

The relatively recent appearance of influenza A virus (IAV) epidemics in dogs expands our understanding of IAV host range and ecology, providing useful and relevant models for understanding critical factors involved in viral emergence. Here we integrate viral whole-genome sequence analysis and comprehensive surveillance data to examine the evolution of the emerging avian-origin H3N2 canine influenza virus (CIV), particularly the factors driving ongoing circulation and recent increases in incidence of the virus within the United States. Our results provide a detailed understanding of how H3N2 CIV achieves sustained circulation within the United States despite widespread host contact heterogeneity and recurrent epidemic fade-out. Moreover, our findings suggest that the types and intensities of selection pressures an emerging virus experiences are highly dependent on host population structure and ecology and may inhibit an emerging virus from acquiring sustained epidemic or pandemic circulation.
KEYWORDS: influenza, canine influenza, emerging virus, virus evolution, virus host adaptation
ABSTRACT
Avian-origin H3N2 canine influenza virus (CIV) transferred to dogs in Asia around 2005, becoming enzootic throughout China and South Korea before reaching the United States in early 2015. To understand the posttransfer evolution and epidemiology of this virus, particularly the cause of recent and ongoing increases in incidence in the United States, we performed an integrated analysis of whole-genome sequence data from 64 newly sequenced viruses and comprehensive surveillance data. This revealed that the circulation of H3N2 CIV within the United States is typified by recurrent epidemic burst–fade-out dynamics driven by multiple introductions of virus from Asia. Although all major viral lineages displayed similar rates of genomic sequence evolution, H3N2 CIV consistently exhibited proportionally more nonsynonymous substitutions per site than those in avian reservoir viruses, which is indicative of a large-scale change in selection pressures. Despite these genotypic differences, we found no evidence of adaptive evolution or increased viral transmission, with epidemiological models indicating a basic reproductive number, R0, of between 1 and 1.5 across nearly all U.S. outbreaks, consistent with maintained but heterogeneous circulation. We propose that CIV's mode of viral circulation may have resulted in evolutionary cul-de-sacs, in which there is little opportunity for the selection of the more transmissible H3N2 CIV phenotypes necessary to enable circulation through a general dog population characterized by widespread contact heterogeneity. CIV must therefore rely on metapopulations of high host density (such as animal shelters and kennels) within the greater dog population and reintroduction from other populations or face complete epidemic extinction.
IMPORTANCE The relatively recent appearance of influenza A virus (IAV) epidemics in dogs expands our understanding of IAV host range and ecology, providing useful and relevant models for understanding critical factors involved in viral emergence. Here we integrate viral whole-genome sequence analysis and comprehensive surveillance data to examine the evolution of the emerging avian-origin H3N2 canine influenza virus (CIV), particularly the factors driving ongoing circulation and recent increases in incidence of the virus within the United States. Our results provide a detailed understanding of how H3N2 CIV achieves sustained circulation within the United States despite widespread host contact heterogeneity and recurrent epidemic fade-out. Moreover, our findings suggest that the types and intensities of selection pressures an emerging virus experiences are highly dependent on host population structure and ecology and may inhibit an emerging virus from acquiring sustained epidemic or pandemic circulation.
INTRODUCTION
The biological and ecological factors that enable newly introduced viruses to replicate and spread efficiently to cause sustained epidemics or pandemics in a new host population are central issues for studies of disease emergence (1, 2). In particular, a newly introduced virus may be adapted suboptimally to the novel host and face many opportunities for epidemic fade-out (2, 3). Hence, there may be natural selection for increased transmission in the new host as well as key roles for individuals that create a large number of secondary infections (“superspreaders”) and for well-connected host subpopulations that take the pathogen from the initially infected individual or small outbreak into the broader population (1, 4). However, for most successful emergent viruses, connections between the various properties of the pathogen and how transmission is favored or reduced by the structure and demographics of the new host population remain obscure.
Influenza A viruses (IAVs) have a complex natural history, with most viruses found as endemic gastrointestinal tract infections of specific bird populations in marine or freshwater environments (5, 6). Occasionally, avian IAVs spill over to infect and spread as respiratory infections within terrestrial avian hosts, such as domestic chickens or turkeys, or within certain mammalian populations. In the last century, pandemic IAVs emerged in humans in 1918 and 2009 after transfer of entire viruses from other hosts, while pandemic variants of these viruses emerged in 1957 and 1968 following replacement of three and two of the viral gene segments from avian virus sources, respectively (7). In addition, there have been many other avian-to-human spillover events or small outbreaks that result in serious infections, but with little or no onward human-to-human transmission (8). Analogous IAV emergence events have occurred in a limited number of other mammalian species, including swine, seals, dogs, horses, and occasionally mink and cats (5). Such events can provide alternative and highly informative models for understanding how IAVs traverse the host range barrier and spread efficiently within those populations.
While the threat of emergent IAVs reaching epidemic or pandemic transmission levels is great, these events appear to be relatively rare given the prevalence of virus in the environment and the presumed frequency of opportunities for viral exposure of novel hosts. Likewise, the barriers that prevent IAVs from emerging in a new host to produced sustained epidemics or pandemics are only partially understood and often oversimplified to a few biological factors, including the host cell receptor (sialic acid) and its linkages, which can influence viral hemagglutinin (HA) binding and/or neuraminidase (NA) cleavage, viral HA stability and pH activation, viral polymerase efficiency, and host innate immune responses (9–13). Traits that overcome some of these barriers may be selected rapidly after virus infection and transmission in a new host, and similar adaptive mutations may also appear after small numbers of passages in experimental animals (10, 14, 15), in embryonated chicken eggs (16), or in cell culture (17). However, complete host adaptation in nature is likely a more prolonged and complex process, and we lack a thorough understanding of the posttransfer evolutionary steps that IAVs must take to produce sustained infection in a new host species. Moreover, exactly how these evolutionary processes are facilitated or hindered by host ecology and population structures remains difficult to define.
The appearance of IAV epidemics in dogs is a relatively recent development that expands our understanding of IAV host range and ecology, making these epidemics useful models for understanding the critical factors involved in successful viral emergence. An H3N8 strain variant of equine influenza virus was the first IAV to cause an epidemic disease in dogs, emerging around 1999 in the southeastern United States (21, 51). That virus, the H3N8 canine influenza virus (CIV), circulated continuously among U.S. dogs for over a decade but was eventually restricted to only a few small geographic pockets and now appears to be reduced to very low levels (18). The second CIV, the avian-origin H3N2 CIV, emerged in dogs in China or South Korea around 2005 and then spread within China and South Korea and to Thailand (19, 20). The H3N2 CIV was first introduced into the United States in February 2015 (22) and has since caused an ongoing epidemic of disease. Because each of the CIV epidemics ultimately arose from a single cross-species transfer event—the H3N8 subtype from a mammalian intermediate host and the H3N2 subtype directly from an avian reservoir—they provide informative and relatively simple comparative models for understanding how IAVs emerge and spread in new hosts.
To better understand how an IAV is able to establish itself in new host populations and the determinants of this process, we analyzed the evolution and epidemiology of H3N2 CIV during its emergence and spread, particularly among dogs in the United States. In particular, we addressed how the U.S. H3N2 CIV epidemic is situated within the context of global H3N2 CIV circulation and evolution and what has driven sustained transmission and periodic epidemic bursts of the virus within the United States.
RESULTS
We initially analyzed sequences from 138 different H3N2 CIVs (excluding partial segment sequences), among which complete coding region sequences of all eight segments were available for 113. Over half (64 viruses) of these 113 viruses were sequenced as part of this study. Sequencing provided complete (∼99% coverage) genome sequences, which were missing only the last 12 or 13 bases of the terminal noncoding sequences that were covered by the primers used to amplify the genomic segments (see reference 47 and Materials and Methods). The viruses sequenced in this study included clinical submissions obtained throughout the first H3N2 CIV epidemic in the United States, with most samples obtained from dogs in Chicago and nearby regions of Illinois as well as in the neighboring state of Indiana, and from the second epidemic that involved animals infected mainly in the southeastern states of Florida and Georgia, as well as California, Kentucky, Ohio, Texas, and Minnesota. As a background, we also determined the sequences of two viruses from central China, collected in 2015, and one from South Korea, collected in 2016. Sequences of viruses collected from infected dogs that had been brought directly from southern China to Los Angeles in March 2017 were also obtained. As the latter sequences represent a transient outbreak in Los Angeles and were clearly of Chinese origin, they were grouped with other Chinese viruses for the purpose of this study.
Global circulation of H3N2 CIV.
To provide a larger context for the U.S. H3N2 CIV epidemic and to gain an understanding of the degree of intersubtype reassortment occurring in these viruses, we first inferred phylogenies for individual genomic segments. While H3N2 CIV has been known to reassort with IAV strains from other hosts (48–50), these events have not been shown to produce dog-to-dog transmissible viruses. We therefore excluded segments resulting from reassortment with other non-H3N2 CIV IAVs. As no single avian virus has been identified as a direct ancestor of H3N2 CIV, we rooted the individual gene segment phylogenies on the most closely related avian virus sequence in the databases for each respective segment (for PB2, A/duck/Beijing/40/04 [H3N8]; for PB1, A/mallard/Huadong/S/2005 [H5N1]; for PA, A/migratory duck/Hong Kong/MP2553/2003 [H8N4]; for HA, A/duck/Korea/JS53/2004 [H3N2]; for NP, A/duck/Jiangsu/4/2010 [H3N6]; for NA, A/duck/Korea/JS53/2004 [H3N2]; for M1, A/avian/Israel/320/2001 [H6N2]; and for NS1, A/chicken/Nanchang/7-010/2000 [H3N6]) (Fig. 1A). Tree topologies remained generally consistent across all genomic segment major coding regions, with South Korean and Chinese lineages bifurcating and remaining distinct after around 2009 and all circulating U.S. viruses nested within the South Korean clade. As reported previously (19), a few isolated regional intersubtype reassortants exist within South Korean and Chinese geographic groups; however, our analysis revealed that none of these reassortments led to sustained canine transmission. Furthermore, no reassortants were detected among the U.S. viruses. Taken together, these observations suggest that reassortment in H3N2 CIVs, be it with other H3N2 CIVs or with IAVs of different subtypes and hosts, typically results in evolutionary dead ends. This also enabled us to analyze concatenated major coding regions of each viral genome in a single phylogeny (Fig. 1B).
FIG 1.

Evolutionary relationships of H3N2 CIV sequences. Black branches on the phylogenies represent early emergent viruses isolated in Asia between 2006 and 2007, while branches leading to enzootic viruses isolated from different geographic regions are color coded as follows: green, China; blue, South Korea; and red, United States. Red triangles show viruses of Chinese origin that formed a transient and locally contained outbreak in Los Angeles, CA, in March 2017. Horizontal branch lengths are drawn to scale (nucleotide substitutions per site). (A) Individual segment phylogenies for all complete coding regions from all H3N2 CIV sequences available. Each segment tree is rooted on the closest related avian IAV sequence in the database. Identical sequences were collapsed into single branches for purposes of clarity. (B) Phylogeny of concatenated major reading frames from each genome segment for all nonreassortant viruses circulating in Asia and the United States (see inset). Bootstrap values for key nodes are indicated.
Molecular epidemiology of CIV in the United States, 2015 to 2017.
We next aimed to determine the cause of sustained circulation and periodic increases of incidence of the virus within the United States. As noted above, segment phylogenies of U.S. viruses confirmed the absence of reassortment in the sequences from the U.S. outbreaks such that we based our analysis on phylogenies of concatenated major coding regions (Fig. 1B). Notably, this analysis revealed multiple incursions into the United States from Asia and a series of clade expansions and fade-outs.
As previously reported (22) and confirmed in more detail here, the first outbreak in the United States was clearly initiated by the introduction of a single virus closely related to viruses from South Korea that were collected in early 2015 (Fig. 1B, inset). Once introduced to the United States, that virus spread quickly through many of the animal shelters and kennels in the city of Chicago and in nearby regions and states. The sequences of viruses from around Chicago fell into two distinct clades, defined here as an early midwestern clade (MW2015) that arose from the initially introduced virus and a second midwestern clade (MW2016) that emerged around March 2016. Interestingly, a satellite outbreak nested within the MW2015 clade occurred in the southeastern United States (namely, Georgia, Florida, and North Carolina) in late June 2015 but appeared to have died out by early August 2015. Likewise, both of the larger Midwest clades eventually appeared to experience rapid fade-out, with no viruses recovered from MW2015 and MW2016 after June 2016 and February 2017, respectively. This suggests that each of these groups of viruses has either gone extinct or is circulating at very low levels in isolated populations of dogs, despite once dominating the clinical sampling.
An additional large clade of CIV sequences (denoted SE2017) was identified and associated with a large outbreak of virus in the southeastern United States during May 2017 and continuing for a number of months in Georgia, Florida, Texas, Alabama, and Kentucky. Our phylogenetic analysis clearly established that this outbreak represents a second introduction into the United States, most likely from South Korea, in early 2017 (Fig. 1B, inset). It should be noted that a single SE2017 virus (A/canine/California/84315/2017 [H3N2]), highly similar to the other SE2017 viruses but occupying a more divergent phylogenetic position, was recovered from a dog in California with an unknown prior travel history. At present, viruses belonging to the SE2017 clade overwhelmingly dominate clinical sampling in the United States.
An additional transient outbreak in the United States was seen in Los Angeles, CA, in early 2017, within a group of dogs imported directly from China, although that outbreak was controlled soon after by quarantine of the infected dogs. As expected, these viruses were closely related to those from southern China (Fig. 1B). The fact that this outbreak has not been sampled since 24 April 2017 suggests that it has gone extinct, again demonstrating the burst–fade-out evolutionary dynamics of this virus within the United States following a regional introduction.
Rates of genome sequence evolution in H3N2 CIV.
The cross-species transfer of H3N2 CIV to dogs, its subsequent spread in China and South Korea, and its eventual transfer to the United States to initiate separate outbreaks allowed us to compare the genomic evolutionary rates throughout different stages and scenarios of viral emergence.
We first performed a regression of root-to-tip genetic distances on the phylogenetic tree for concatenated full-genome sequences against sampling dates (Fig. 2). This revealed a strongly linear accumulation of nucleotide divergence over time (R2 = 0.97). Given this strong clock-like evolution, we next analyzed the rates in more detail by using a Bayesian Markov chain Monte Carlo method implemented in the BEAST package, version 1.8.2 (23). Overall, H3N2 CIV evolved at a mean rate of 1.38 × 10−3 substitutions/site/year during the 11 years since it was first reported in dogs (95% highest posterior density [HPD] = 1.211 × 10−3 to 1.548 × 10−3 substitutions/site/year), according to the best-fitting model (constant population size, strict clock). These rates are similar to those previously determined for Asian H3N2 CIV (19) and are at the lower end of the 95% HPD intervals reported for various IAVs circulating in avian and human hosts (24, 25). Such consistent and relatively low rates of genomic evolution (among IAVs) suggest that the evolutionary dynamics of H3N2 CIV are largely shaped by the background mutation rate.
FIG 2.

Clock-like nature of H3N2 CIV evolution. Regression of root-to-tip genetic divergence against sampling date was performed based on a phylogeny of concatenated major coding regions of H3N2 CIV sequences from Asia and the United States. Dotted ovals demark the approximate ranges of the MW2015, MW2016, and SE2017 U.S. phylogenetic clades.
Protein-level evolution and natural selection.
Putative or confirmed canine-adaptive mutations that differentiate H3N2 CIV from avian reservoir viruses have been reported previously (19, 26, 27) and likely occurred early in the emergence of these viruses. Interestingly, our analysis reveals that since becoming established in dogs, H3N2 CIV has consistently accumulated more nonsynonymous substitutions per site than those in reservoir H3N2 avian influenza A viruses (AIVs). (Fig. 3). Excluding M1 and NS1, which displayed considerable variance (likely due to their short sequence length), mean ratios of nonsynonymous to synonymous evolutionary changes (dN/dS) for the major coding regions in CIVs ranged from 0.15 to 0.26, whereas in the case of avian H3N2 the equivalent range was 0.03 to 0.10. These elevated mean dN/dS values for CIVs are reflected by numerous lineage-defining amino acid changes that have arisen throughout the genome during its circulation in Asia, and even within the short period of spread within the United States (Table 1; Fig. 4). Within the United States, a few of these changes were identified to have potential phenotypic consequences due to their position in or proximity to functional or antigenic protein domains. These consist of changes in HA, including one close to the receptor-binding site (at position 219) and one at an antigenic site (position 188); changes in NA, near the enzymatic active site (positions 155 and 222); and changes within the NS1 sequence (at position 218), resulting in a premature stop codon and an N-terminal truncation present among viruses in the SE2017 clade.
FIG 3.
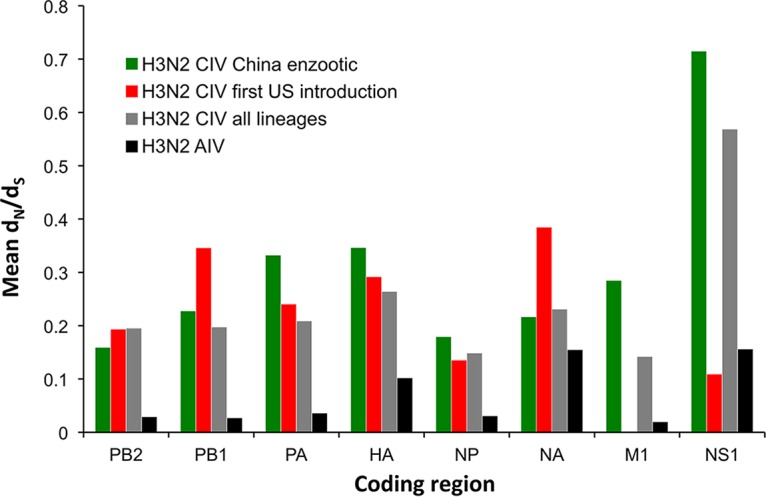
Mean dN/dS values for major coding regions of H3N2 CIV and H3N2 AIV. All H3N2 AIV sequences available in the database were included. Only full genomes of nonreassorted CIVs were analyzed. NS1 contains a portion of the NS2 alternative reading frame. It should be noted that estimates for the shorter segments (M1 and NS1) show variance, especially among U.S. viruses that have circulated for a short period and accumulated only a small number of nucleotide substitutions.
TABLE 1.
Amino acid diversity among H3N2 CIVs in the United Statesa

Consensus coding changes in each genomic segment (by position) that differentiate various viral groups of interest in this study. Residues that differ from those seen in the first U.S. outbreak of virus (Chicago 2015 outbreak) are shown in bold.
FIG 4.

Amino acid diversity among H3N2 CIVs in the United States. Clade consensus-level segregating amino acid positions across major coding regions in H3N2 CIV that differentiate various virus groups of interest in this study are shown. Numbering for each major reading frame starts at 1 on the first amino acid, with the exception of HA, which is numbered according to the standard H3 numbering scheme. See Table 1 for more details.
Despite this abundance of amino acid diversity, an analysis of selection pressures at individual amino acid sites, while often inadequate at the level of selective sweeps, did not reveal positive selection in any U.S. CIV lineage. A similar lack of detectable adaptive evolution was observed among the CIVs circulating within Asia, with the only exceptions being positive selection of a 2-amino-acid insertion in the NA stalk (detected with the SLAC method in HyPhy), characterized previously (26), and at HA residue 453 (detected with FUBAR; posterior probability = 0.98), which is of unknown phenotypic consequence. In addition, we found no evidence that any of the observed amino acid changes increased transmission rates of virus within the United States as determined by epidemiological analysis (see below).
Epidemiological analysis of CIV transmission in the United States.
One of the factors that determine the success of newly emerged viruses is their ability to be transmitted in the new host population. We used diagnostic sample data from three different centers which receive viruses from throughout the United States, as well as more regionally, to track the spread of CIVs and to understand their transmission characteristics at different stages of the epidemics. For the U.S. outbreaks, we had comprehensive coverage of diagnostic sampling for CIV over the entire outbreak as well as sample collection dates and postal codes (5-digit ZIP codes) to determine the spatial-temporal distribution of outbreaks across the entire United States (Fig. 5; see Movie S1 in the supplemental material). Importantly, both negative and positive quantitative PCR (qPCR) diagnostic testing results were included in this analysis, showing the intensity of testing in each region.
FIG 5.

Spatiotemporal incidence of H3N2 CIV infection in the United States. Samples included those collected throughout the United States between March 2015, soon after the first introduction of virus, and September 2017. Blue marks indicate samples that were tested for CIV but were negative, while red marks indicate samples that tested positive for H3N2 CIV. Yellow bars on the incidence-by-date histogram (lower panel) indicate periods of elevated incidence. An animated version of this map is included in Movie S1 in the supplemental material.
In total, surveillance efforts processed 33,435 samples, of which 1,718 were positive, with an average (± standard deviation [SD]) of 1,078 ± 532 samples processed per month, of which 55 ± 70 were positive. From the distribution of the positive and negative samples versus time and ZIP code in the United States (Fig. 6A), we identified nine spatially and temporally separate outbreaks, which comprised the majority of viruses in the surveillance data (Fig. S1). Four of these outbreaks occurred in 2015, one in 2016, and four in 2017, with additional viruses circulating at low levels primarily within the Midwest states. Six of the nine epidemiologically defined outbreaks were represented in the sequence data, and phylodynamics analysis revealed that the epidemiologically defined outbreaks were consistent with the underlying phylogenetic topology of the virus as it circulated in the United States, with each outbreak or cluster of outbreaks associated with a monophyletic clade of viruses (Fig. 6B). Interestingly, we found no outbreaks associated with the MW2016 clade. While outbreaks not represented in the sequence data (e.g., Cincinnati 2015) may be associated with viruses that fall into this clade, it appears that the MW2016 clade is dominated by viruses circulating at low but persistent levels in the Midwest United States between March 2016 and February 2017. Overall, this analysis emphasizes the burst–fade-out dynamics of phylogenetically distinct viruses circulating throughout the United States, with additional low levels of viruses circulating primarily within the Midwest United States.
FIG 6.

Phylodynamics analysis of major outbreaks occurring during the circulation of H3N2 CIV in the United States. (A) Spatiotemporal incidence of the virus and sequencing coverage of the U.S. epidemic. Black ticks represent samples testing positive for CIV by qRT-PCR, gray ticks represent negative tests, and triangles represent sequenced viruses, with all samples separated by collection date (horizontal axis) and ZIP code (vertical axis). Epidemiologically defined outbreak clusters are demarked by colored boxes, and sequenced virus markers are colored accordingly. (B) Phylogenetic structure of U.S. epidemiological outbreaks based on concatenated segment major reading frames (as in Fig. 1B, inset). Branch tips (triangles) are colored by epidemiologically defined outbreaks as in panel A. Open node circles indicate bootstrap proportions of >98%.
Despite distinct phylogenetic histories, nearly all U.S. outbreaks exhibited similar R0 values of around 1.0 to 1.5 (Fig. 7), based on epidemiological modeling. Such levels of transmission alone would suggest the potential to establish an endemic virus in dogs, at least by comparison with human IAVs (28). However, the repeated pattern of sporadic outbreaks interspersed with frequent fade-outs (or singleton cases in the surveillance data) suggests that these viruses are generally relatively poorly transmissible among dogs across different metapopulation networks. Specifically, while epidemic growth rates appear to be sufficient for pandemic spread in dense and/or well-connected host populations (likely animal shelters and kennels in the case of CIV in the United States), the sparse connectivity of these populations acts a barrier to pandemic formation.
FIG 7.
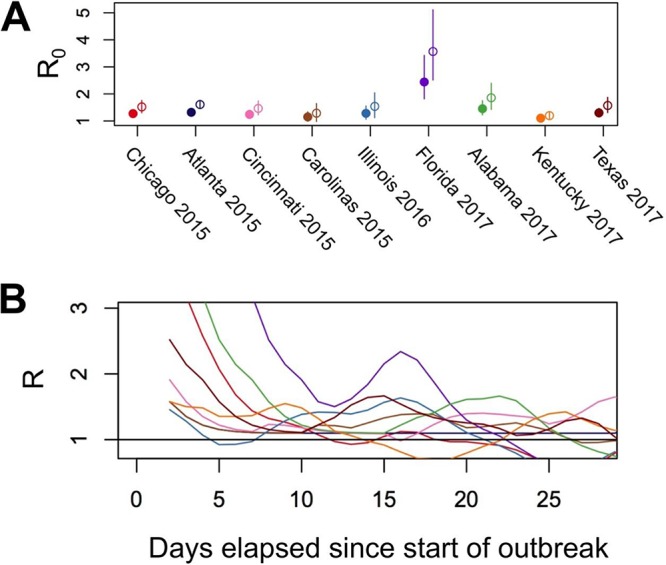
Transmission rates among different U.S. outbreaks. (A) R0 estimates for the different U.S. outbreaks based on epidemiological analysis. Estimates were performed using a gamma-distributed serial interval for both short (mean = 3.5 days; SD = 1.5 days; solid dots) and long (mean = 7 days; SD = 6 days; open dots) assumed serial interval periods. Error bars represent 95% credible intervals. (B) Estimates of the variation in the rate of transmission over the course of each outbreak.
Interestingly, the Florida 2017 outbreak appeared to produce a relatively large number of secondary infections, on average (R0 of ∼2.5 to 3.5) (Fig. 7). However, differences in the R0 values and rates of secondary infections did not differ significantly between any of the epidemiologically defined outbreaks when the 95% confidence intervals were considered (Fig. 7; Fig. S2). Furthermore, viral sequences recovered from the Florida 2017 outbreak were identical to those from other concurrent outbreaks in other southeastern states (Fig. 1B and 6B) that exhibited lower R0 values. This suggests that the elevated transmission rates observed were likely transient and/or due to random variation in the stochastic process of disease spread rather than due to viral adaptation to canine hosts.
DISCUSSION
We show here that the U.S. dog population has experienced multiple, periodic introductions of Asian-origin H3N2 CIV since 2015, resulting in hierarchically structured outbreak bursts of various sizes, geographic locations, and durations. In this way, the emergence of H3N2 CIV and its evolutionary and epidemiological patterns within the United States are similar to those seen previously for other viruses that have emerged in new hosts, with persistence in a small number of locations combined with outbreaks and epidemics that persisted for various periods (18, 29, 30). However, despite the multiple introductions of these viruses into the United States after approximately a decade of circulation in Asia and the accumulation of numerous protein-level changes, our data suggest that H3N2 CIV has been unable to overcome the barriers associated with producing large-scale incursion into the general dog population. We found no clear or consistent evidence for direct codon-specific adaptive evolution in H3N2 CIV, such that the elevated mean dN/dS values most likely result from relaxed selective constraints in dogs compared to those in birds, and perhaps from the occurrence of recurrent population bottlenecks that allow the accumulation of slightly deleterious amino acid changes. Moreover, the fact that elevated mean dN/dS values were observed across all major coding regions is inconsistent with positive selection for host-adaptive mutations, which would be expected to result in more targeted genomic changes. Whatever the cause, these results illustrate the challenge inherent in trying to determine adaptive protein changes through simple comparisons of viral genome sequences, and further experimental work is needed to clarify any functional nuances of the coding changes described in this study.
There are about 80 million dogs in the United States, or over one-fourth the size of the human population. Critically, however, in comparison to the human population, the dog population differs in significant aspects of its structure and in the levels of the close and prolonged physical contacts that facilitate the transmission of IAVs. In the United States, both the H3N8 and H3N2 CIVs appear to sustain transmission only in unusually well-connected populations of dogs, such as in animal shelters and kennels in large cities (18, 31). This contact heterogeneity, in combination with increased vaccination and control measures, likely contributed to the reduced prevalence of H3N8 CIV in recent years, and similar barriers to large-scale and sustained transmission patterns may exist in other highly structured host populations that have experienced outbreaks of IAVs, such as seals in rookeries, mink in fur farms, and domestic horses.
Given this type of population structure, significantly increased transmission rates of IAVs might best be accomplished by the acquisition of traits that increase the likelihood of a virus reaching a new metapopulation. For instance, among dogs, prolonged environmental persistence of canine parvovirus (32), and thus a great capacity for indirect transmission, appears to be key to its pandemic spread (33). Changes in environmental survival are less commonly considered for IAVs due to their labile viral envelope. However, alternative adaptive traits, such as an increased incubation time or duration of virus shedding by an infected dog, are plausible and may produce similar epidemiological outcomes. Critically, however, we propose that it is unlikely that such traits would be selected for in CIVs spreading in animal shelters, where there may be fewer barriers to efficient transmission given the close and frequent contact between hosts, and in this way, H3N2 CIV may currently be confined to an evolutionary cul-de-sac.
It is too early to know whether this mode of circulation necessarily predestines the H3N2 CIV to a fate similar to that of the H3N8 CIV epidemic, which has largely faded out in recent years. Many similarities between these epidemics do exist, including a multiyear presence in one or two densely populated cities (New York for H3N8 CIV and Chicago for H3N2 CIV) and additional, transient outbreaks that occurred throughout the country, largely confined to shelter and kennel populations. However, our results have highlighted potentially critical differences. Whereas the H3N8 CIV U.S. epidemic arose from a cross-species transmission event that occurred within the United States, the H3N2 CIV U.S. epidemic is clearly driven by multiple introductions from an Asian-origin source population of virus also circulating in dogs. It is likely that some or all of the introductions of H3N2 CIV into the United States occurred through the transfer of rescued dogs from Asia, which come from populations that appear to be infected endemically (34–36). As there is no formal quarantine requirement for dogs being brought to the United States or barriers that would stop the introduction of most canine respiratory pathogens, including IAVs, we may continue to see incursions of H3N2 CIV into the United States. Further studies comparing the evolutionary trajectories of the two CIVs, as well as a more detailed understanding of the evolutionary dynamics of H3N2 CIV in Asia and of IAV epidemics in other mammalian populations, are needed to identify other critical events in the emergence and long-term establishment of these viruses in different host population contexts.
MATERIALS AND METHODS
Sample collection.
No animal samples were collected directly for the purposes of this study. Rather, samples were obtained under standard passive diagnostic procedures, using samples submitted to various diagnostic laboratories to determine the etiological agent(s) causing respiratory disease in dogs. Viral RNAs were extracted from nasal or pharyngeal swabs collected from dogs showing signs of respiratory infection and screened in a quantitative reverse transcription-PCR (qRT-PCR) using primers detecting the M protein gene from any IAVs (37, 38). Typing of positive samples was performed by retesting using typing primers (39) to confirm the H3 and N2 genotypes of the outbreak viruses. While the sampling distribution is heterogeneous, arising from the complex decision-making processes of individual providers (whether to submit or not), the spatiotemporal coverage of sampling (positive and negative samples) indicates relatively strong coverage for a passive surveillance network of an emerging infection.
Viral sequences.
All samples sequenced in this study originated from nasal or pharyngeal swabs collected directly from infected dogs and stored in virus transport medium. A small number of samples were passaged once in either Madin-Darby canine kidney (MDCK) epithelial cells (NBL-2; ATCC CCL-34) (1 sample) or embryonated chicken eggs (12 samples) obtained from a commercial vendor. Fertilized eggs were inoculated with virus after 10 days of incubation at 37°C, and allantoic fluid was harvested after an additional 5-day incubation at 37°C. All fertilized eggs were terminated before hatching.
An effort was made to sequence all viruses received from diagnostic centers for which initial diagnostic threshold cycle (CT) values were sufficient for full-genome amplification (which we found to be in the range of <∼25). Due to limited resources, samples from unique times and/or geographic locations were occasionally given priority over samples representing redundant spatiotemporal coverage. Viral RNA was extracted using a QIAamp viral RNA minikit, and cDNA was generated with the SuperScript III OneStep RT-PCR system with Platinum Taq DNA polymerase (Invitrogen). The entire genome of each virus was amplified using common primers that recognize the terminal sequences of the fragments (underlined regions) (GTTACGCGCCAGCGAAAGCAGG and GTTACGCGCCAGTAGAAACAAGG) in 36 to 40 rounds of PCR. Viral cDNA was purified with a 0.45× volume of AMPure XP beads (Beckman Coulter), and 1 ng input DNA was used to construct barcoded sequencing libraries with a NexteraXT kit. Libraries were multiplexed, sequences were determined using Illumina MiSeq 2 × 250 sequencing, and consensus sequences were determined for each genomic segment. Additional H3N2 CIV sequences were obtained from GenBank (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html), along with the closest non-canine-virus sequences in the databases for each gene segment as identified by nucleotide BLAST searches (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Phylogenetic analysis.
Consensus sequence editing, alignment, and phylogenetic analyses were performed with Geneious v9.0.5 (40). Each gene segment was trimmed to contain only its major open reading frame, aligned using MAFFT v7.222 (41), and either analyzed separately or concatenated with all other genomic segment sequences from the same virus. No passage-specific mutations were evident when sequences from passaged virus were compared to those obtained directly from original nasal wash material, and all sequences, regardless of passage history, were therefore used in our analyses. For individual segment phylogenies, segments from intersubtype reassortant viruses and H3N2 viruses isolated from dogs but containing segments from non-canine-virus hosts were excluded. For concatenated full-genome phylogenies, both inter- and intrasubtype reassortant viruses, as determined by RDP4 analysis (42) and individual segment phylogeny comparisons, were excluded. This resulted in a total of 76 full genomes analyzed. Total sequence alignment lengths were as follows (in nucleotides [nt]): PB2, 2,277; PB1, 2,268 to 2,271; PA, 2,148; HA, 1,698; NP, 1,494; NA, 1,407 to 1,413; M1, 756; and NS1, 690. Concatenation of major coding regions from the eight segments yielded a total consensus alignment length of 12,741 to 12,747 nt. Phylogenetic relationships among the sequences were determined using the maximum likelihood (ML) method available in PhyML (43), employing a general time-reversible (GTR) substitution model, gamma-distributed (Γ) rate variation among sites, and bootstrap resampling (1,000 replications). All trees were rooted with the earliest and most basal H3N2 CIV isolate available (A/canine/Guangdong/1/2006 [H3N2]).
To assess the degree of clock-like structure in the data, we performed a regression of root-to-tip genetic distances against dates of sampling by using the ML tree described above and the TempEst program (44). Day and year values were utilized for all samples for which this information was available. For samples lacking the exact day of collection, the first of the month was used. As root-to-tip analysis revealed a strong molecular clock (see Results), we next conducted a more detailed analysis of evolutionary rates by using the Bayesian Markov chain Monte Carlo method implemented in the BEAST package, version 1.8.2 (23). Accordingly, rates of nucleotide substitution were estimated by assuming a GTR+ Γ nucleotide substitution model and a strict molecular clock (following the root-to-tip regression) under both constant population and Bayesian skyline demographic models. Each analysis was run for 100 million generations, with a 10% burn-in and with statistical uncertainty reflected in values for the 95% highest posterior density (HPD).
Sequence variation and selection analyses.
Majority consensus sequences were generated for each monophyletic outbreak or cluster of outbreaks within the United States. Consensus sequences were aligned and segregating sites recorded. Positions of segregating sites on phylogenetic trees were confirmed manually and using the Treesub program (https://github.com/tamuri/treesub). Codon-specific and mean ratios per segment of the numbers of nonsynonymous and synonymous substitutions per rate (dN/dS) for various groups of viruses were estimated using the SLAC and FUBAR methods in the HyPhy package, available through the DATAMONKEY Web interface (45).
Epidemiological analysis and modeling of transmission and outbreaks.
We used detailed data from the first U.S. outbreak to define the epidemiological parameters of the virus in the dog population. Results were obtained for nasal swab specimens submitted for testing to a commercial diagnostic laboratory (Idexx Corporation, Sacramento, CA) which received samples from throughout the United States, and occasionally from sources in South Korea, as well as to the veterinary diagnostic laboratories at Cornell University and the University of Wisconsin. All results were classified by postal code (ZIP code), allowing the location to be determined with high accuracy, especially within the cities, as well as by the date the sample was collected. These data were used along with the phylogeny of the viruses to track the spread of the virus in the United States, including that of the different variants. As expected, many of the viruses were found within the vicinity of Chicago, IL, while others were found as single or small groups of samples from other regions of the United States, or in some cases were sampled from larger outbreaks. These data were used to infer the epidemic curve of the virus overall and for nine spatially and temporally contiguous outbreaks and to estimate the epidemiological parameters of its spread by use of standard methods. Specifically, we used the “exponential growth” approach to estimating R0 as described by Wallinga and Lipsitch (46) and implemented in the R package R0, using case count time series from the time of the first case to the time of peak incidence. These time series were smoothed by back filtering from date of submission to date of infection, using a gamma distribution with a mean of 3 days and a standard deviation of 1 day, which dampened fluctuations in the raw case count time series due to most laboratory tests being submitted on weekdays. A second U.S. outbreak was seen to initiate with the introduction of a second virus from South Korea in early 2017, and we sequenced samples from Georgia, Florida, Texas, Kentucky, Indiana, Illinois, and Minnesota up until November 2017. As all viruses sequenced after May 2017 were very similar and fell into a single clade, we assumed that any positive samples from those states after May 2017 were from the second introduction and used the more limited information about this outbreak to define the epidemiological parameters and compare these data to those from the first outbreak.
Accession number(s).
All sequences produced as part of this study were submitted to the NCBI, and accession numbers are reported in Table S1 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wendy Weichert, Renee Anderson, Brittany Chilson, and Jayeeta Dutta for their technical support on this project.
Christian Leutenegger is employed by the commercial company Idexx Laboratories, Inc. The contribution of this author included resource acquisition for many samples sequenced here, demographic data related to the samples, and assistance in drafting the manuscript. At no point did the affiliation with Idexx Inc. influence the study design, interpretation of results, or conclusions drawn.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00323-18.
REFERENCES
- 1.Geoghegan JL, Holmes EC. 2017. Predicting virus emergence amid evolutionary noise. Open Biol 7:170189. doi: 10.1098/rsob.170189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parrish CR, Holmes EC, Morens DM, Park E-C, Burke DS, Calisher CH, Laughlin CA, Saif LJ, Daszak P. 2008. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev 72:457–470. doi: 10.1128/MMBR.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woolhouse MEJ, Haydon DT, Antia R. 2005. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol Evol 20:238–244. doi: 10.1016/j.tree.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM. 2005. Superspreading and the effect of individual variation on disease emergence. Nature 438:355–359. doi: 10.1038/nature04153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parrish CR, Murcia PR, Holmes EC. 2015. Influenza virus reservoirs and intermediate hosts: dogs, horses, and new possibilities for influenza virus exposure of humans. J Virol 89:2990–2994. doi: 10.1128/JVI.03146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon S-W, Webby RJ, Webster RG. 2014. Evolution and ecology of influenza A viruses. Curr Top Microbiol Immunol 385:359–375. doi: 10.1007/82_2014_396. [DOI] [PubMed] [Google Scholar]
- 7.Horimoto T, Kawaoka Y. 2001. Pandemic threat posed by avian influenza A viruses. Clin Microbiol Rev 14:129–149. doi: 10.1128/CMR.14.1.129-149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CDC. 2017. Examples of human infections with avian influenza A viruses with possible limited, non-sustained human-to-human transmission. https://www.cdc.gov/flu/avianflu/h5n1-human-infections.htm.
- 9.Cauldwell AV, Long JS, Moncorgé O, Barclay WS. 2014. Viral determinants of influenza A virus host range. J Gen Virol 95:1193–1210. doi: 10.1099/vir.0.062836-0. [DOI] [PubMed] [Google Scholar]
- 10.Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki Y, Ito T, Suzuki T, Holland RE, Chambers TM, Kiso M, Ishida H, Kawaoka Y. 2000. Sialic acid species as a determinant of the host range of influenza A viruses. J Virol 74:11825–11831. doi: 10.1128/JVI.74.24.11825-11831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taubenberger JK, Kash JC. 2010. Influenza virus evolution, host adaptation and pandemic formation. Cell Host Microbe 7:440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russier M, Yang G, Rehg JE, Wong S-S, Mostafa HH, Fabrizio TP, Barman S, Krauss S, Webster RG, Webby RJ, Russell CJ. 2016. Molecular requirements for a pandemic influenza virus: an acid-stable hemagglutinin protein. Proc Natl Acad Sci U S A 113:1636–1641. doi: 10.1073/pnas.1524384113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y. 2012. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486:420–428. doi: 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keleta L, Ibricevic A, Bovin NV, Brody SL, Brown EG. 2008. Experimental evolution of human influenza virus H3 hemagglutinin in the mouse lung identifies adaptive regions in HA1 and HA2. J Virol 82:11599–11608. doi: 10.1128/JVI.01393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Widjaja L, Ilyushina N, Webster RG, Webby RJ. 2006. Molecular changes associated with adaptation of human influenza A virus in embryonated chicken eggs. Virology 350:137–145. doi: 10.1016/j.virol.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Tamura D, Nguyen HT, Sleeman K, Levine M, Mishin VP, Yang H, Guo Z, Okomo-Adhiambo M, Xu X, Stevens J, Gubareva LV. 2013. Cell culture-selected substitutions in influenza A(H3N2) neuraminidase affect drug susceptibility assessment. Antimicrob Agents Chemother 57:6141–6146. doi: 10.1128/AAC.01364-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalziel BD, Huang K, Geoghegan JL, Arinaminpathy N, Dubovi EJ, Grenfell BT, Ellner SP, Holmes EC, Parrish CR. 2014. Contact heterogeneity, rather than transmission efficiency, limits the emergence and spread of canine influenza virus. PLoS Pathog 10:e1004455. doi: 10.1371/journal.ppat.1004455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu H, Hughes J, Murcia PR. 2015. Origins and evolutionary dynamics of H3N2 canine influenza virus. J Virol 89:5406–5418. doi: 10.1128/JVI.03395-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song D, Kang B, Lee C, Jung K, Ha G, Kang D, Park S, Park B, Oh J. 2008. Transmission of avian influenza virus (H3N2) to dogs. Emerg Infect Dis 14:741–746. doi: 10.3201/eid1405.071471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crawford PC, Dubovi EJ, Castleman WL, Stephenson I, Gibbs EPJ, Chen L, Smith C, Hill RC, Ferro P, Pompey J, Bright RA, Medina M-J, Group IG, Johnson CM, Olsen CW, Cox NJ, Klimov AI, Katz JM, Donis RO. 2005. Transmission of equine influenza virus to dogs. Science 310:482–485. doi: 10.1126/science.1117950. [DOI] [PubMed] [Google Scholar]
- 22.Voorhees IEH, Glaser AL, Toohey-Kurth K, Newbury S, Dalziel BD, Dubovi EJ, Poulsen K, Leutenegger C, Willgert KJE, Brisbane-Cohen L, Richardson-Lopez J, Holmes EC, Parrish CR. 2017. Spread of canine influenza A(H3N2) virus, United States. Emerg Infect Dis 23:1950–1957. doi: 10.3201/eid2312.170246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Holmes EC. 2006. Avian influenza virus exhibits rapid evolutionary dynamics. Mol Biol Evol 23:2336–2341. doi: 10.1093/molbev/msl102. [DOI] [PubMed] [Google Scholar]
- 25.Westgeest KB, Russell CA, Lin X, Spronken MIJ, Bestebroer TM, Bahl J, van Beek R, Skepner E, Halpin RA, de Jong JC, Rimmelzwaan GF, Osterhaus ADME, Smith DJ, Wentworth DE, Fouchier RAM, de Graaf M. 2014. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J Virol 88:2844–2857. doi: 10.1128/JVI.02163-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Y, Xie X, Zhao Y, Kalhoro DH, Lu C, Liu Y. 2016. Enhanced replication of avian-origin H3N2 canine influenza virus in eggs, cell cultures and mice by a two-amino acid insertion in neuraminidase stalk. Vet Res 47:53. doi: 10.1186/s13567-016-0337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang G, Li S, Blackmon S, Ye J, Bradley KC, Cooley J, Smith D, Hanson L, Cardona C, Steinhauer DA, Webby R, Liao M, Wan X-F. 2013. Mutation tryptophan to leucine at position 222 of haemagglutinin could facilitate H3N2 influenza A virus infection in dogs. J Gen Virol 94:2599–2608. doi: 10.1099/vir.0.054692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biggerstaff M, Cauchemez S, Reed C, Gambhir M, Finelli L. 2014. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 14:480. doi: 10.1186/1471-2334-14-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauch CT, Lloyd-Smith JO, Coffee MP, Galvani AP. 2005. Dynamically modeling SARS and other newly emerging respiratory illnesses: past, present, and future. Epidemiology 16:791. doi: 10.1097/01.ede.0000181633.80269.4c. [DOI] [PubMed] [Google Scholar]
- 30.Lau MSY, Dalziel BD, Funk S, McClelland A, Tiffany A, Riley S, Metcalf CJE, Grenfell BT. 2017. Spatial and temporal dynamics of superspreading events in the 2014-2015 West Africa Ebola epidemic. Proc Natl Acad Sci U S A 114:2337–2342. doi: 10.1073/pnas.1614595114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pecoraro HL, Bennett S, Huyvaert KP, Spindel ME, Landolt GA. 2014. Epidemiology and ecology of H3N8 canine influenza viruses in US shelter dogs. J Vet Intern Med 28:311–318. doi: 10.1111/jvim.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saknimit M, Inatsuki I, Sugiyama Y, Yagami K. 1988. Virucidal efficacy of physico-chemical treatments against coronaviruses and parvoviruses of laboratory animals. Jikken Dobutsu 37:341–345. [DOI] [PubMed] [Google Scholar]
- 33.Parrish CR, Kawaoka Y. 2005. The origins of new pandemic viruses: the acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu Rev Microbiol 59:553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- 34.Su S, Cao N, Chen J, Zhao F, Li H, Zhao M, Wang Y, Huang Z, Yuan L, Wang H, Zhang G, Li S. 2012. Complete genome sequence of an avian-origin H3N2 canine influenza A virus isolated in farmed dogs in southern China. J Virol 86:10238. doi: 10.1128/JVI.01601-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su S, Chen Y, Zhao F-R, Chen J-D, Xie J-X, Chen Z-M, Huang Z, Hu Y-M, Zhang M-Z, Tan L-K, Zhang G-H, Li S-J. 2013. Avian-origin H3N2 canine influenza virus circulating in farmed dogs in Guangdong, China. Infect Genet Evol 19:251–256. doi: 10.1016/j.meegid.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Lee C, Song D, Kang B, Kang D, Yoo J, Jung K, Na G, Lee K, Park B, Oh J. 2009. A serological survey of avian origin canine H3N2 influenza virus in dogs in Korea. Vet Microbiol 137:359–362. doi: 10.1016/j.vetmic.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 37.Shu B, Wu K-H, Emery S, Villanueva J, Johnson R, Guthrie E, Berman L, Warnes C, Barnes N, Klimov A, Lindstrom S. 2011. Design and performance of the CDC real-time reverse transcriptase PCR swine flu panel for detection of 2009 A (H1N1) pandemic influenza virus. J Clin Microbiol 49:2614–2619. doi: 10.1128/JCM.02636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. 2002. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 40:3256–3260. doi: 10.1128/JCM.40.9.3256-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Khan MI, Khan M, Măndoiu I. 2013. Neuraminidase subtyping of avian influenza viruses with PrimerHunter-designed primers and quadruplicate primer pools. PLoS One 8:e81842. doi: 10.1371/journal.pone.0081842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 44.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2:vew007. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pond SLK, Frost SDW. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 46.Wallinga J, Lipsitch M. 2007. How generation intervals shape the relationship between growth rates and reproductive numbers. Proc Biol Sci 274:599–604. doi: 10.1098/rspb.2006.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 48.Hong M, Na W, Yeom M, Park N, Moon H, Kang B-K, Kim J-K, Song D. 2014. Complete genome sequences of H3N2 canine influenza virus with the matrix gene from the pandemic A/H1N1 virus. Genome Announc 2:e01010-14. doi: 10.1128/genomeA.01010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee IH, Le TB, Kim HS, Seo SH. 2016. Isolation of a novel H3N2 influenza virus containing a gene of H9N2 avian influenza in a dog in South Korea in 2015. Virus Genes 52:142–145. doi: 10.1007/s11262-015-1272-z. [DOI] [PubMed] [Google Scholar]
- 50.Song D, Moon H-J, An D-J, Jeoung H-Y, Kim H, Yeom M-J, Hong M, Nam J-H, Park S-J, Park B-K, Oh J-S, Song M, Webster RG, Kim J-K, Kang B-K. 2012. A novel reassortant canine H3N1 influenza virus between pandemic H1N1 and canine H3N2 influenza viruses in Korea. J Gen Virol 93:551–554. doi: 10.1099/vir.0.037739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson TC, Bromfield CR, Crawford PC, Dodds WJ, Gibbs EPJ, Hernandez JA. 2012. Serological evidence of H3N8 canine influenza-like virus circulation in USA dogs prior to 2004. Vet J 191:312–316. doi: 10.1016/j.tvjl.2011.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.