSV), and the emerging viruses Ebola virus and Middle East respiratory syndrome coronavirus (MERS-CoV).
MATERIALS AND METHODS
Cell lines and plasmids.
HEK293T cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). The following reagents were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl indicator cells, which stably express large amounts of CD4 and CCR5, along with endogenously expressed CXCR4, from John C. Kappes and Xiaoyun Wu; the Panel of Global HIV-1 Env Clones, which contains 12 envelope clones as reference strains representing the global AIDS epidemic, from David Montefiori; a panel of molecular clones for producing infectious HIV-1 isolates, including pNL4-3 from Malcolm Martin, pLAI.2 from Keith Peden, pSG3.1 from Sajal Ghosh, Beatrice Hahn, and George Shaw, pYK-JRCSF from Irvin S. Y. Chen and Yoshio Koyanagi, and p89.6 from Ronald G. Collman; the HIV-2 ST infectious molecular clone from Beatrice Hahn and George Shaw; and the HIV-2 ROD infectious molecular clone (pROD10) from the Centre for AIDS Reagents, NIBSC, United Kingdom. Two plasmids encoding SIV Env (pSIVpbj-Env and pSIV239-Env) were kindly provided by Jianqing Xu at the Shanghai Public Health Clinical Center and Institutes of Biomedical Sciences, Fudan University, Shanghai, China.
Peptide synthesis and lipid conjugation.
Peptides were synthesized using a standard solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) method as described previously (19). A template peptide contains a C-terminal lysine residue with a 1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl (Dde) side chain-protecting group, which enables the conjugation of a fatty acid that requires a deprotection step in a solution of 2% hydrazine hydrate–N,N-dimethylformamide. All peptides were acetylated at the N terminus and amidated at the C terminus. They were purified by reverse-phase high-performance liquid chromatography (HPLC) to >95% homogeneity and were characterized by mass spectrometry. The concentrations of the peptides were measured by UV absorbance and a theoretically calculated molar extinction coefficient based on the tryptophan and tyrosine residues. The cytotoxicities of the peptide inhibitors were determined on TZM-bl cells using the CellTiter 96 AQueous One Solution cell proliferation assay (Promega) as described previously (24).
Determination of the α-helicities and binding stabilities of fusion inhibitors.
The secondary structures of peptide fusion inhibitors and their interactions with a target surrogate were determined by CD spectroscopy according to protocols described previously (44, 45). Briefly, an inhibitor was diluted in phosphate-buffered saline (PBS) (pH 7.2) and incubated at 37°C for 30 min in the presence or absence of an equal molar concentration of the NHR-derived target mimic peptide N39. CD spectra were acquired on a Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 270 nm at room temperature. The spectra were corrected by subtracting a solvent blank. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity [θ] at 222 nm by the value expected for 100% helix formation (−33,000° · cm2 · dmol−1). Thermal denaturation was performed by monitoring the ellipticity change at 222 nm from 20°C to 98°C at a rate of 2°C/min, and Tm (melting temperature) was defined as the midpoint of the thermal unfolding transition.
Inhibitory activities of fusion inhibitors on HIV-1, HIV-2, and SIV isolates.
The inhibitory activities of fusion inhibitors against a panel of replication-competent HIV-1 isolates (NL4-3, LAI, SG3, JR-CSF, 89.6, R3A), HIV-2 isolates (ROD, ST), and SHIV isolates (SF162P3, 1157) was assessed as described previously (20). Briefly, viral stocks were prepared by transfecting viral molecular clones into HEK293T cells. Culture supernatants were harvested at 48 h posttransfection, and 50% tissue culture infectious doses (TCID50) in TZM-bl cells were determined. One hundred TCID50 of a virus was used to infect TZM-bl cells in the presence or absence of serially 3-fold diluted inhibitors. Cells were harvested 2 days postinfection and were lysed in reporter lysis buffer, and luciferase activity was measured.
The inhibitory activities of inhibitors on a panel of HIV-1 and SIV pseudoviruses were measured by single-cycle infection assays described previously (20, 23). Briefly, a pseudovirus was generated via the cotransfection of HEK293T cells with an Env-expressing plasmid and a backbone plasmid encoding an Env-defective, luciferase-expressing HIV-1 genome (pSG3Δenv). Similarly, culture supernatants were harvested 48 h after transfection, and TCID50 were determined in TZM-bl cells. Peptide inhibitors were prepared in 3-fold dilutions, mixed with 100 TCID50 of a virus, and then incubated for 1 h at room temperature. The mixture was added to TZM-bl cells (104/well), and the cells were incubated for 48 h at 37°C. Luciferase activity was measured.
Ex vivo anti-HIV activities of fusion inhibitors in rhesus macaques.
The ex vivo antiviral activities of lipopeptide inhibitors were evaluated as described previously (19, 20). In brief, LP-40, LP-50, or LP-51 (3 mg/kg) was subcutaneously administered to six Chinese rhesus macaques (three males and three females aged 3.5 to 4.5 years and weighing 4 to 5 kg) who were previously used for evaluating the in vivo anti-HIV activities of T-20 and LP-19. Similarly, serum samples of macaques were harvested before injection (0 h) and after injection (1, 2, 4, 6, 8, 12, 18, 24, 36, 48, 60, and 72 h). The inhibitory activities of all the sera were determined with the HIV-1NL4-3 isolate in a single-cycle infection assay. The 50% effective concentration was defined as the fold serum dilution that inhibited 50% of virus infection.
Therapeutic efficacies of LP-50 and LP-51 in SHIV-infected rhesus monkeys.
The in vivo therapeutic efficacies of inhibitors were evaluated in SHIV-infected rhesus monkeys as described previously (20). For this purpose, 21 adult Chinese rhesus macaques were screened to be negative for SIV, herpes B virus, and simian T-lymphotropic virus. SHIVSF162P3 (obtained through the AIDS Reagent Program, NIAID, NIH) was expanded on macaque peripheral blood mononuclear cells (PBMCs), and the TCID50 was determined. Macaques were intravenously inoculated with 1,000 TCID50 of virus and were randomly assigned to 4 treatment groups. For the treatment of acutely SHIV infected monkeys (groups F, G, and H), 1 ml of a 0.9% saline solution, LP-50 (3 mg/kg), or LP-51 (3 mg/kg) was subcutaneously injected once daily for 4 weeks beginning on day 11 after virus inoculation. For the treatment of chronic infection, macaques (J0 to J5) were subcutaneously administered 1 ml of LP-51 at 3 mg/kg or 1 mg/kg once daily for 4 weeks. Plasma viral loads were determined by a quantitative real-time reverse transcription-PCR (qRT-PCR) assay as described previously (20). The limit of detection was 100 copy equivalents of RNA per ml of plasma. Triplicate test reactions were performed for each sample.
Ethics statement.
The protocols for the ex vivo studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Beijing Institute of Microbiology and Epidemiology (protocol 2016-15). The protocols for the in vivo studies were approved by the IACUC at the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (protocol ILAS-VL-2015-004). All monkeys were housed and fed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility. The study of animals was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Sciences (46) to ensure personnel safety and animal welfare. All animals were anesthetized with ketamine hydrochloride (10 mg/kg) prior to the procedures. The experiments were performed in a biosafety level 3 laboratory.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81630061, 81473255) and the CAMS Innovation Fund for Medical Sciences (2017-I2M-1-014).
REFERENCES
- 1.Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 2.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 3.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 4.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 6.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H; MOTIVATE Study Teams. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 359:1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fätkenheuer G, Nelson M, Lazzarin A, Konourina I, Hoepelman AI, Lampiris H, Hirschel B, Tebas P, Raffi F, Trottier B, Bellos N, Saag M, Cooper DA, Westby M, Tawadrous M, Sullivan JF, Ridgway C, Dunne MW, Felstead S, Mayer H, van der Ryst E; MOTIVATE Study Teams. 2008. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N Engl J Med 359:1442–1455. doi: 10.1056/NEJMoa0803154. [DOI] [PubMed] [Google Scholar]
- 8.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A 91:9770–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M; TORO 1 Study Group. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 10.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 11.Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 72:986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenberg ML, Cammack N. 2004. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother 54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 13.Lalezari JP, Bellos NC, Sathasivam K, Richmond GJ, Cohen CJ, Myers RA Jr, Henry DH, Raskino C, Melby T, Murchison H, Zhang Y, Spence R, Greenberg ML, DeMasi RA, Miralles GD; T1249-102 Study Group. 2005. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J Infect Dis 191:1155–1163. doi: 10.1086/427993. [DOI] [PubMed] [Google Scholar]
- 14.Eron JJ, Gulick RM, Bartlett JA, Merigan T, Arduino R, Kilby JM, Yangco B, Diers A, Drobnes C, DeMasi R, Greenberg M, Melby T, Raskino C, Rusnak P, Zhang Y, Spence R, Miralles GD. 2004. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J Infect Dis 189:1075–1083. doi: 10.1086/381707. [DOI] [PubMed] [Google Scholar]
- 15.Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, Thaler S, Stiegler G, Welker R, von Laer D. 2001. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J Virol 75:3038–3042. doi: 10.1128/JVI.75.6.3038-3042.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, Baum C, Choi I, Alexandrov A, von Laer D. 2004. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol 78:568–575. doi: 10.1128/JVI.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A 106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Augusto MT, Hollmann A, Castanho MA, Porotto M, Pessi A, Santos NC. 2014. Improvement of HIV fusion inhibitor C34 efficacy by membrane anchoring and enhanced exposure. J Antimicrob Chemother 69:1286–1297. doi: 10.1093/jac/dkt529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong H, Wu X, Su Y, He Y. 2016. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 30:1187–1196. doi: 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 20.Chong H, Xue J, Xiong S, Cong Z, Ding X, Zhu Y, Liu Z, Chen T, Feng Y, He L, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2017. A lipopeptide HIV-1/2 fusion inhibitor with highly potent in vitro, ex vivo, and in vivo antiviral activity. J Virol 91:e00288-17. doi: 10.1128/JVI.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashkenazi A, Viard M, Unger L, Blumenthal R, Shai Y. 2012. Sphingopeptides: dihydrosphingosine-based fusion inhibitors against wild-type and enfuvirtide-resistant HIV-1. FASEB J 26:4628–4636. doi: 10.1096/fj.12-215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wexler-Cohen Y, Shai Y. 2009. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog 5:e1000509. doi: 10.1371/journal.ppat.1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Zhang X, Ding X, Chong H, Cui S, He J, Wang X, He Y. 2018. Exceptional potency and structural basis of a T1249-derived lipopeptide fusion inhibitor against HIV-1, HIV-2, and simian immunodeficiency virus. J Biol Chem 293:5323–5334. doi: 10.1074/jbc.RA118.001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding X, Zhang X, Chong H, Zhu Y, Wei H, Wu X, He J, Wang X, He Y. 2017. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: implication for viral entry and inhibition. J Virol 91:e00831-17. doi: 10.1128/JVI.00831-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu M, Kim PS. 1997. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J Biomol Struct Dyn 15:465–471. doi: 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- 26.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, Fujii N. 2002. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew Chem Int Ed Engl 41:2937–2940. doi:. [DOI] [PubMed] [Google Scholar]
- 27.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 28.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A 104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. 2008. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A 105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peisajovich SG, Gallo SA, Blumenthal R, Shai Y. 2003. C-terminal octylation rescues an inactive T20 mutant: implications for the mechanism of HIV/simian immunodeficiency virus-induced membrane fusion. J Biol Chem 278:21012–21017. doi: 10.1074/jbc.M212773200. [DOI] [PubMed] [Google Scholar]
- 31.Wexler-Cohen Y, Shai Y. 2007. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J 21:3677–3684. doi: 10.1096/fj.07-8582com. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Lu H, Niu J, Xu Y, Wu S, Jiang S. 2005. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem 280:11259–11273. doi: 10.1074/jbc.M411141200. [DOI] [PubMed] [Google Scholar]
- 33.Kliger Y, Gallo SA, Peisajovich SG, Muñoz-Barroso I, Avkin S, Blumenthal R, Shai Y. 2001. Mode of action of an antiviral peptide from HIV-1. Inhibition at a post-lipid mixing stage. J Biol Chem 276:1391–1397. doi: 10.1074/jbc.M004113200. [DOI] [PubMed] [Google Scholar]
- 34.Muñoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. 1998. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol 140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorizate M, de la Arada I, Huarte N, Sanchez-Martinez S, de la Torre BG, Andreu D, Arrondo JL, Nieva JL. 2006. Structural analysis and assembly of the HIV-1 gp41 amino-terminal fusion peptide and the pretransmembrane amphipathic-at-interface sequence. Biochemistry 45:14337–14346. doi: 10.1021/bi0612521. [DOI] [PubMed] [Google Scholar]
- 36.Jiang S, Lin K, Strick N, Neurath AR. 1993. HIV-1 inhibition by a peptide. Nature 365:113. [DOI] [PubMed] [Google Scholar]
- 37.Yuan W, Craig S, Si Z, Farzan M, Sodroski J. 2004. CD4-induced T-20 binding to human immunodeficiency virus type 1 gp120 blocks interaction with the CXCR4 coreceptor. J Virol 78:5448–5457. doi: 10.1128/JVI.78.10.5448-5457.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, Hunter E, Doms RW. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc Natl Acad Sci U S A 99:16249–16254. doi: 10.1073/pnas.252469399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Derdeyn CA, Decker JM, Sfakianos JN, Zhang Z, O'Brien WA, Ratner L, Shaw GM, Hunter E. 2001. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J Virol 75:8605–8614. doi: 10.1128/JVI.75.18.8605-8614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. doi: 10.1128/JVI.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinn K, Traboni C, Penchala SD, Bouliotis G, Doyle N, Libri V, Khoo S, Ashby D, Weber J, Nicosia A, Cortese R, Pessi A, Winston A. 2017. A first-in-human study of the novel HIV-fusion inhibitor C34-PEG4-Chol. Sci Rep 7:9447. doi: 10.1038/s41598-017-09230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chong H, Yao X, Sun J, Qiu Z, Zhang M, Waltersperger S, Wang M, Cui S, He Y. 2012. The M-T hook structure is critical for design of HIV-1 fusion inhibitors. J Biol Chem 287:34558–34568. doi: 10.1074/jbc.M112.390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chong H, Qiu Z, Su Y, Yang L, He Y. 2015. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. AIDS 29:13–21. doi: 10.1097/QAD.0000000000000498. [DOI] [PubMed] [Google Scholar]
- 44.Xiong S, Borrego P, Ding X, Zhu Y, Martins A, Chong H, Taveira N, He Y. 2017. A helical short-peptide fusion inhibitor with highly potent activity against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus. J Virol 91:e01839-16. doi: 10.1128/JVI.01839-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chong H, Yao X, Qiu Z, Qin B, Han R, Waltersperger S, Wang M, Cui S, He Y. 2012. Discovery of critical residues for viral entry and inhibition through structural insight of HIV-1 fusion inhibitor CP621-652. J Biol Chem 287:20281–20289. doi: 10.1074/jbc.M112.354126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. The National Academies Press, Washington, DC. doi: 10.17226/12910. [DOI] [Google Scholar]