

Abstract
Oxidative stress is arguably the most common mechanism in the toxicology of environmental agents, unifying the action of broad classes of physichochemically disparate environmental pollutants, including oxidant gases, organic compounds, particulate surfaces, and metal ions. As advances in redox biology identify previously unrecognized targets for disruption by exposure to xenobiotics, redox toxicology has emerged as a new field of investigation. Environmental contaminants can induce oxidative stress on cells through mechanisms that are direct, indirect or involve the disruption of metabolic or bioenergetic processes that are regulated by thiol redox switches. Live-cell imaging has proven to be a powerful approach to the study of environmental oxidative stress. Cells are equipped with multiple complementary energy-dependent systems for maintaining redox homeostasis in the face of environmental oxidative stress.
Keywords: Oxidative stress, live cell imaging, xenobiotic, toxicology, environmental exposure, thiols, glutathione, thioredoxin, peroxiredoxin
Just What is Oxidative Stress, Anyway, and Is It Always Bad?
The term “oxidative stress” is used in toxicology to refer to a range of pathologic conditions and reactions that together constitute a departure from a baseline homeostatic reductive state in the cell or tissue. Accordingly, the presence of elevated concentrations of reactive species (typically-but not exclusively-oxygen or nitrogen species) is taken as evidence of oxidative stress. Oxidative stress can also refer to the formation of increased levels of oxidized biomolecules, including macromolecules such as fatty acids, proteins and nucleic acids, but also small molecular weight peptides or antioxidants such as glutathione, ascorbate, and tocopherol. The adduction of biomolecules, as in the generation of 6-hydroxy guanine from acrolein adducted to the nucleotide guanine is also taken to evince oxidative stress [1]. The term oxidative stress may also apply to the chemical reactions involving the generation of these oxidized or adducted biomolecules. Examples of this are seen in the oxidation of fatty acids or quinones, which can involve single electron transfer reactions, thus generating free radicals in reactions that progress through initiation, propagation and termination stages (Figure 1). In cases where single electron transfer reactions are sustained by repeated cycles or reduction and oxidation of transition metals or quinones, the term “redox cycling” may be invoked [2]. A more recently accepted marker of oxidative stress is the activation of signaling pathways that are known to transduce the effects of exposure to oxidizing toxicants s to cellular responses such as gene expression [3-5]. The most common example of this is the Keap1/Nrf2/ARE pathway [6]. Evidence of oxidative stress extends beyond a demonstration of the activation of the effects of this pathway (e.g., nuclear translocation of Nrf2), to also include downstream readouts such as an increase in the expression of specific genes containing ARE (antioxidant response element) consensus sequences such as HO-1 and NQO1 [7,8]. Entire patterns of gene expression may be taken as indicative of a response to a stimulus initiated by oxidative stress as utilized in a number of “oxidative stress” gene array assays.
Figure 1.

All Roads Lead to H2O2: A broad range of physichochemically disparate environmental agents induce elevations in intracellular H2O2 through multiple mechanisms. The ubiquitous photochemical air pollutant ozone (O3) is a potent oxidizer that reacts readily with alkenyl groups in membrane fatty acids to produce primarily aldehydes and H2O2, as well as lesser quantities of reducible products such as lipid hydroperoxides. Certain organic compounds such redox active quinones (Q) can undergo single electron reduction, catalyzed enzymatically or by reaction with extracellular or intracellular cofactors, to produce semiquinone free radicals that can donate an electron to diatomic oxygen to produce superoxide (O2.-). In addition, certain nanoscale particulate surfaces such as those of elemental carbon particles (EC) are known to have carbon-centered radicals that similarly reduce oxygen to form O2.-. Transition metals with two or more adjacent valence states (e.g., Fe, Cu, V, Ni) may be oxidized by O2 and form O2.- and be re-reduced in a cyclical manner by compounds such as ascorbate. Many environmental agents, including quinones and metal ions, impair mitochondrial respiration where mitochondrial complexes I and III are known sources of O2.-. O2.- is a short lived species, undergoing rapid spontaneous or enzymatically catalyzed dismutation to form H2O2. Certain regulatory proteins such as the protein tyrosine phosphatases (PTP) are subject to reversible redox modification by H2O2 wherein the thiolate anion (-S-) on the cysteine is oxidized to the sulfenic form (inactivating the PTP). Redox of direct electrophilic PTP inactivation leads to a loss of signaling quiescence.
An integrating kinetic definition that encompasses the dynamic and multi-themed nature of oxidative stress is the accumulation of reactants, reactions and/or outcomes at a rate that exceeds the capacity of cellular or tissue defense mechanisms to counter them, leading to a transient or permanent loss of homeostasis. While each of the applications of the term oxidative stress described above can be rationalized, the breadth of meanings spanning generation of reactants, reactions, formation of effectors, and outcomes has, in the aggregate, the unfortunate effect of devolving the specificity of the concept. The remedy is to encourage the adoption of more specific terms and promote the awareness of oxidative stress as an umbrella concept under which its many manifestations may be found.
An emerging concept in toxicological oxidative stress takes its cues from advances in the field of redox biology, where it is increasingly understood that cells generate and employ reactive species in important biological processes. Cells have sophisticated systems to utilize low levels of reactive species to modify the activity of a diverse range of regulatory functions, from suppressing signaling to apportioning glucose utilization for maintaining quiescence, growth, differentiation and programmed cell death [9,10]. Clearly, the oxidative processes involved in maintaining and executing these functions fulfill an essential physiological role and should be considered to be distinct from the adverse meaning of oxidative stress as defined above. As the appreciation of the importance of redox reactions in biology has grown, so has the notion that these sophisticated biological processes represent potentially critical targets for disruption by toxicants, thus giving rise to the nascent field of redox toxicology.
Is There Anything that Does Not Induce Oxidative Stress?
Oxidative stress is arguably the most common mechanistic feature in toxicology. Indeed, the sheer diversity of physichochemical properties of xenobiotics that are known to induce oxidative stress suggests that it is a universal toxicological mechanism. However, an important mechanistic distinction must be made between agents that induce oxidative stress directly, compounds whose oxidative potential requires the participation of cellular factors, and those xenobiotics that are not themselves oxidative but can induce an oxidative response from the cell (more on the last two types in the following section). Still, even considering only environmental agents that are primary oxidants, the heterogeneity is striking. The list of environmental toxicants that are directly oxidative includes gaseous pollutants such as ozone [11], Cl2 [12] and phosgene [13]. In addition, entire classes of organic electrophiles exemplified by acrolein [14], aromatic quinines [15] and isoprenesact through oxidative reactions [16], while heavy metal ions of zinc [17], lead [18], mercury [19] and cadmium [20] are capable of coordinating with sulfhydryl groups on peptides and proteins, promoting their oxidation. An additional persuasive argument of the universality of oxidative stress in toxicology is the paradoxical observation that even agents that are used to activate antioxidant pathways appear to also cause oxidative stress [21-23]. Oxidative stress may not be the primary mechanism of toxicity for all xenobiotic exposures; however, given the number and variety of oxidative reactions that xenobiotics can undergo or induce, one is hard-pressed to provide an example of a xenobiotic exposure that does not involve some level of oxidative stress.
Sometimes the Cells Just Makes Things Worse for Itself
A number of environmental toxicants, again of widely varying physichochemical composition, induce oxidative stress through mechanisms that depend on interaction with the cell or cellular components. Moreover, it is possible to distinguish between varying levels of cellular participation required for the oxidative stress induced by environmental agents. A relatively passive interaction is the reduction of metal ions, such as Fe3+ and V5+, by cell-derived reductants such as ascorbate, the ability of the resulting Fe2+ or V4+ ions to transfer an electron to O2 produces the reactive oxygen species O2.-, which is rapidly dismutated to H2O2 either spontaneously or enzymatically by superoxide dismutase. The interaction of Fe2+ (Fenton reaction) or O2.- (Haber-Weiss reaction) and H2O2 produces OH., unequivocally one of the most reactive oxidants known [24].
Ghio et al. [23] have reported a fascinating mechanism that represents the active involvement of the cell in the oxidative stress of a xenobiotic exposure. The oxidative stress is initiated by the cell in response to the abstraction of metal cations (specifically iron cations) from the cell by metal-coordinating particulate surfaces such as silinol groups on silica particles. In what can only be characterized as an unfortunately misguided response, the cell uses NADH oxidoreductase to generate and release O2.- extracellularly in an apparent attempt to reduce extracellular ferric iron (Fe3+) to its ferrous (Fe2+) state in order to import it. A similar enzyme-induced oxidative stress is observed when environmental quinones are converted to oxidative species through an active cellular process. For example, 1,2-NQ undergoes single electron reduction by cytochrome p450 reductase in an NADPH-dependent reaction that generates the semi-quinones, a radical that can reduce O2 to O2.- spontaneously [15].
Inhibition of mitochondrial respiration by disparate environmental agents such as acrolein, carbon monoxide, paraquat, Cd2+, and Zn2+, can be seen as inducing a self-inflicted type of oxidative stress, as they induce increased release of partially reduced oxygen species (e.g., superoxide) from Complexes I and III [25]. The most extreme form of cell-initiated oxidative stress may be the oxidative burst of phagocytes, such as macrophages and neutrophils, engulfing relatively inert materials like polystyrene beads or long fibers such as asbestos which leads to frustrated phagocytosis, a process which can be a significant source of oxidative stress to surrounding cells [26,27].
Sitting Ducks: Thiols as Redox Targets
The toxicity of heavy metals such as lead, mercury and cadmium has long been known to be underlain by their affinity for sulfhydryl groups [28]. In fact, a number of antioxidant proteins, such as metallothionein and albumin, contain multiple sulfhydryl residues that serve as a sink for heavy metals ions, thereby sparing essential protein thiols. A more recent appreciation of the importance of sulfhydryls in toxicology has been prompted by the identification of redox-switch proteins that utilize the cysteine thiol as a pivotal physiological regulatory mechanism that appears to control virtually every function of the cell [29]. The central redox reaction involved is the reversible oxidation of the thiol (-SH) side group of specific cysteine residues to the sulfenic form (-SOH) (Figure 2). Not every cysteine residue is equally susceptible to sulfenylation, as this modification appears to be limited to specific regulatory proteins. Although thiol sulfenylation requires deprotonation to the thiolate (-S-), pKa is not the only determinant of the tendency of specific thiols to be oxidized, but rather involves other structural factors as well [30,31]. Intracellularly, sulfenylation is effected by attack of the thiolate anion by H2O2 in what is suspected to be an enzyme catalyzed reaction [32,33], creating multiple potential pathways for toxicological disruption by xenobiotic oxidative stressors. Sulfenylation typically leads to the downregulation of the function of a protein. The classic example is the sulfenylation of the essential catalytic cysteine in protein tyrosine phosphatases, which results in the suppression of dephosphorylation tone and allows kinases to work unopposed during signaling [34,35]. A key feature of redox regulation of proteins through sulfenylation is its reversibility, which may be attributed to thioredoxin (Trx) -mediated regeneration of the thiol directly [36], or the formation of a disulfide which is then reduced by glutaredoxin (Grx) [29]. However, the sulfenic group also acts as a nexus for further modification of the cysteinyl sulfur which could involve oxidation to higher states such the sulfinic (-SO2H) or the sulfonic (-SO3H) forms, the latter considered an irreversible modification. The formation of a sulfenamide by intramolecular cyclization between the sulfenic group and a vicinal amino group, as occurs in PTP1B, has been proposed to represent a protective mechanism to prevent hyper-oxidation to the higher sulfinic and sulfonic forms [29]. Alternatively, sulfenic groups can form mixed disulfides by glutathionylation or condensation with another thiol to form the structurally important disulfide bridge [37,38]. H2O2-dependent sulfenylation of the regulatory proteins PTP1B and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was recently reported in cells exposed to the air pollutant 1,2-NQ [39]. Given that the concentrations of H2O2 involved in physiological signaling are low relative to those that can be induced by xenobiotic exposure, the potential for toxicological disruption of protein sulfenylation would seem considerable.
Figure 2.
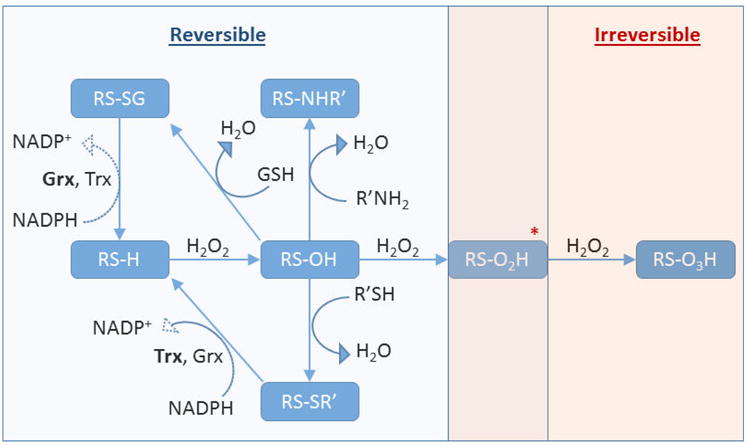
Cysteine Posttranslational Modifications. Protein thiols (RS-H) can undergo oxidation to the sulfenic acid (RS-OH), which then can react with intracellular glutathione to form a mixed disulfide (RS-SG) or with another protein moiety such as another thiol to form a disulfide bond (RS-SR') or an amine to form a sulfenamide (RS-NHR'). These modifications are reversible and can be reduced back to the thiol, enzymatically, at the expense of NADPH. The sulfenic acid can be further oxidized to the irreversible modifications sulfinic acid (RS-O2H) and sulfonic acid (RS-O3H)*. It has been demonstrated that the hyperoxidized sulfinic acid in the peroxiredoxin can be enzymatically reduced to the sulfenic acid at the expense of ATP, and as such it is possible that other unidentified proteins may utilize a reversible sulfinic acid in their function.
Watching it Happen
One of the most exciting developments in the field of redox biology in recent years has been the introduction of a new generation of fluorogenic sensors that enable real-time monitoring of oxidative endpoints with unprecedented sensitivity and specificity. These sensors have found considerable utility in studies of xenobiotic-induced oxidative stress. Small molecule sensors such as PF6-AM and DAF-FM DA are trappable fluorogenic dyes that can be used to conveniently monitor intracellular concentrations of H2O2and NO, respectively [40]. Genetically-encoded sensors developed through modifications of green fluorescent protein (GFP), as in the case of the roGFP family [41,42], can monitor the intracellular glutathione status; while chimeric fusions of GFP or its variant yellow fluorescent protein (YFP) and sensory proteins, such as the H2O2 -sensing bacterial transcription factor Oxy-R in the case of HyPer and its variants, can measure intracellular H2O2 levels [43-46].
roGFP is a sensor of the glutathione redox potential (Egsh) that functions by equilibrating with the intracellular GSH/GSSG pool through the intervention of Grx. A fusion of Grx and roGFP that exhibits faster response kinetics also exists [47]. The HyPer family of sensors respond to H2O2 with sub-micromolar sensitivity. Members of both the roGFP and HyPer family of sensors are dynamic, meaning that they report bidirectional changes in Egsh or H2O2 concentration, respectively. These sensors have two excitation maxima, making them ratiometric and, therefore, are insensitive to artifacts that commonly afflict fluorescence microscopy studies (e.g., photobleaching, compartmentalization). Although vastly superior to obsolete reporters such as the “ROS” sensor dichlorofluorohydrazine diacetate [48,49], the new generation of fluorogenic sensors do have performance limitations that require special consideration when used in toxicological applications, where strongly reactive xenobiotics could induce potential off-target effects. The HyPer family of sensors in particular is afflicted by a pronounced sensitivity to changes in pH, although an excellent control for this exists in the form of an H2O2-insensitive variant called SyPher which differs from HyPer by a single amino acid and retains pH responsiveness [50]. With proper validation, the high spatial and temporal resolution afforded by live-cell microscopy makes the current class of small molecule and genetically-encoded sensors powerful tools for the elucidation of causal relationships in xenobiotic-induced oxidative stress.
The Defenders: The Glutathione and Thioredoxin Systems and Their Allies
The roles of antioxidants in the protection against oxidative stress has long been a subject of investigation. The high reaction rate constants of peroxisomal catalase (K ∼107 M-1 s-1) [51] and the much higher activity of cytosolic and mitochondrial superoxide dismutase (K ∼109 M-1 s-1) [52] are understood to provide important protective functions through the dismutation reaction of hydrogen peroxide and superoxide, respectively. However, their roles in physiological redox reactions may be secondary to those of the enzymes of the glutathione and thioredoxin systems described below.
The importance of the tripeptide glutathione (GSH) in the detoxification of xenobiotics such as acetaminophen, chloroform, and paraquat is also well appreciated. Present in millimolar concentrations in most cell types, GSH and its oxidized form (GSSG) are the predominant redox pair in the cell [53]. From a teleological perspective, it is perhaps not surprising that an intricate system of enzymes has evolved to synthesize, transport, maintain, and regulate GSH. It is likewise fitting that the expression of the enzymes involved in these processes are themselves subject to the activity of Nrf2, a pivotal switch in the response to oxidative stress [54]. Resting cells and tissues maintain a very high (100:1) GSH/GSSG ratio through the action of glutathione reductase (GR), at the expense of NADPH derived from the pentose phosphate pathway.
The glutathione-S-transferases (GST) are phase II enzymes that catalyze the glutathionylation of xenobiotics, most famously acetaminophen, but also as naphthalene, aflatoxin, DDT, and many other environmentally relevant agents [55]. The selenocysteine-containing glutathione peroxidases (GPx) mediate the reduction of peroxides to alcohols and are found in a variety of intracellular and extracellular compartments. By lowering the pKa, the substitution of Se for S in the selenocysteine creates a better nucleophile in the attack of the peroxide group. The attack generates a selenic acid, moiety (-SeOH, the analog of sulfenic acid, -SOH) which then binds to a molecule of GSH, forming a mixed seleno-sulfide that then condenses with a second molecule of GSH to produce GSSG and regenerate the enzyme [56] (Figure 3). While all members of the GPx family have specificity for H2O2, Gpx4 can also reduce organic hydroperoxides, including fatty acids attached to phospholipids. Another important member of the GSH system is Grx, an enzyme with cysteinyl thiol that reacts directly with the oxidized thiol in a protein substrate and is then reduced non-enzymatically by GSH. Grx is believed to have an important role in the restoration of glutathionylated proteins substrates [57].
Figure 3.

The Defenders and Their Reactions. A, The catalytic site of glutathione peroxidases (GPx) uses selenocysteine. Relative to sulfur, the reduced pKa of selenium (Se) makes it more readily ionizable at physiological pH. In the GPx peroxidation reaction the selenite anion (Se-) is oxidized by a peroxide to the selenic acid form (-SeOH), reducing the peroxide to a hydroxide in the process. The selenic acid group in GPx-SeOH can be glutathionylated, giving the mixed selenide-sulfide Se-SG, which can be attacked by a second glutathione (GSH) molecule to generate oxidized glutathione (GSSG) and restore the enzyme to its starting reduced form. B, Glutaredoxin (Grx) mediates reversible transfer reactions of glutathione involving glutathionylated protein targets. C, Peroxiredoxins (Prx) are abundant oligomeric enzymes increasingly recognized as important in the regulation of intracellular peroxide levels. Prx of the 2-Cys type have a peroxidatic (P) cysteine that become sulfenylated by a peroxide. The resulting sulfenic acid (-SOH) on the peroxidatic cysteine, condenses with the resolving (R) cysteine, generating a disulfide (-S-S-). Alternatively, the sulfenylated Prx can be oxidized further by reacting with a second peroxide to generate the sulfinic form (-SOOH) on the peroxidatic cysteine. Sulfiredoxin (Srx) can reverse this overoxidation by reducing the Prx-SOOH back to Prx-SOH in an ATP-dependent reaction. Thioredoxin (Trx) use a thiol-disulfide exchange reaction that reduces a protein disulfide on Prx to generate two thiols (the starting Prx form in this illustration) while two cysteines in the Trx molecule are themselves oxidized to a disulfide. The regeneration of Trx is effected by thioredoxin reductase (TrxR), which like GPx, is a selenocysteine-bearing enzyme that uses NADPH rather than GSH as a reductant. Glutathione reductase (GR) also uses NADPH to reduce GSSG back to GSH. Thus, both the thioredoxin and glutathione systems utilize NADPH derived from the pentose phosphate pathway (PPP), and are ultimately dependent on the availability of glucose as an energy source.
The cytosolic and mitochondrial polypeptides in the Trx family support metabolic functions as well as the reduction of disulfides on a wide variety of target proteins through an exchange mechanism in which the Trx thiols become oxidized to a disulfide. Thioredoxin reductase (TrxR, another selenocysteine protein), then uses NADPH as a source of reducing equivalents to reduce the disulfide in oxidized Trx back to thiols [58]. The most important role of Trx in redox biology may be acting as the reductive co-factor for the peroxiredoxins (Prx), a family of enzymes with specificity for both H2O2 and lipid hydroperoxides [59]. Most Prx have two cysteines but Prx6 is a one-cysteine form that is intriguing because it also acts as a phospholipase A2 to remove oxidized fatty acids from the sn-2 position of phospholipids. Under conditions of persistent oxidative stress, Prx can accumulate intracellularly and their cysteines can become hyperoxidized to the sulfinic form (-SOOH). Sulfiredoxin (Srx) is a specialized enzyme that can reduce the Prx active site from the sulfinic acid to its sulfenic form, consuming an ATP in the process [60]. Whether Srx is also involved in the reduction of other protein sulfinics is not currently known, and this reaction currently represents the end of the line in the repair of oxidative damage to proteins.
Highlights.
Oxidative stress is a commonly cited mechanistic feature in the toxicology of environmental agents
Advances in redox biology suggest new targets for disruption of cellular homeostasis by environmental agents
Bioenergetic and metabolic processes in cells can contribute to the oxidative stress of toxic agents
Live-cell imaging offers unparalleled spatiotemporal resolution, sensitivity and specificity needed for redox toxicology studies
Acknowledgments
Disclaimer: This article was reviewed by the National Health and Environmental Effects Research Laboratory, U.S. EPA, and approved for publication. Approval does not signify that the contents reflect the views of the agency nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- 1.Nechev LV, Kozekov ID, Brock AK, Rizzo CJ, Harris TM. DNA adducts of acrolein: site-specific synthesis of an oligodeoxynucleotide containing 6-hydroxy-5,6,7,8-tetrahydropyrimido[1,2-a]purin-10(3H)-one, an acrolein adduct of guanine. Chem Res Toxicol. 2002;15:607–613. doi: 10.1021/tx010181y. [DOI] [PubMed] [Google Scholar]
- 2.Charrier JG, Anastasio C. Rates of Hydroxyl Radical Production from Transition Metals and Quinones in a Surrogate Lung Fluid. Environ Sci Technol. 2015;49:9317–9325. doi: 10.1021/acs.est.5b01606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Wages PA, Silbajoris R, Speen A, Brighton L, Henriquez A, Tong H, Bromberg PA, Simmons SO, Samet JM. Role of H2O2 in the oxidative effects of zinc exposure in human airway epithelial cells. Redox Biol. 2014;3:47–55. doi: 10.1016/j.redox.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kodavanti UP, Thomas R, Ledbetter AD, Schladweiler MC, Shannahan JH, Wallenborn JG, Lund AK, Campen MJ, Butler EO, Gottipolu RR, et al. Vascular and cardiac impairments in rats inhaling ozone and diesel exhaust particles. Environ Health Perspect. 2011;119:312–318. doi: 10.1289/ehp.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo J, Xu Y, Ji W, Song L, Dai C, Zhan L. Effects of exposure to benzo[a]pyrene on metastasis of breast cancer are mediated through ROS-ERK-MMP9 axis signaling. Toxicol Lett. 2015;234:201–210. doi: 10.1016/j.toxlet.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 6.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu ML, Layne MD, Yet SF. Heme oxygenase-1 in environmental toxin-induced lung disease. Toxicol Mech Methods. 2012;22:323–329. doi: 10.3109/15376516.2012.666685. [DOI] [PubMed] [Google Scholar]
- 8.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10*.Bindoli A, Rigobello MP. Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal. 2013;18:1557–1593. doi: 10.1089/ars.2012.4655. [DOI] [PubMed] [Google Scholar]
- 11**.Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta. 2016;1860:2771–2781. doi: 10.1016/j.bbagen.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Winder C. The toxicology of chlorine. Environ Res. 2001;85:105–114. doi: 10.1006/enrs.2000.4110. [DOI] [PubMed] [Google Scholar]
- 13.Li W, Pauluhn J. Phosgene-induced acute lung injury (ALI): differences from chlorine-induced ALI and attempts to translate toxicology to clinical medicine. Clin Transl Med. 2017;6:19. doi: 10.1186/s40169-017-0149-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moghe A, Ghare S, Lamoreau B, Mohammad M, Barve S, McClain C, Joshi-Barve S. Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci. 2015;143:242–255. doi: 10.1093/toxsci/kfu233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15*.Kumagai Y, Shinkai Y, Miura T, Cho AK. The chemical biology of naphthoquinones and its environmental implications. Annu Rev Pharmacol Toxicol. 2012;52:221–247. doi: 10.1146/annurev-pharmtox-010611-134517. [DOI] [PubMed] [Google Scholar]
- 16.Lin YH, Arashiro M, Clapp PW, Cui T, Sexton KG, Vizuete W, Gold A, Jaspers I, Fry RC, Surratt JD. Gene Expression Profiling in Human Lung Cells Exposed to Isoprene-Derived Secondary Organic Aerosol. Environ Sci Technol. 2017 doi: 10.1021/acs.est.7b01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Wu W, Bromberg PA, Samet JM. Zinc ions as effectors of environmental oxidative lung injury. Free Radic Biol Med. 2013;65:57–69. doi: 10.1016/j.freeradbiomed.2013.05.048. [DOI] [PubMed] [Google Scholar]
- 18.Vaziri ND. Mechanisms of lead-induced hypertension and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2008;295:H454–465. doi: 10.1152/ajpheart.00158.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahboob M, Shireen KF, Atkinson A, Khan AT. Lipid peroxidation and antioxidant enzyme activity in different organs of mice exposed to low level of mercury. J Environ Sci Health B. 2001;36:687–697. doi: 10.1081/PFC-100106195. [DOI] [PubMed] [Google Scholar]
- 20.Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, et al. Cadmium stress: an oxidative challenge. Biometals. 2010;23:927–940. doi: 10.1007/s10534-010-9329-x. [DOI] [PubMed] [Google Scholar]
- 21.Mentor S, Fisher D. Aggressive Antioxidant Reductive Stress Impairs Brain Endothelial Cell Angiogenesis and Blood Brain Barrier Function. Curr Neurovasc Res. 2016 doi: 10.2174/1567202613666161129113950. [DOI] [PubMed] [Google Scholar]
- 22*.Teodoro JS, Rolo AP, Palmeira CM. The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicol Mech Methods. 2013;23:297–302. doi: 10.3109/15376516.2012.759305. [DOI] [PubMed] [Google Scholar]
- 23.Singh F, Charles AL, Schlagowski AI, Bouitbir J, Bonifacio A, Piquard F, Krahenbuhl S, Geny B, Zoll J. Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim Biophys Acta. 2015;1853:1574–1585. doi: 10.1016/j.bbamcr.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 24**.Halliwell B, Gutteridge JM. Free Radicals in Biology and Medicine. Fifth. Oxford University Press; 2015. [Google Scholar]
- 25.Harris PS, Roy SR, Coughlan C, Orlicky DJ, Liang Y, Shearn CT, Roede JR, Fritz KS. Chronic ethanol consumption induces mitochondrial protein acetylation and oxidative stress in the kidney. Redox Biol. 2015;6:33–40. doi: 10.1016/j.redox.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mossman BT, Marsh JP. Evidence supporting a role for active oxygen species in asbestos-induced toxicity and lung disease. Environ Health Perspect. 1989;81:91–94. doi: 10.1289/ehp.898191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mossman BT, Hansen K, Marsh JP, Brew ME, Hill S, Bergeron M, Petruska J. Mechanisms of fibre-induced superoxide release from alveolar macrophages and induction of superoxide dismutase in the lungs of rats inhaling crocidolite. IARC Sci Publ. 1989:81–92. [PubMed] [Google Scholar]
- 28.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 29**.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.Poole LB, Schoneich C. Introduction: What we do and do not know regarding redox processes of thiols in signaling pathways. Free Radic Biol Med. 2015;80:145–147. doi: 10.1016/j.freeradbiomed.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31**.Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. doi: 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone JR. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch Biochem Biophys. 2004;422:119–124. doi: 10.1016/j.abb.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 33**.Winterbourn CC, Hampton MB. Redox biology: signaling via a peroxiredoxin sensor. Nat Chem Biol. 2015;11:5–6. doi: 10.1038/nchembio.1722. [DOI] [PubMed] [Google Scholar]
- 34**.Meng TC, Tonks NK. Analysis of the regulation of protein tyrosine phosphatases in vivo by reversible oxidation. Methods Enzymol. 2003;366:304–318. doi: 10.1016/s0076-6879(03)66023-4. [DOI] [PubMed] [Google Scholar]
- 35.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 36**.Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49:7748–7755. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rehder DS, Borges CR. Possibilities and pitfalls in quantifying the extent of cysteine sulfenic acid modification of specific proteins within complex biofluids. BMC Biochem. 2010;11:25. doi: 10.1186/1471-2091-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39*.Wages PA, Lavrich KS, Zhang Z, Cheng WY, Corteselli E, Gold A, Bromberg P, Simmons SO, Samet JM. Protein Sulfenylation: A Novel Readout of Environmental Oxidant Stress. Chem Res Toxicol. 2015;28:2411–2418. doi: 10.1021/acs.chemrestox.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Wages PA, Cheng WY, Gibbs-Flournoy E, Samet JM. Live-cell imaging approaches for the investigation of xenobiotic-induced oxidant stress. Biochim Biophys Acta. 2016;1860:2802–2815. doi: 10.1016/j.bbagen.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 42*.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 43**.Bogdanova YA, Schultz C, Belousov VV. Local Generation and Imaging of Hydrogen Peroxide in Living Cells. Curr Protoc Chem Biol. 2017;9:117–127. doi: 10.1002/cpch.20. [DOI] [PubMed] [Google Scholar]
- 44.Ermakova YG, Bilan DS, Matlashov ME, Mishina NM, Markvicheva KN, Subach OM, Subach FV, Bogeski I, Hoth M, Enikolopov G, et al. Red fluorescent genetically encoded indicator for intracellular hydrogen peroxide. Nat Commun. 2014;5:5222. doi: 10.1038/ncomms6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malinouski M, Zhou Y, Belousov VV, Hatfield DL, Gladyshev VN. Hydrogen peroxide probes directed to different cellular compartments. PLoS One. 2011;6:e14564. doi: 10.1371/journal.pone.0014564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 47**.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal. 2010;13:621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 48*.Winterbourn C. Current methods to study reactive oxygen species--pros and cons. Preface Biochim Biophys Acta. 2014;1840:707. doi: 10.1016/j.bbagen.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 49*.Winterbourn CC. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim Biophys Acta. 2014;1840:730–738. doi: 10.1016/j.bbagen.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Matlashov ME, Bogdanova YA, Ermakova GV, Mishina NM, Ermakova YG, Nikitin ES, Balaban PM, Okabe S, Lukyanov S, Enikolopov G, et al. Fluorescent ratiometric pH indicator SypHer2: Applications in neuroscience and regenerative biology. Biochim Biophys Acta. 2015;1850:2318–2328. doi: 10.1016/j.bbagen.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogura Y, Yamazaki I. Steady-state kinetics of the catalase reaction in the presence of cyanide. J Biochem. 1983;94:403–408. doi: 10.1093/oxfordjournals.jbchem.a134369. [DOI] [PubMed] [Google Scholar]
- 52.Forman HJ, Fridovich I. Superoxide dismutase: a comparison of rate constants. Arch Biochem Biophys. 1973;158:396–400. doi: 10.1016/0003-9861(73)90636-x. [DOI] [PubMed] [Google Scholar]
- 53.Van Laer K, Hamilton CJ, Messens J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid Redox Signal. 2013;18:1642–1653. doi: 10.1089/ars.2012.4964. [DOI] [PubMed] [Google Scholar]
- 54.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 55.Seidegard J, Ekstrom G. The role of human glutathione transferases and epoxide hydrolases in the metabolism of xenobiotics. Environ Health Perspect. 1997;105 Suppl 4:791–799. doi: 10.1289/ehp.105-1470052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 57.Kalinina EV, Chernov NN, Novichkova MD. Role of glutathione, glutathione transferase, and glutaredoxin in regulation of redox-dependent processes. Biochemistry (Mosc) 2014;79:1562–1583. doi: 10.1134/S0006297914130082. [DOI] [PubMed] [Google Scholar]
- 58.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 59*.Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci. 2015;40:435–445. doi: 10.1016/j.tibs.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]