

Abstract
Gangliosides are sialic acid-containing glycosphingolipids recognized to play essential role in biological processes. Both the glycan and lipid structures influence their biological function, and thus necessitate their determination as intact molecular species. To our knowledge, no multiplexed method for intact gangliosides currently exists. In this paper, we aimed to demonstrate an approach for isobaric labeling of intact gangliosides. Specifically, we carried out the rapid, chemoselective oxidation of sialic acid side chain in common ganglioside core structures using NaIO4 followed by ligation with a carbonyl-reactive isobaric tandem mass tag (TMT) reagent, and subsequent RPLC-MS/MS analysis. Attachment of the isobaric label was observed to improve the ionization efficiency of complex gangliosides using electrospray ionization. Fragmentation of the resulting [M+2H]2+ ions of TMT-labeled gangliosides provided information-rich spectra containing fragments from the glycan, lipid, and TMT reporter ions. This facile approach enabled simultaneous quantification of up to six samples as well as identification of glycan and lipid compositions in a single injection. As a proof-of-concept, using porcine brain total ganglioside extracts pooled at known ratios, we obtained overall sample-to-sample precision of <12% RSD and mean error of <10%. This showcased the great promise and feasibility of this strategy for high-throughput analysis of intact gangliosides in biological extracts.
Table of Contents
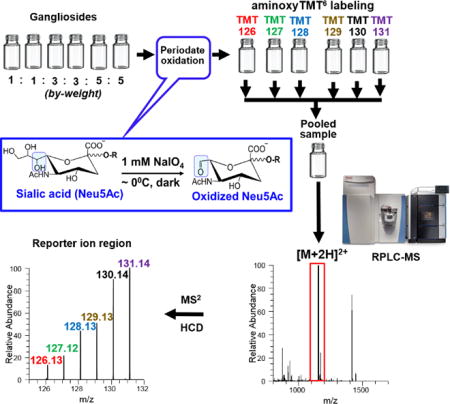
Gangliosides are glycosphingolipids composed of a ceramide backbone and a glycan head group that carries at least one sialic acid moiety. In mammals, the most abundant form of the latter is N-acetyl neuraminic acid (Neu5Ac) (Fig.1a)1,2. These ubiquitous molecules are recognized to play important role in biological processes, and perturbation of their metabolism is often associated with human diseases. Despite this, they remain relatively underexplored due to their compositional and structural complexity. In general, the overall biological function of glycosphingolipids is influenced by both the structures of the lipid and glycan, thus they warrant to be determined as intact molecular species3–5.
Figure 1.

Representative structure of a ganglioside (GM1a d18:1/18:0) (A). The structure of aminoxyTMT isobaric label (B) and the chemoselective oxidation of sialic acid side chain using NaIO4 (C). During CID or HCD, the bond shown in (B) breaks to generate the reporter ion. Numbering of carbon atoms in the Neu5Ac used throughout this text is also shown.
Traditional analysis of gangliosides involves cleavage of the lipid from the glycan head group followed by gas chromatography–mass spectrometry (GC-MS)6. In recent years, the use of liquid chromatography–mass spectrometry (LC-MS) based approaches namely reversed-phase (RPLC)7–10, and hydrophilic interaction (HILIC)11–15 as well as chip-based technologies16,17 have made it possible to analyze intact gangliosides18,19. Despite these developments, quantification of these biomolecules has remained a challenging task due to limited availability of standards, variability of ionization efficiency across ganglioside classes, and throughput. To this end, relative quantification is still considered the most practical approach for these biomolecules20.
Isobaric labeling is one promising technique for relative quantification using MS/MS-capable instruments. Here, analytes are covalently linked with isobaric tags through a reactive functional group, such as aldehyde. Isobaric label reagents are multiple versions of the same molecule which only differ in the number or site of isotopic elemental distribution21,22. Upon mass-selection and fragmentation of labeled compounds in an MS/MS experiment, reporter ions are generated whose intensity reflects the relative abundance of the analytes. This has been shown to minimize potential errors caused by variability of ionization process as well as run-to-run instrument response since samples are ionized simultaneously in a single injection. This technique has also been shown to greatly improve measurement accuracy, precision, and throughput23,24. In this respect, stable isotope labeling for multiplexed relative quantification in proteomics, metabolomics, and glycomics has been well demonstrated. In glycomics, multiplexing is commonly exploited using carbonyl-reactive isobaric mass tags22,25,26 such as aminoxy tandem mass tag (aminoxyTMT) reagent21,27,28. The latter contains a carbonyl-reactive moiety, a mass normalizer, and a reporter ion group, which fragments upon ion activation through collision-induced dissociation (CID), higher-energy collisional dissociation (HCD) or electron-transfer dissociation (ETD)21,27,28 (Fig. 1b). To use this reagent, glycans should contain a free carbonyl group at the reducing terminus to enable its attachment through a stable oxime bond21. Thus, enzymatic or chemical modification is commonly employed, e.g. glycopeptides/glycoproteins are digested with peptide N-glycosidase F (PNGase F)21,22,27,28 to release N-glycans. If glycolipids are desired to be labeled with aminoxyTMT, treatment with endoglycoceramidases (EDCase)29 would be necessary. Not only this technique eliminates the lipid structural information, but also the use of enzyme requires longer sample preparation time which typically takes ~18 hours.
Selective oxidation of vicinal diols using sodium metaperiodate (NaIO4) has been known in organic chemistry for decades30–32 and was utilized for labeling of live cells through bioconjugation of cell-surface sialic acids31,33,34. Under properly controlled conditions, Neu5Ac side chain can be selectively cleaved to form aldehyde at C-7 position leaving the remainder of the molecule unaltered (Fig.1c)31,33,34. In this paper, we applied this approach to label gangliosides with aminoxyTMT reagent (Scheme 1) followed by RPLC-MS/MS analysis. This facile strategy enabled the simultaneous analysis of ganglioside molecular species from multiple samples in one injection.
Scheme 1.

Reactions involved in the oxidation and isobaric labeling technique implemented in this study. Shown here is GM1a d18:1/18:0 as an ex-ample. Chemoselective oxidation of sialic acid side chain to generate aldehyde functional group (A). The afforded product undergoes aminoxyTMT labeling at the reactive aldehyde via oxime bond formation (B).
MATERIALS AND METHODS
Materials and reagents
Detailed list of materials and reagents is provided in the supporting information. Structure and systematic name of gangliosides discussed in the text are provided in Supporting Information Table S1.
Chemoselective oxidation
The protocol described by Hermanson was adopted with modifications31. Briefly, aliquots equivalent to 1.0 nmol to 5.0 nmol of individual ganglioside standards or porcine brain ganglioside extract (1.9 µg to 9.4 µg) were transferred to glass vials from 1.0 µg/µL stock solutions in chloroform/methanol/water (CHCl3/CH3OH/H2O, 2:1:0.1, v/v), and dried under a stream of nitrogen (N2) gas. The dried samples were reconstituted in 250 µL of oxidation buffer, 0.1 M CH3COONa/CH3COOH (pH ~5.5) and then added with 12.5 µL of 20 mM NaIO4 prepared in the same buffer. After vortexing for five seconds, the vial was kept in an ice bath (~0°C) in the dark. After incubation for 60 minutes, the reaction was quenched by adding 2.5 µL of 100 mM aqueous glycerol, incubated for another 60 minutes and then subjected to solid-phase extraction (SPE) to remove salts and dried to eliminate by-products of the oxidation process.
SPE clean-up
A 1.0 mL capacity Isolute C18 SPE cartridge (Biotage) was activated using 2.5 mL of CH3OH and conditioned with 2.5 mL of CH3OH/H2O (10/90, v/v). The oxidized ganglioside was loaded in the cartridge and desalted by washing five times with 1.0 mL of H2O. Finally, the oxidized gangliosides were eluted using 1.0 to 3.0 mL of CHCl3/CH3OH/H2O (5:5:0.5, v/v), dried under N2 or vacuum, and kept at −20°C until further processing.
Isobaric labeling
Labeling using aminoxyTMT was performed following manufacturer’s instructions with minor modifications. Briefly, the labeling reagent was dissolved in 200 µL of 95% CH3OH with 0.1% CH3COOH, vortexed, and mixed with oxidized gangliosides for 10 mins at room temperature. The solvent was removed under vacuum followed by re-dissolving the sample in 200 µL of 95% CH3OH and vortexed for 10 min to ensure complete labeling. Thereafter, the sample was dried under N2 or vacuum. Excess labeling reagent was scavenged by adding 100 µL of 10% acetone in 95% CH3OH/H2O, pooled, dried under N2 and finally reconstituted in LC-MS mobile phase.
LC-MS/MS analysis
A binary Acquity UPLC system (Waters, Milford, MA) coupled with LTQ Orbitrap XL (Thermo Scientific, Bremen, Germany) was used to monitor the progress of chemoselective oxidation of ganglioside standards. For TMT-labeling experiments, a Vanquish UHPLC system (Thermo Scientific) coupled with Q Exactive HF (Thermo Scientific) was employed. In either case, a Cortecs C18 column (2.6 mm ID×100 mm, 1.6 µm) (Waters) and the following mobile phases were used35,36: A (60/40 CH3CN/H2O with 10 mM NH4COOH and 0.1% HCOOH), B (90/10 isopropanol/CH3CN with 10 mM NH4COOH and 0.1% HCOOH). The gradient was 30% B (0 min), 50% B (1 min), 70% B (7 min), 99% B (13 min), 30% B (13.1 min), and column equilibration for additional 2 min at 30% B. The column was maintained at 40°C and a flow rate of 350 µL/min. The injection volume was 5.0 µL.
Both mass spectrometers were operated using heated electrospray ionization (HESI) source in positive and negative ion modes. Since native gangliosides ionize more efficiently as negative ions17, oxidation reaction was monitored in ESI− mode, while TMT-labeled gangliosides are characterized in the ESI+ mode. In LTQ Orbitrap XL, the following parameters were used: Spray voltage 3.5 kV, vaporizer temperature 300°C, sheath gas 40 arb. units (au), auxiliary gas 20 au, sweep gas 1 au, ion transfer capillary temperature 300°C, tube lens voltage 100V, and resolution setting of 100,000. The Q Exactive HF settings were spray voltage 3.0 kV, vaporizer temperature 400°C, sheath gas 20 au, auxiliary gas 5 au, sweep gas 1 au, ion transfer capillary temperature 350°C, S-lens RF voltage 50V, and resolution 120,000. Fragmentation of TMT-labeled gangliosides was performed using parallel reaction monitoring (PRM) of pre-determined inclusion list. The following settings were used for HCD fragmentation: maximum injection time 100 ms, automatic gain control (AGC) 1e5, and resolution of 15,000.
RESULTS AND DISCUSSION
Optimization of oxidation conditions
We found that the optimum condition to oxidize Neu5Ac side chain to C-7 aldehyde involves incubation of the sample with 1 mM NaIO4 for 60 min in the dark at ~ 0°C, followed by quenching with glycerol, and further incubation for 60 min. Prolonged incubation prior to quenching resulted to a decline in the yield of the C-7 aldehyde which probably due to side reactions. Performing the reaction at room temperature led to formation of side products that could affect the downstream labeling steps. It also appeared that the use of CH3COONa/CH3COOH buffer is an important component during the oxidation process. Changing the solvent to purely organic or purely aqueous resulted to low yields and even side reactions involving opening of the susceptible neutral sugar rings at the non-reducing terminus (data not shown). We monitored the reaction progress using RPLC-MS/MS with LTQ Orbitrap XL as detector. Based on accurate mass data, conversion of the C-7 hydroxyl group of the model compound GM1a d18:1/18:0 to C-7 aldehyde was judged to be complete with nearly negligible side products (Fig. 2a–b). This result demonstrates that the target species has been efficiently converted to aldehyde-containing product. Inefficient or incomplete oxidation can be easily identified as manifested by the presence of other products resulting from the stepwise cleavage of the C-8 and C-9, which can be determined in a straightforward manner based on the mass difference of 14 Da, 18 Da, and 30 Da from the precursor ion, for example when the reaction is carried out at suboptimum conditions; these side products are speculated to originate from geminal diol intermediates which result from well-known hydration of aldehydes in aqueous medium (Supporting Information Fig. S1). It is of note that in GD1a, the Neu5Ac connected to the terminal Gal unit oxidized more efficiently than the one connected to the internal Gal (Supporting Information Fig. S3d). Furthermore, no degradation of labile Neu5Ac was observed both during oxidation and ionization in HESI. Based on the peak area of the substrate before and after oxidation, gangliosides were oxidized with an efficiency of at least ~ 90% across different classes investigated (vide infra). Reuter et al.37 showed that the Neu5Ac in GM1a ganglioside is hardly oxidizable; however, using our method, we observed highly efficient oxidation of this analyte. Notably, their method uses lower periodate: substrate molar ratio (10:1) while ours uses high enough periodate: substrate ratio (200:1) leading to efficient oxidation to take place but low enough to prevent unwanted side reactions to occur.
Figure 2.

Full scan MS spectrum of unoxidized (A) and oxidized (B) ganglioside GM1a d18:1/18:0 acquired using LTQ Orbitrap XL demonstrating almost negligible side reactions. (C) Representative extracted ion chromatograms (EIC’s) of GM1a d18:1/18:0 substrate before oxidation (panel 1), residual unoxidized substrate left after incubation with 1mM NaIO4 (panel 2), and the resulting product of oxidation (panel 3). Guide to symbols:
 : aldehyde at C-7 position of Neu5Ac side chain; Δ: absolute mass error; ■: tentatively assigned as sodium formate adduct, [M+HCOO−+Na+-H]− based on accurate mass measurement and isotopic distribution. Structure and systematic name of gangliosides are shown in Supporting Information Table S1. Full scan mass range of other gangliosides subjected to NaIO4 oxidation is shown in Supporting Information Fig. S3 & S4.
: aldehyde at C-7 position of Neu5Ac side chain; Δ: absolute mass error; ■: tentatively assigned as sodium formate adduct, [M+HCOO−+Na+-H]− based on accurate mass measurement and isotopic distribution. Structure and systematic name of gangliosides are shown in Supporting Information Table S1. Full scan mass range of other gangliosides subjected to NaIO4 oxidation is shown in Supporting Information Fig. S3 & S4.
Chemoselectivity of oxidation
While the widely accepted mechanism of IO4− oxidation involves the cleavage of vicinal diols, other structural motifs could also be oxidized31,32. For example, it is possible that common structural features in glycosphingolipids such as cis- or trans- diols in the neutral carbohydrate ring, and hydroxyl group adjacent to carbonyl in the fatty acyl chain could all be affected by IO4− oxidation. Thus, we subjected representative glycosphingolipids possessing these common structural features under the same conditions we employed for gangliosides. Specifically, we investigated GlcCer d18:1/18:0, Sulfatide d18:1/18:0, Gb3Cer d18:1/18:0, and GalCer d18:1/18:0(2ROH). Absolute abundance and peak area comparion between NaIO4-treated and control (only in oxidation buffer) show that effects of dilute NaIO4 to these motifs are negligible and remained intact under the conditions optimized for gangliosides (Supporting Information Fig. S2). Minimal difference in peak area and absolute intensity could be attributed to sample handling such as transfer from one container to the next and SPE steps which we consider to be acceptable and suffice to show that the oxidation process under these conditions are not detrimental to these motiffs. This validates previous studies about the selectivity of mild periodate oxidation31,33,34.
Oxidation of different ganglioside classes
To test the applicability of the oxidation process to different classes of ganglioside, we used the optimum condition to oxidize commercially available standards individually. Our results show that this procedure is applicable to different classes of ganglioside such as monosialogangliosides (contain one Neu5Ac) GM1a, GM2 and GM3, disialogangliosides (contain two Neu5Ac) GD1a, GD1b, GD2, and GD3, and trisialoganglioside (contain three Neu5Ac) GT1b (Fig. 3 and Supporting Information Fig. S3). These constitute the predominant mammalian core structures38 that are commonly analyzed both qualitatively and quantitatively in milk8,9, biofluids (cerebrospinal fluid, serum and plasma)10,12,15,39,40, and tissues14,15,41. It is noteworthy that when more than one Neu5Ac is present, the number of oxidizable Neu5Ac is dependent on how the C-7, C-8, and C-9 are used for glycosidic bonding or O-acetylation. Our results show that C-7 aldehyde is produced only when C-8 is free, consistent with the known mechanism of IO4− oxidation32. For example, GD1a has two oxidizable Neu5Ac whereas GD1b, GD2, and GD3 only have one since their Neu5Ac’s are connected through α(2–8) linkage (Supporting Information Table S1)42.
Figure 3.

Extracted ion chromatograms (EIC) of individual ganglioside standards oxidized using 1 mM NaIO4 demonstrating its applicability across different core structures. Asterisk (
) on the Neu5Ac symbol indicates aldehyde at C-7 position. Corresponding total ion chromatograms and full mass ranges are shown in Supporting Information Fig. S3 & S4.
To verify that aldehyde group is generated at the C-7 position of Neu5Ac side chain, we compared the fragmentation pattern of oxidized and unoxidized analytes. An observed neutral loss of 62.04 Da (−C2H6O2) from the [M-H]− ion or 31.02 Da from the corresponding [M-2H]2− ion confirmed that C-8 and C-9 of sialic acid have been cleaved off. For unoxidized analyte, using GM1a d18:1/18:0 as a model, Yn ions due to glycosidic bond cleavages and ceramide backbone were observed (annotated according to nomenclature proposed by Domon and Costello43, and Ann and Adams44). More importantly, loss of 291.0891 Da from the precursor ion, characteristic of Neu5Ac, generated the Y2β fragment at m/z 1253.7796, (Fig. 4a). When subjected to IO4− oxidation, instead of 291.0891, loss of 229.0571 Da was observed to yield the same Y2β fragment while majority of the ions remained the same for both analytes suggesting that loss of C2H6O2 originates from Neu5Ac (Fig. 4b). Moreover, the B1β fragment (m/z 290.0888) known as Neu5Ac marker8,13,15,17 is absent in the oxidized species, instead the corresponding m/z 228.0519 was observed (cf. Fig. 4a–b). Based on its accurate mass, this ion was assigned to be due to C9H10NO6 (Neu5Ac B1β ion - C2H6O2) with an absolute mass error of 0.83 ppm. This further confirms that IO4− oxidation took place selectively at the Neu5Ac portion of the molecule and that aldehyde is generated at C-7 position consistent with the results of earlier reports33,34. Fragmentation of precursor ions in negative mode of oxidized and unoxidized gangliosides generated peaks that are isobaric to TMT reporter ions (Fig. 4a–b, inset). By comparing individual carbohydrate standards, we deduced that m/z 131.03 originates from Hexose (Gal or Glc) fragment while m/z 126.05 originates mainly from Neu5Ac with minor contribution from GalNAc. Surprisingly, we also observed that m/z 126.05 appears in positive ionization polarity, which could pertain to species having the same molecular formula but differ in mass by ~2 electrons, so-called “twin ions”45 (cf. Fig. 4a–b and Fig. 5d). However, only m/z 126.05 in the TMT reporter ion region was observed after aminoxyTMT labeling upon fragmentation of [M+2H]2+ ions as discussed below. Since quantification using isobaric tag is carried out almost exclusively in positive ion mode21,27,28, the presence of these interfering ions in negative mode does not jeopardize the utilization of this technique.
Figure 4.

Negative ESI-MS/MS spectra of unoxidized (A) and oxidized (B) ganglioside GM1a d18:1/18:0 acquired using Q Exactive HF. Asterisk (
) on the Neu5Ac symbol indicates aldehyde at C-7 position. Insets show the magnified m/z 126–131 region. Δ indicates absolute mass error. Fragments are labeled according to Domon and Costello nomenclature43. Structure and systematic name of gangliosides are shown in Supporting Information Table S1.
Figure 5.

EIC of representative oxidized ganglioside GT1b d18:1/18:0 (A) before labeling (panel 1), residual oxidized species left after labeling (panel 2) and the resulting aminoxyTMT0 labeled product (panel 3) where notable enhancement in ionization is apparent. Representative MS/MS spectrum of the [M+2H]2+ of aminoxyTMT0-labeled GM1a d18:1/18:0 (B). The TMT reporter ion region of (B) is magnified to show the presence of interfering peak from GalNAc (m/z 126.0550) and its baseline resolution with the reporter ion m/z 126.1280; data were acquired at 15,000 resolution setting at MS2 level using Q Exactive HF (C); Δ indicates absolute mass error. Also observed is the contribution of the isotopic [M+1] peak of the aminoxyTMT label at m/z 127.1315. Reporter ion region of the [M+H]+ of unlabeled GM1a d18:1/18:0 (D) (for reference, the full MS/MS fragmentation of [M+H]+ ion of the unlabeled ganglioside is shown in Supporting Information Fig. S6b). This indicates that the most significant interfering ion for the analysis of gangliosides using the aminoxyTMT labeling in positive ion mode is the GalNAc-derived oxonium ion as described in the present work.
Isobaric labeling of ganglioside standards
Commercially available carbonyl-reactive aminoxyTMT0 was used to label the oxidized individual standard gangliosides GM1a, GM2, GM3, GD1a, GD1b, GD2, GD3, and GT1b (Supporting Information Fig. S5). We observed the TMT-labeled species as [M+2H]2+ ions in positive ion mode, and [M-H]− in the negative ion mode. Based on the peak area of unlabeled gangliosides before and after reaction with aminoxyTMT0, efficiency of labeling was estimated to be greater than 95%. This highly efficient labeling is attributed to the rapid oxime bond formation using aminoxy functional group in the isobaric tag21. Also, compared to unlabeled gangliosides, significant increase in ionization efficiency in the positive mode was observed (Fig. 5a). This is expected since the aminoxyTMT label made the analyte relatively more hydrophobic and enhanced its proton affinity. Zhou et al.28 and Zhong et al.27 showed that TMT-labeled glycans could exist in multiple charge states with varying number of proton and sodium adducts during ionization in ESI. In contrast, our mobile phase35 which is supplemented with 10 mM NH4COOH allowed ionization of TMT-labeled gangliosides as mostly in protonated form with minimal (<30% of the intensity of the protonated form) to no detectable sodium adducts in some species, thus allowing better quantification. Although gangliosides ionize better as negative ions8, the TMT reporter ions were only observed in the positive ion mode27,28,46, hence, the latter was used for the fragmentation of TMT-labeled gangliosides.
Successful ligation of aminoxyTMT0 to the oxidized monosialoganglioside GM1a was confirmed from the HCD fragmentation of [M+2H]2+ ions measured at a resolution setting of 15,000 FWHM at the MS2 level. We observed highly abundant B1β fragment at m/z 526.2871 (theoretical formula: C24H40N5O8, absolute mass error = 1.09 ppm) corresponding to the C-7 aldehyde linked to TMT via oxime bond. The high abundance of this fragment could be attributed to the lability of the Neu5Ac moiety during collisional dissociation. These results suggest that the aminoxyTMT0 label was successfully attached to the C-7 aldehyde of the oxidized Neu5Ac unit. The presence of B1β fragment in conjunction with reporter ions is useful as diagnostic markers for the straightforward identification of TMT-labeled ganglioside (Fig. 5b). Also, information about the composition of the lipid backbone is easily identified from Y0 or dehydrated Y0 (Y0’) and O” ions, the latter being diagnostic of the long chain base7,13. For instance, the Y0’ ion (m/z 548.5413, theoretical formula: C36H70NO2, absolute mass error = 1.23 ppm) in Fig. 5b indicates a lipid composition of 36:1 (total carbon: number of unsaturation), which could correspond to either d18:1/18:0, d20:1/16:0, or d16:1/20:0 and so on. The presence of the O” ion in the MS/MS spectrum at m/z 264.2684 (theoretical formula: C18H34N, absolute mass error = 2.69 ppm) facilitated the unequivocal assignment of the lipid moiety as d18:1/18:0. Likewise, GM1a d20:1/18:0 was identified using the m/z 292.2995 (O” ion, theoretical formula: C20H38N, absolute mass error = 3.11 ppm) resulting from double dehydration of the eicosasphingosine (d20:1) long chain base (Supporting Information Fig. S6a). More complex gangliosides GD1a, GD1b and GT1b were also successfully tagged with aminoxyTMT (vide infra). Like GM1a, diagnostic fragment B1β (or B1α) along with reporter ion was observed indicating that the aminoxyTMT0 reagent was attached to the C-7 aldehydes (Supporting Information Fig. S7a–c).
While previous studies27,28 employed sodiated adduct to obtain high intensity reporter ions, our results show that for TMT-labeled gangliosides, high enough intensity of reporter ions can also be obtained using [M+2H]2+ as precursor ions by applying relatively lower collision energy. This difference from their study and ours can be attributed to the type of analytes and how the TMT label is connected to the molecule. It was suggested by Zhou et al.28 that the fragment ion yield of the TMT reporter ions is related to degree of branching, molecular weight, and strength of the reporter ion bond. In this respect, the ligation of TMT to the labile Neu5Ac could have aided the cleavage of reporter ions more easily. We found that by varying the normalized collision energy (NCE) between 20 to 35 arb. units, different levels of structural information can be obtained ranging from the glycan, lipid, and TMT reporter ions; increasing the NCE value yields higher reporter ion intensity and consequently lower signals due to glycan fragmentation (Supporting Information Fig. S8). Also, one potential interference for the reporter ion m/z 126.13 is the isobaric oxonium ion (m/z 126.05) derived from Neu5Ac and GalNAc fragment47. The resolution we employed at the MS/MS level, R = 15,000 FWHM is more than sufficient to distinguish these two fragments (Fig. 5c), like those previously achieved27,28. Taken together, this indicates a need for a high-resolution mass spectrometer in order for this technique to be useful.
Isobaric labeling of total ganglioside extract from porcine brain
The developed strategy using aminoxyTMT0 was applied to porcine brain total ganglioside extract. The major classes that comprise ~97% of gangliosides in mammalian brain include GM1a, GD1b, GD1a, and GT1b, in decreasing abundance, respectively38. We successfully labeled these gangliosides and confirmed their identity based on accurate mass (Supporting Information Table S2) and HCD fragmentation data (Supporting Information Fig. S7). Two Neu5Ac (one terminal, and one internal) in GD1a are oxidizable and only one in GD1b (Supporting Information Table S1). This results to different products of oxidation which could be potentially used to differentiate them. However, the internal Neu5Ac in GD1a is less exposed towards oxidation which is oxidized more slowly than the terminal Neu5Ac. Due to this differential reactivity, GD1a could contribute certain amount of oxidation products isomeric to the product of GD1b upon oxidation that co-elute during RPLC separation and could be co-isolated for fragmentation (Supporting Information Fig. S3d and S4c). Hence, the use of MS data alone could overestimate GD1b. Since GD1b has only one oxidizable Neu5Ac unit, we verified that it does not generate oxidation product isomeric to that of GD1a, thus the latter can be effectively quantified using the present approach. To quantitate GD1b using the described technique, appropriate separation method is thus recommended. Using RPLC, separation of GD1a and GD1b isomers is not possible7, but in our preliminary experiments using HILIC mode, these two gangliosides and their oxidation products were easily distinguishable (data not shown). This issue is not observed in other structures included in this study that contain multiple Neu5Ac units. This result highlights the potential of chemoselective oxidation followed by TMT labeling of gangliosides in mixtures; and application of this strategy in HILIC mode or ion mobility spectrometry48 would be a fertile ground for future method development.
Oxidation and labeling of gangliosides in total lipids extract
To investigate whether oxidation followed by TMT labeling workflow can be used even without prior isolation and purification of gangliosides, we directly oxidized total porcine brain lipid extract containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidic acid (PA) spiked with 1.0 to 5.0 nmol of porcine brain ganglioside mixture. During IO4− oxidation and subsequent TMT labeling, no appreciable interference from the other lipid species was observed. However, slight ion suppression was apparent which probably caused by phospholipids due to competitive process during ionization. For more reliable quantification, sample purification is recommended especially when low abundant ganglioside species are sought.
Feasibility for multiplexed relative quantification
To test the practical utility of our technique, using separate preparations of porcine brain gangliosides extract in two different ratios by weight, chemoselective oxidation and labeling using aminoxyTMT6 reagents were performed (Fig. 6a). We noted that this approach can be applied to limited amounts of ganglioside extract as low as 1.0 µg of starting material even in the presence of total lipids extract. One-to-one volumetric ratio of the resulting solutions were combined and analyzed by RPLC-MS/MS. Using a pre-determined inclusion list for HCD fragmentation in PRM mode we extracted the intensities of reporter ions, calculated the experimental ratios and compared them to the theoretical value for GM1a d18:1/18:0, GM1a d20:1/18:0, GD1a d18:1/18:0, GD1a d20:1/18:0, GT1b d18:1/18:0 and GT1b d20:1/18:0 (Fig. 6b). We obtained a mean error of 5.68% and 8.42%, and mean relative standard deviation (RSD) of 4.88% and 11.29% for theoretical ratios 1:1:1:1:1:1 and 1:1:3:3:5:5, respectively, indicating the feasibility of this technique for reliable relative quantification of multiple samples (Fig. 6c–f). Collectively, these results highlight the potential of IO4− oxidation followed by TMT labeling for relative quantification of ganglioside at the molecular species level using tandem mass spectrometry.
Figure 6.

Workflow employed to demonstrate feasibility of multiplexed experiment using porcine brain extract (A). Resulting EIC of major ganglioside components in porcine brain labeled with aminoxyTMT6 (B). Representative reporter ion region of aminoxyTMT6-labeled GD1a d20:1/18:0 in porcine brain extract mixed in 1:1:1:1:1:1 (C) and 1:1:3:3:5:5 (E) ratios by-weight for the channels indicated. Calculated intensity ratios plotted as overall mean + standard deviation (n=3, triplicate preparations, each injected twice) relative to arbitrary reference channel, TMT-127 (D and F). Δ indicates absolute mass error.
CONCLUSION
In this paper, we demonstrated a strategy to label major mammalian ganglioside core structures to allow the use of commercially available isobaric mass tag reagent without removing the lipid backbone. Under conditions described in this paper, NaIO4 is chemoselective to the C-7 side chain of Neu5Ac to form aldehyde functional group without affecting other structural motifs. The oxidizability of Neu5Ac depends on the availability of free hydroxy at C-8, which could be exploited to aid the structural characterization of the connectivity of Neu5Ac in the glycan moiety. Attachment of the isobaric label is straightforward and found to be advantageous to increase the ionization efficiency of complex gangliosides. The high-resolution, accurate mass measurements afforded the baseline resolution of interfering ions that are isobaric to TMT reporter ions. Multiplexed analysis is an efficient and sensitive technique for relative quantification of gangliosides. Using this methodology, not only can we analyze six samples simultaneously, but also determine both the glycan and lipid compositions. This promising approach can be used to compare gangliosides composition across different samples, biological treatments, or temporal studies involving longitudinal research design. Moreover, as this strategy is selective to sialic acid group, it could also be extended to other biomolecules of interest such as glycopeptides or others that commonly possess sialic acids. Future application of this novel strategy to answer relevant biological questions is expected.
Supplementary Material
Acknowledgments
This work was partially supported by grants from the National Institute of General Medical Sciences (GM 104678) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK114345) of the National Institutes of Health. The authors are also grateful for Triad Mass Spectrometry Facility of the UNCG Department of Chemistry and Biochemistry and Dr. Daniel Todd.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Structures of gangliosides, systematic names, detailed list of materials and reagents, total ion chromatograms, and representative MS/MS spectra (PDF).
The authors declare no financial interests.
References
- 1.Schnaar RL. J. Mol. Biol. 2016;428:3325–3336. doi: 10.1016/j.jmb.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fong BY, Ma L, Khor GL, van der Does Y, Rowan A, McJarrow P, MacGibbon AK. J. Agric. Food Chem. 2016;64:6295–6305. doi: 10.1021/acs.jafc.6b02200. [DOI] [PubMed] [Google Scholar]
- 3.Krengel U, Bousquet PA. Front. Immunol. 2014;5:325. doi: 10.3389/fimmu.2014.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrientos RC, Vu N, Zhang Q. J. Am. Soc. Mass Spectrom. 2017;28:2330–2343. doi: 10.1007/s13361-017-1772-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oikawa N, Matsubara T, Fukuda R, Yasumori H, Hatsuta H, Murayama S, Sato T, Suzuki A, Yanagisawa K. PLoS One. 2015;10:e0121356. doi: 10.1371/journal.pone.0121356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argia T, Yu RK, Miyatake T. J. Lipid. Res. 1984;25:1096–1101. [PubMed] [Google Scholar]
- 7.Ikeda K, Shimizu T, Taguchi R. J. Lipid Res. 2008;49:2678–2689. doi: 10.1194/jlr.D800038-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Lee H, German JB, Kjelden R, Lebrilla CB, Barile D. J. Agric. Food Chem. 2013;61:9689–9696. doi: 10.1021/jf402255g. [DOI] [PubMed] [Google Scholar]
- 9.Sørensen LK. Rapid Comm. Mass Spectrom. 2006;20:3625–3633. doi: 10.1002/rcm.2775. [DOI] [PubMed] [Google Scholar]
- 10.Huang Q, Zhou X, Liu D, Xin B, Cechner K, Wang H, Zhou A. Anal. Biochem. 2014;455:26–34. doi: 10.1016/j.ab.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda K, Taguchi R. Rapid Comm. Mass Spectrom. 2010;24:2957–2965. doi: 10.1002/rcm.4716. [DOI] [PubMed] [Google Scholar]
- 12.Garcia A, Chavez J, Mechref Y. J. Chromatogr. B. 2014;947–948:1–7. doi: 10.1016/j.jchromb.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 13.Masson EA, Sibille E, Martine L, Chaux-Picquet F, Bretillon L, Berdeaux O. J. Lipid Res. 2015;56:1821–1835. doi: 10.1194/jlr.D060764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong B, Norris C, Lowe E, McJarrow P. Lipids. 2009;44:867–874. doi: 10.1007/s11745-009-3327-1. [DOI] [PubMed] [Google Scholar]
- 15.Hajek R, Jirasko R, Lisa M, Cifkova E, Holcapek M. Anal. Chem. 2017;89:12425–12432. doi: 10.1021/acs.analchem.7b03523. [DOI] [PubMed] [Google Scholar]
- 16.Zamfir A, Vukelić Ž, Bindila L, Peter-Katalinić J, Almeida R, Sterling A, Allen M. J. Am. Soc. Mass Spectrom. 2004;15:1649–1657. doi: 10.1016/j.jasms.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Lee H, Lerno LA, Jr, Choe Y, Chu CS, Gillies LA, Grimm R, Lebrilla CB, German JB. Anal. Chem. 2012;84:5905–5912. doi: 10.1021/ac300254d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farwanah H, Kolter T. Metabolites. 2012;2:134–164. doi: 10.3390/metabo2010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sisu E, Flangea C, Serb A, Rizzi A, Zamfir AD. Electrophoresis. 2011;32:1591–1609. doi: 10.1002/elps.201100067. [DOI] [PubMed] [Google Scholar]
- 20.Zhou S, Tello N, Harvey A, Boyes B, Orlando R, Mechref Y. Electrophoresis. 2016;37:1489–1497. doi: 10.1002/elps.201600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahne H, Neubert P, Kuhn K, Etienne C, Bomgarden R, Rogers JC, Kuster B. Anal. Chem. 2012;84:3716–3724. doi: 10.1021/ac300197c. [DOI] [PubMed] [Google Scholar]
- 22.Yang S, Wang M, Chen L, Yin B, Song G, Turko IV, Phinney KW, Betenbaugh MJ, Zhang H, Li S. Sci. Rep. 2015;5:17585. doi: 10.1038/srep17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atwood JA, Cheng L, Alvarez-Manilla G, Warren NL, York WS, Orlando R. J. Proteome Res. 2008;7:367–374. doi: 10.1021/pr070476i. [DOI] [PubMed] [Google Scholar]
- 24.Rauniyar N, Yates JR. J. Proteome Res. 2014;13:5293–5309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S, Yuan W, Yang W, Zhou J, Harlan R, Edwards J, Li S, Zhang H. Anal. Chem. 2013;85:8188–8195. doi: 10.1021/ac401226d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Y, Jiao J, Bin Z, Zhang Y, Yang P, Lu H. Chem. Comm. 2015;51:772–775. doi: 10.1039/c4cc08086f. [DOI] [PubMed] [Google Scholar]
- 27.Zhong X, Chen Z, Snovida S, Liu Y, Rogers JC, Li L. Anal. Chem. 2015;87:6527–6534. doi: 10.1021/acs.analchem.5b01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou S, Hu Y, Veillon L, Snovida SI, Rogers JC, Saba J, Mechref Y. Anal. Chem. 2016;88:7515–7522. doi: 10.1021/acs.analchem.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujitani N, Takegawa Y, Ishibashi Y, Araki K, Furukawa J-i, Mitsutake S, Igarashi Y, Ito M, Shinohara Y. J. Biol. Chem. 2011;286:41669–41679. doi: 10.1074/jbc.M111.301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thaysen-Andersen M, Larsen MR, Packer NH, Palmisano G. RSC Adv. 2013;3:22683–22705. [Google Scholar]
- 31.Hermanson GT. Bioconjugate Techniques (3) Academic Press; San Diego, US: 2013. [Google Scholar]
- 32.Sudalai A, Khenkin A, Neumann R. Org. Biol. Chem. 2015;13:4374–4394. doi: 10.1039/c5ob00238a. [DOI] [PubMed] [Google Scholar]
- 33.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Nat. Meth. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramya TNC, Weerapana E, Cravatt BF, Paulson JC. Glycobiology. 2013;23:211–221. doi: 10.1093/glycob/cws144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narvaez-Rivas M, Zhang Q. J. Chromatogr. A. 2016;1440:123–134. doi: 10.1016/j.chroma.2016.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narvaez-Rivas M, Vu N, Chen GY, Zhang Q. Anal. Chim. Acta. 2017;954:140–150. doi: 10.1016/j.aca.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reuter G, Schauer R, Szeiki C, Kamerling JP, Vliegenthart JF. Glycoconjugate J. 1989;6:35–44. doi: 10.1007/BF01047888. [DOI] [PubMed] [Google Scholar]
- 38.Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Physiol. Rev. 2014;94:461–518. doi: 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuller M, Duplock S, Hein LK, Rigat BA, Mahuran DJ. Anal. Biochem. 2014;458:20–26. doi: 10.1016/j.ab.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 40.Gu J, Tifft CJ, Soldin SJ. Clin. Biochem. 2008;41:413–417. doi: 10.1016/j.clinbiochem.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 41.Robu AC, Vukelić Ž, Schiopu C, Capitan F, Zamfir AD. Anal. Biochem. 2016;509:1–11. doi: 10.1016/j.ab.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Schengrund C. Trends Biochem. Sci. 2015;40:397–406. doi: 10.1016/j.tibs.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 43.Domon B, Costello C. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 44.Ann Q, Adams J. J. Am. Soc. Mass Spectrom. 1992;3:260–263. doi: 10.1016/1044-0305(92)87010-V. [DOI] [PubMed] [Google Scholar]
- 45.Ferrer I, Thurman EM. Anal. Chem. 2005;77:3394–3400. doi: 10.1021/ac0485942. [DOI] [PubMed] [Google Scholar]
- 46.Afiuni-Zadeh S, Rogers JC, Snovida SI, Bomgarden RD, Griffin TJ. Biotechniques. 2016;60:186–188. 190, 192–186. doi: 10.2144/000114402. [DOI] [PubMed] [Google Scholar]
- 47.Yu J, Schorlemer M, Gomez Toledo A, Pett C, Sihlbom C, Larson G, Westerlind U, Nilsson J. Chem. Eur. J. 2016;22:1114–1124. doi: 10.1002/chem.201503659. [DOI] [PubMed] [Google Scholar]
- 48.Wojcik R, Webb I, Deng L, Garimella S, Prost S, Ibrahim Y, Baker E, Smith R. Int. J. Mol. Sci. 2017;18:183. doi: 10.3390/ijms18010183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.