

Abstract
Dynamic modulation of lipid membrane curvature can be achieved by a number of peripheral protein binding mechanisms such as hydrophobic insertion of amphipathic helices and membrane scaffolding. Recently, an alternative mechanism was proposed in which crowding of peripherally bound proteins induces membrane curvature through steric pressure generated by lateral collisions. This effect was enhanced using intrinsically disordered proteins that possess high hydrodynamic radii, prompting us to explore whether membrane bending can be triggered by the folding—unfolding transition of surface-bound proteins. We utilized histidine-tagged human serum albumin bound to Ni-NTA- DGS containing liposomes as our model system to test this hypothesis. We found that reduction of the disulfide bonds in the protein resulted in unfolding of HSA, which subsequently led to membrane tubule formation. The frequency of tubule formation was found to be significantly higher when the proteins were unfolded while being localized to a phase-separated domain as opposed to randomly distributed in fluid phase liposomes, indicating that the steric pressure generated from protein unfolding can drive membrane deformation. Our results are critical for the design of peripheral membrane protein-immobilization strategies and open new avenues for exploring mechanisms of membrane bending driven by conformational changes of peripheral membrane proteins.
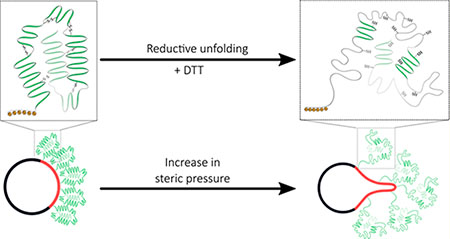
INTRODUCTION
Generation of membrane curvature is essential to cell function. Important cellular processes, such as cell signaling, vesicle trafficking, membrane fusion, endocytosis, and many others,1,2 depend on protein structural dynamics at the membrane interface and coupled membrane responses. The interplay between membrane geometry and protein structural dynamics is complex. Dynamin, for example, assembles into helical polymers at necks of budding vesicles and directly participates in membrane-remodeling events through a GTP hydrolysis- dependent conformational change.3 Alternatively, the membrane interface may induce the folding of intrinsically disordered proteins.4 For example, α-synuclein folds into an α-helical structure from an initially disordered random coil upon membrane binding.5 This transition has a strong influence on membrane structure, resulting in membrane thinning6 and induction of membrane curvature.7 For these reasons, the effect of membrane bound proteins on shaping membranes has been of immense interest and the subject of intense study. Several classic protein-mediated membrane bending mechanisms have been proposed in the literature.2,8,9 The hydrophobic insertion mechanism postulates that insertion of amphipathic helices causes the membrane to bend by generating defects between phosphate head groups, whereas scaffolding proteins are thought to generate membrane curvature by molding it into the shape of theprotein.8
A new membrane bending mechanism has been proposed recently in which crowding of peripherally bound proteins locally confined on the membrane surface induces curvature independent of protein scaffolding or membrane insertion.10,12 Stachowiak and co-workers reported that confining His-tagged GFP on membrane domains generates enough steric pressure from lateral protein collisions to drive membrane bending.12 Chen and co-workers used pipet aspiration to quantitatively study the ability of protein crowding to generate membrane curvature under controlled membrane tension.11 Similar behavior was observed when intrinsically disordered proteins were confined to the liquid- disordered domain of phase separated liposomes.13 A recent report suggests lateral pressure from protein crowding can stretch and bend the membrane and lead to membrane fission.14 Additionally, theoretical analysis has shown that crowded membrane-bound proteins generate substantial pressure at membrane surfaces.15 These observations suggest crowding of proteins locally confined on the membrane surface, such as a region of membrane phase separation, may function as a general mechanism of membrane bending regardless of the specific protein structures involved. While the efficacy of protein crowding in bending membranes still remains a subject of debate,16 the coupled effects of protein crowding and protein conformational changes on membrane geometry have not been investigated despite the dynamic nature of protein structures. Consequently, the goal of the present study was to investigate whether a transient change in protein conformation could induce membrane curvature.
Our approach was simple in concept: tightly bind a protein to the membrane surface with protein binding lipid (Ni-NTA-DGS), and then unfold it and probe the effect on membrane curvature. But, this was difficult in practice, since most protein unfolding methods directly perturb the membrane, including temperature,17 denaturants such as guanidine hydrochloride,18 and acid-induced unfolding.19 We chose His-tagged human serum albumin20 as a model for this study because the helical structure of HSA is stabilized by 17 disulfide bridges.21 Reduction of these disulfide bonds by 1,4-dithiothreitol (DTT)22 triggers unfolding of the protein and increases the Stokes radius by 1.2 fold,23,24 but DTT by itself does not perturb the membrane structure. We have used DTT-triggered unfolding of HSA to induce membrane bending, tubulation, and budding in model membrane systems.
2. EXPERIMENTAL SECTION
2.1. Materials
POPC, DPhPC, DPPC, cholesterol and Ni-NTA- DGS were purchased from Avanti Polar Lipids (Alabaster, AL). Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethy-lammonium salt (Texas Red DHPE) and Alexa 488 C3 maleimide were purchased from ThermoFisher Scientific (Waltham, MA). His-tagged human serum albumin was obtained from ACRO Biosystems (Newark, DE). Dithiothreitol (DTT) was purchased from Roche (Branford, CT). Human serum albumin, sucrose, glucose, uranyl acetate, methanol, acetic acid (glacial, ≥99.85%), and tris(2-carboxyethyl) phosphine hydrochloride (TCEP) were obtained from Sigma-Aldrich (St. Louis, MO). MOPS, NaCl, and Amicon Ultra-0.5 mL centrifugal filters (100 kDa MWCO) were obtained from EMD Millipore (Billerica, MA). Tris-HCl was obtained from J.T. Baker-Avantor Performance Materials (Center Valley, PA). ITO coated glass slides (100 ohm/sq) were obtained from Nanocs (New York, NY). Horizontal ATR germanium crystal (45° angle) and multiple reflection ATR accessory units were obtained from Pike Technologies (Madison, WI).
2.2. Large Unilamellar Liposome and Giant Unilamellar Vesicle Preparations.
Nonphase-separated liposomes (NPSL) contain POPC plus 10−30 mol % Ni-NTA-DGS. Composition for phase-separated liposomes (PSL) was chosen according to published composition25 and contains 12.5 mol % fluid phase lipid (DPhPC plus 10–30 mol % Ni-NTA-DGS), 40 mol % DPPC, and 47.5 mol % cholesterol. The overall concentration of Ni-NTA-DGS is smaller in PSL than in NPSL. For instance, the overall Ni-NTA-DGS concentration in 20 mol % Ni-NTA lipid doped 1 mM PSL, where the Ni-NTA modified lipid partitions preferentially into the liquid- disordered domain, is 0.025 mM. In comparison, 1 mM 20 mol % Ni- NTA lipid doped NPSL contains 0.2 mM overall Ni-NTA-DGS. Large unilamellar vesicles (LUV, referred to as liposomes) were prepared by extrusion. Lipid mixtures in chloroform solutions were dried on the inner wall of a small flask using compressed air and were then lyophilized overnight under vacuum at−42 °C. Lipid cakes were rehydrated to a final concentration of 2 mM with MOPS buffer (20 mM MOPS, 100 mM NaCl, pH 7.2) for 1 h at room temperature for NPSL or at 60 °C for PSL and were vortexed occasionally. The resulting solutions were extruded through a 100 nm pore size single polycarbonate membrane (Whatman/GE Healthcare) 20 times to produce a clear solution of liposomes.
Giant unilamellar vesicles (referred to as GUV) were prepared according to a published protocol.26 A thin film of lipid was first deposited onto an ITO coated slide and dried under vacuum for 2 h. The lipid film was then rehydrated with 350 mM sucrose solution (~350 mOsm) to a 1 mM final lipid concentration. Electroformation was performed at room temperature for nonphase-separated GUV (NPS GUV) and at 60 °C for phase-separated GUV (PS GUV). PS GUV were slow cooled overnight, and NPS GUV were cooled to room temperature before experiments. Small quantities of fluorescent lipid probes (0.05 mol % Texas Red DHPE) were used to label the liquid-disordered phase in PS GUV (DPhPC) and NPS GUV (POPC).
2.3. Tryptophan Fluorescence Measurement of HSA with and without DTT.
Human serum albumin solution (20 μM) was prepared with Tris HCl buffer. To unfold the protein, it was incubated with 200 mM DTT for 2 h at room temperature. Concentration was verified with absorbance at 280 nm using a Nanodrop 2000 spectrophotometer (ThermoFisher Scientific, Waltham, MA). Fluorescence measurements were taken on a FluoroMax spectrophotometer (Horiba Scientific, Edison, New Jersey) with a 1 cm fluorescence cuvette. Data were collected using 2 nm excitation and emission slit widths, by exciting tryptophan at 295 nm with an integration time of 1 s over a total range of 285–550 nm.
2.4. Circular Dichroism.
Secondary structure of 20 μM human serum albumin after incubation with 200 mM DTT for 2 h was confirmed with CD spectroscopy. CD spectra were measured on a JASCO J-810 spectrophotometer (Jasco, Inc., Easton, MD). Samples were transferred to a quartz cuvette with a 1 mm path length. The scanning range of the instrument was set between 190 and 260 nm at a rate of 100 nm/min with a 2 s response time and 2 nm spectral bandwidth. Helical contents in folded and unfolded HSA were calculated using mean residue ellipticity at 222 nm:
where θ is the observed ellipticity (mdeg), c is the protein concentration, l is the cuvette path length, M is the molecular weight of protein, and n is the number of peptide bonds. Percent helicity is calculated as follows:27
where MRE222nm is mean residue ellipticity at 222 nm, 4000 is the mean residue ellipticity of β-character and random coil at 222 nm, and 33000 is the mean residue ellipticity of pure helical character at 209 nm
2.5. Equilibrium Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR) of HSA in solution.
ATR-FTIR measurements were performed with a Varian 660 FTIR spectrophotometer (Agilent, Santa Clara, CA) coupled to a liquid nitrogen cooled mercury cadmium telluride (MCT) detector. The internal reflection element was an 80 × 10 × 4 mm trapezoidal germanium ATR plate with an aperture angle of 45°. The Ge plate was rinsed with detergent and ethanol before ATR cell assembly. A water spectrum was taken and used as the background for subsequent measurements. Then, ATR-FTIR spectra were obtained for solutions of 100 μM HSA in water and for 100 μM HSA with 1 mM DTT in water after 2 h incubation. Each spectrum was obtained with an average of 400 scans at 2 cm−1 resolution.
2.6. ATR-FTIR of HSA on Supported Lipid Bilayer.
The germanium ATR reflective element was first rinsed with detergent and ethanol and then subjected to plasma cleaning for 15 min. The crystal was then treated with 1 M H2SO4 for 15 min. A background of the germanium ATR element and water was collected before introducing the lipids. To form the supported lipid bilayer, 2 mM PSL doped with 30 mol % Ni-NTA-DGS in fluid phase lipid was prepared at 60 °C, added to the crystal while PSL was still at 60 °C, and allowed to incubate for 30 min. Water was flushed through the cell to rinse away any excess vesicles, and the supported lipid bilayer spectrum was obtained.28,29 The supported lipid bilayer spectrum was saved and used as a background spectrum for subsequent measurements. His- tagged human serum albumin (1 μM) was added to the bilayer and allowed to bind to Ni-NTA for 15 min, and then the sample spectrum was taken. Afterward, 1 M EDTA was added to the crystal in order to unbind His-tagged human serum albumin from the bilayer. Then, the bilayer was flushed with water in order to remove proteins, and 1 M NiCl2 was added to the bilayer (final concentration 100 mM) in order to recharge Ni-NTA-DGS. After NiCl2 was flushed away with copious amount of water, a spectrum was taken to ensure removal of previously bound proteins from the bilayer. Then, 1 μM His-tagged human serum albumin and 10 mM DTT, preincubated together for 2 h at room temperature, were added to the ATR cell. Each spectrum was obtained with an average of 400 scans at 2 cm−1 resolution.
2.7. Quantification of Bound HSA Concentration on Liposomes.
Fluorescence measurements were taken on a FluoroMax spectrophotometer (Horiba Scientific, Edison, New Jersey) with a 1 cm fluorescence cuvette. Data were collected using 2 nm excitation and emission slit widths, by exciting the fluorophore at 488 nm with an integration time of 1 s over a total range of 285—550 nm. A calibration curve was first generated by measuring fluorescence intensities of 0.05, 0.1, 0.3, 0.5, 0.7, 0.9, and 1 μM of Alexa 488- label ed His-tagged HSA at 525 nm (Figure S9). Then, 1 mM of PSL and NPSL with 20 mol % Ni-NTA- DGS in the liquid-disordered phase were each incubated with 1 μM Alexa 488-labeled His-tagged HSA. The mixture was allowed to incubate for 30 min and then was filtered with an Amicon centrifugal filter unit with a molecular cutoff weight of 100 kDa at 14000g for 10 min to remove any unbound protein. Following centrifugation, the amount of Alexa 488-labeled His-tagged HSA in the flow through and in collected liposomes were quantified by measuring fluorescence intensities at 525 nm.
2.8. Giant Unilamellar Vesicles Tubulation Assay.
PS GUV and NPS GUV (~ mM) were incubated with 1 μM Alexa 488- labeled His-tagged HSA with or without 4 mM DTT for 2 h. The GUV solutions were then diluted twice in 20 mM MOPS and 100 mM NaCl buffer (osmolarity adjusted with glucose to ∼ 320 mOsm) prior to imaging at room temperature. Vesicles were observed using a confocal laser scanning microscope, a 60x oil-immersion objection, and a PMT detector on a Nikon Confocal C2+ microscope (Melville, NY). Vesicles were excited with a laser at 488 nm and/or at 561 nm. Confocal images and movies were analyzed with IMAGE J software (National Institutes of Health, Bethesda, MD).
2.9. Modeling of Steric Pressure Generated by Protein Unfolding on Giant Unilamellar Liposomes.
We used a previously established thermodynamic model of membrane steric pressure12 to compare our experimental results with the predicted effect of steric pressure on membrane bending. To compute the steric pressure generated by the bound protein layer before and after unfolding, we used the Carnahan—Starling equation of state for hard disks.12,30 The intrinsic assumption of this model is that the proteins are considered as spherical, non-attracting disks with uniform dimensions. Stokes radii values used for the calculation (native HSA r ~ 3.52 nm and reduced unfolded HSA r ~ 4.22 nm) were based on results obtained from size exclusion chromatography.23 Considering the membrane—protein interface as a two-dimensional interface, the Carnahan—Starling equation can be described as follows.
The pressure is denoted by p, described in terms of free energy per unit area. The protein height is denoted by σ, and fraction of membrane area covered by the protein is denoted by η. and T denote the Boltzmann constant and temperature, respectively.
In our experiments, we modified the Ni-NTA loading density to modulate dissociation constants of the protein binding to the membrane surface. While the exact HSA binding constants were not determined directly, a reasonable assumption is that the values will fall within 1 nM to 5 μM from previously published results.31,32 The binding constants will affect the fraction of protein bound to the surface, in turn affecting the fraction coverage. The fraction of proteins bound to the surface was estimated as follows.
Here, θ denotes the fraction of proteins bound to the surface. [HSA] denotes the concentration of the protein solution, and Kd denotes the dissociation constant of 6x His-HSA binding to the Ni-NTA-DGS liposome surface. The number of liposomes in solution and overall available membrane surface area were determined by assuming the headgroup area of ~0.71 nm2 for phosphatidylcholine lipids.33
3. RESULTS AND DISCUSSION
Reductive Unfolding of HSA.
We optimized conditions for reductive unfolding of HSA using multiple spectroscopic probes, including tryptophan fluorescence, circular dichroism (CD) spectroscopy, ATR-FTIR (Figure 1), and dynamic light scattering (DLS) (Figure S1A, B). Addition of DTT to a solution of HSA caused the tryptophan fluorescence to blue- shift (Figure 1A), consistent with previous work that demonstrated this unusual signature of HSA unfolding.23,34 Time-dependent tryptophan fluorescence measurements were performed to determine the incubation time necessary for protein unfolding (Figure S1D). The CD spectrum also indicates some loss of the helix bands at 208 and 222 nm in the presence of DTT (Figure 1B), clear evidence of HSA unfolding while retaining ~60% helical character. The infrared amide I band, arising from C=O stretching vibrations of polypeptide backbone carbonyls, is an indicator of secondary and tertiary structural changes due to its sensitivity to hydrogen bonding and to vibrational coupling. The second derivative of the amide I band is particularly sensitive to protein aggregation, showing sharp minima at 1620 and 1680 cm−1 due to formation of β aggregates.35 The second derivative spectrum of HSA (Figure 1C) highlights subcomponents of the broad amide I absorbance, with minima at 1630 and 1650 cm−1 assigned to solvated and buried helices, respectively.36 The minimum at 1650 cm−1 dominates the second derivative spectrum as expected for the predominantly α-helical HSA, while a minimum at 1630 cm−1 indicates some solvated helix structure. Importantly, the well-established signatures of antiparallel β-sheet structures characteristic of protein aggregation are not observed for the reduced, unfolded protein, indicating that it is not aggregated.
Figure 1.
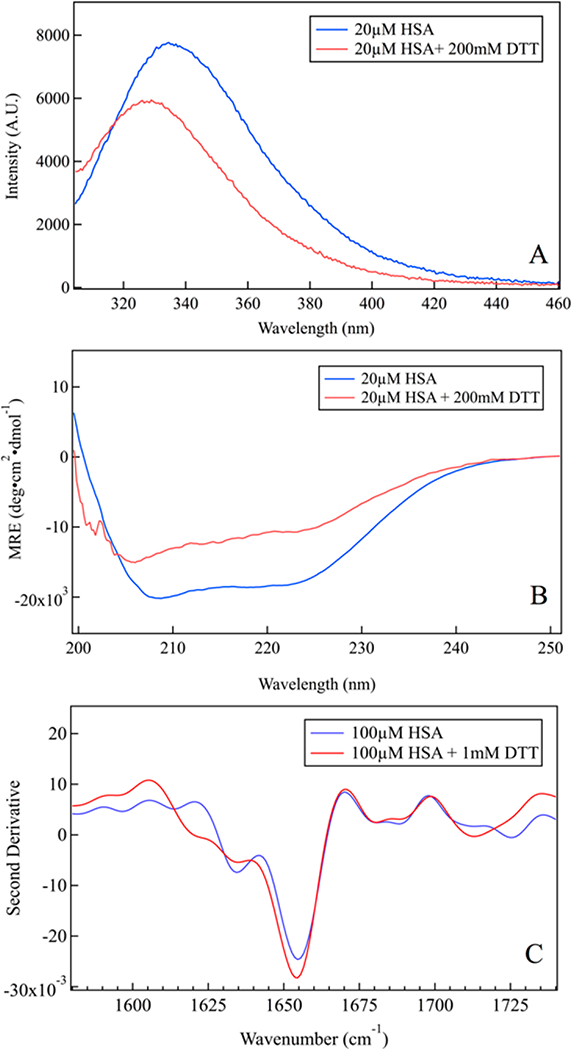
Characterization of HSA unfolding at room temperature. (A) Tryptophan fluorescence blue-shifts upon reduction by DTT, characteristic of HSA unfolding. (B) Far-UV CD spectrum of folded HSA shows minima at 208 and 222 nm, characteristic of a-helical proteins; the helix peaks decrease on addition of DTT, indicating substantial unfolding of HSA. (C) Second derivative ATR-FTIR spectrum shows a decrease of the helix bands and no aggregation of unfolded HSA.
Effect of HSA Unfolding on Large Unilamellar Liposomes.
Phase-separated liposomes (PSL) with coexisting liquid-disordered (DPhPC) and liquid-ordered (DPPC) phases were used to investigate the effect of protein unfolding on membrane bending and to compare to liquid-disordered (POPC) nonphase-separated liposomes (NPSL) as a control. Nonphase-separated liposomes (NPSL) are composed of POPC (+20 mol % Ni-NTA-DGS), whereas phase-separated liposomes are composed of 12.5 mol % fluid phase lipid (DPhPC + 20 mol % Ni-NTA-DGS), 40 mol % DPPC, and 47.5 mol % cholesterol. Thus, Ni-NTA-DGS occupies 2.5 mol % of the total lipid composition in PSL and makes up 20 mol % of the total lipid composition in NPSL. The composition chosen for phase-separated liposomes corresponds to a region in the DPhPC/DPPC/cholesterol phase diagram where the separation of liquid-ordered and liquid-disordered phases is distinct and persists up to high temperature (up to 45 °C) (Figure 2A).12,37 DPhPC and POPC have similar bending rigidities (~10−20J) as reported previously.38,39 HSA was first bound to the liposome surface via interaction of its His-tag with Ni-NTA-functionalized DGS lipid head groups (Kd ~ nM)40 present at 20 mol % in the liquid-disordered phase of lipid membrane and then unfolded by DTT.
Figure 2.
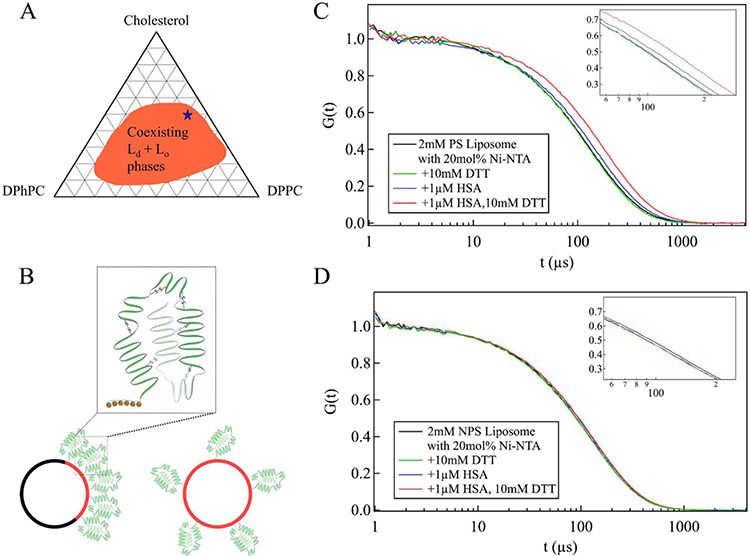
Effect of protein unfolding on lipid membranes. (A) Ternary phase diagram of DPhPC, DPPC, and cholesterol at 25 °C.12,37 A blue star indicates selected lipid composition. (B) Depiction of distribution of liquid-disordered phase, Ni-NTA-DGS, and bound proteins in PSL (left) and NPSL (right). In PSL, liquid-disordered (Ld, red) and liquid-ordered (Lo, black) phases coexist, with Ld occupying one side of the liposome. Thus, His-tagged HSA bound to Ni-NTA-DGS is concentrated on one side of the liposome. In NPSL, His-tagged HSA bound to Ni-NTA-DGS is distributed throughout the entire surface of the liposome. Normalized DLS autocorrelation functions of (C) 2 mM PSL and (D) 2 mM NPSL with 20 mol % Ni-NTA-DGS in the liquid-disordered domain, incubated with HSA (blue traces) or HSA with DTT (red traces).
Ni-NTA-DGS lipid partitions preferentially into the liquid- disordered phase of PSL, due to its unsaturated hydrocarbon tail, thus resulting in an increase in local protein concentration of the bound His-tagged HSA only in the liquid-disordered phase. In contrast, the control 2 mM NPSL consisted of a single phase, liquid-disordered lipid (POPC), which led to random distribution of Ni-NTA-DGS and, hence, the His- tagged HSA over the full surface area of the vesicle. Because of the difference in distribution of His-tagged HSA, we expect a higher local protein density in PSL over NPSL (Figure 2B) by maintaining the same doping level of Ni-NTA-DGS lipid in the liquid-disordered phase.
Dynamic light scattering was used (1) to determine the autocorrelation function decay times of free liposomes and HSA bound liposomes, (2) to compare the change in decay time of folded HSA relative to reductively unfolded HSA bound to liposomes, and (3) to compare effects of HSA binding and HSA unfolding on PSL versus NPSL (Figure 2C, D). The DLS autocorrelation decay time is related to the liposome diffusion time and thus serves as a qualitative measure of liposome tubulation.
PSL showed a small increase in decay time upon binding of HSA at a 20 mol % Ni-NTA-DGS doping level in the liquid- disordered phase (Figure 2C, blue trace). Upon unfolding of HSA on PSL, a significantly larger increase in decay time was observed (Figure 2C, red trace). In contrast, NPSL showed very little change in decay time upon protein binding and protein unfolding (Figure 2D). We interpret the substantial increase in decay time for PSL upon protein unfolding to an increase in the diffusion time as a result of membrane bending and tubulation. We expect the formation of tubules to cause more solvent drag and consequently to slow down diffusion of the liposomes through solution. A similar effect is not observed in NPSL; therefore, we conclude that tubulation does not occur in this case. We attribute the lack of tubulation to the lower local density of bound protein that results from dispersing the His-tagged HSA uniformly over the entire liposome surface. A slight increase in decay time was observed for NPSL with bound, unfolded HSA (Figure 2D, red trace), which is not surprising since the hydrodynamic radius of free, unbound HSA increases substantially upon unfolding (Figure S1A, B).
To investigate how coupling protein unfolding to protein crowding affects membrane curvature, we varied the percentage of Ni-NTA-DGS (10–30 mol % in the liquid- disordered phase) to change the number of binding sites available to His-tagged HSA. Similar to the previous experiments, we first bound HSA to the membrane and then unfolded the protein with DTT. Figure 3A shows the effects of varying the doping level of Ni-NTA-DGS in the liquid- disordered phase of PSL in the presence of native HSA and reductively unfolded HSA (Figure S3 shows the corresponding G(t) curves. Table S2 shows decay time values). Decay times shown were obtained by fitting autocorrelation functions to a single exponential. Insignificant differences in autocorrelation decay times with native and unfolded HSA were observed for 10 mol % Ni-NTA-DGS doped PSL. These results suggest that the crowding effect generated by concentrating either native or unfolded HSA on the liquid-disordered phase is small at a lower doping level. In contrast, at a higher doping level (30 mol % Ni-NTA-DGS), the crowding effect appears to saturate, such that there is a smaller difference in autocorrelation decay times between folded and unfolded HSA on PSL compared to a doping level of 20 mol % Ni-NTA-DGS. Thus, the effect of crowding appears to be resolved best at the intermediate Ni- NTA-DGS doping level.
Figure 3.
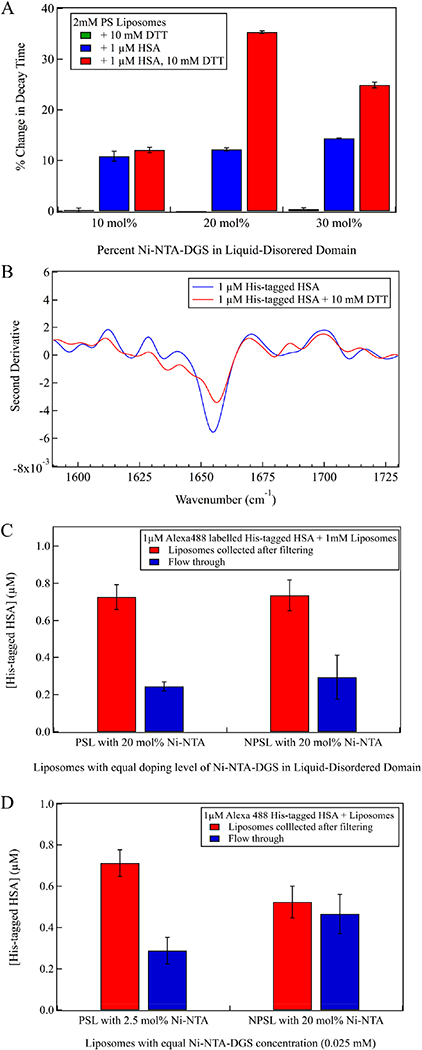
(A) Percent change in autocorrelation decay time of PSL measured after protein incubation for Ni-NTA-DGS doping levels of 10-30 mol % in the liquid-disordered phase. (B) Second derivative IR spectrum of HSA (blue trace) and HSA with DTT (red trace) on phase-separated supported lipid bilayer with 30 mol % Ni-NTA-DGS. (C, D) Fluorescence assay of bound and free HSA concentrations (1 μM total [HSA]) for 1 mM PSL and NPSL liposomes with (C) equal doping levels (20 mol %) of Ni-NTA-DGS in the liquid-disordered domain and (D) equal total Ni-NTA-DGS concentrations (0.025 mM).
Using transmission electron microscopy, we observed that unfolding HSA on lipid membrane is capable of generating highly curved membrane structures in PSL with 20 mol % Ni- NTA-DGS in the liquid-disordered phase, whereas folded HSA causes limited deformation (Figure S5B). In PSL and NPSL with 10 mol % Ni-NTA lipid in the liquid-disordered phase, folded and unfolded HSA did not alter liposome geometry (Figure S5A), which agreed with dynamic light scattering measurements.
Assessing Whether HSA Bound to the Lipid Bilayer Aggregates when Unfolded.
To investigate whether aggregation of unfolded HSA contributes to the observed membrane bending, we performed ATR-FTIR spectroscopy on supported lipid bilayer to probe the protein structure. His- tagged HSA (1 μM) was first bound to supported lipid bilayer (lipid composition remains the same as PSL) and then was unfolded by incubating it with 10 mM DTT. As a control, we performed the same experiment without any DTT. Figure 3B shows second derivative ATR-FTIR spectra of 1 μM His-tagged HSA and 1 μM reductively unfolded His-tagged HSA bound to 30 mol % Ni-NTA-DGS containing phase-separated supported lipid bilayer. In both cases, the characteristic features of aggregated protein at 1620 and 1680 cm−1 are clearly missing. Unfolding of His-tagged HSA in phase- separated lipid systems did not generate aggregates even at the highest Ni-NTA-DGS doping level. C—H stretching bands at 2850 and 2920 cm−1, indicators of bilayer stability, showed binding of HSA and unfolding of HSA had no effect on the lipid bilayer (Figure S4B).29 The C—H stretching region also remained unchanged after the addition of 100 mM NiCl2 (final concentration); therefore, the supported lipid bilayer was not affected by the NiCl2 treatment (Figure S4C).
Quantification of Bound HSA on Liposomes.
The concentrations of protein bound to PSL and NPSL under our experimental conditions were quantified and compared using fluorescence spectroscopy. We attached Alexa 488 dye to His- tagged HSA and measured the fluorescence intensity of Alexa 488 fluorophore at 525 nm. A calibration curve was generated to convert measured fluorescence intensity to protein concentration (Figure S9). Figure 3C shows concentrations of His-tagged HSA bound to 1 mM PSL and 1 mM NPSL with 20 mol % Ni-NTA-DGS in the liquid-disordered phase as quantified by the Alexa 488 fluorescence. It is important to note that the liposome composition was designed to yield 20 mol % Ni-NTA-DGS in the liquid-disordered phase for both PSL and NPSL; therefore, the total Ni-NTA-DGS concentration in PSL is smaller because the fraction of liquid- disordered phase is smaller in this case. Therefore, 1 mM PSL contains 12.5 mol % DPhPC as liquid-disordered phase and, thus, contains 0.025 mM Ni-NTA-DGS (20 mol % of the liquid-disordered phase). In contrast, 1 mM NPSL contains 0.2 mM Ni-NTA-DGS. Since PSL has 8-fold fewer Ni-NTA-DGS than NPSL, PSL should have 8-fold fewer binding sites and the amount of bound protein should be 8-fold less. However, we observed that PSL and NPSL with the same doping level of Ni- NTA-DGS (20 mol %) have a similar amount of protein bound (~1% difference), despite the difference in absolute number of binding sites (Figure 3C). These results demonstrate that the density of HSA bound to the liquid- disordered phase is substantially higher for PSL compared to NPSL.
We repeated the same experiment while maintaining the total concentration of Ni-NTA-DGS at 0.025 mM for both PSL and NPSL (Figure 3D). In this case, PSL contains 2.5 mol % Ni-NTA-DGS in the liquid-disordered domain and NPSL contains 20 mol % Ni-NTA-DGS. The measurement clearly demonstrates that when PSL and NPSL have identical overall concentrations of Ni-NTA-DGS, hence with same number of binding sites, more proteins are bound to PSL (~1.5 fold more) (Figure 3D).
The difference in bound-protein density between PSL and NPSL has been demonstrated quantitatively in previous publications.10,12 Also, previous studies have shown a positive correlation between binding strength of His-tagged HSA and an increase in Ni-NTA-DGS density in the membrane.41 Since the mechanism of interaction between His-tag and Ni-NTA is universal, irrespective of the protein that carries the His-tag, we attribute the difference in bound-protein concentration to multivalency of the His-tag interaction with the Ni-NTA, which enhances its binding affinity.
Effect of HSA Unfolding on Giant Unilamellar Liposomes.
We studied the membrane bending processes due to HSA unfolding with single-vesicle imaging using confocal fluorescence microscopy. First, we exposed electro- formed phase-separated giant unilamellar vesicles (PS GUV) and their nonphase-separated counterparts (NPS GUV, same composition as large unilamellar liposomes) to folded HSA and then induced protein unfolding with DTT. We monitored protein binding and giant liposome geometry changes by confocal fluorescence imaging.
First, the Alexa 488 fluorophore attached to His-tagged HSA via a thiol—maleimide linkage that acts as a reporter to monitor protein binding to the membrane. Second, in order to distinguish the liquid-disordered phases from the liquid- ordered phases, the liquid-disordered phases of PS GUV and NPS GUV were doped with Texas Red DHPE, which is known to partition into the liquid-disordered phase.42 The over-lapping signal on images and intensity profile from Alexa 488 and Texas Red indicated binding of His-tagged HSA to the liquid-disordered phase of GUV (Figure 4A, B). These results also provide additional evidence for partitioning of the Ni- NTA-DGS lipid into the liquid-disordered phase in PS GUV and NPS GUV.
Figure 4.
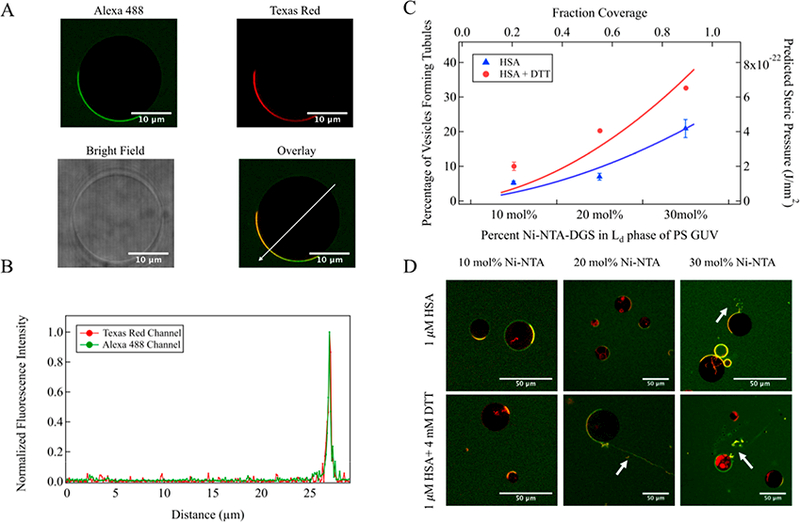
GUV tabulation assay. (A) Confocal images of PSL GUV (+20 mol % Ni-NTA-DGS in fluid domain) incubated with 1 μM Alexa 488-labeled His-tagged HSA. Scale bar, 10 μm long. (B) Fluorescence intensity profile of PS GUV incubated with HSA, measured along the white arrow on the overlaid image. (C) Solid lines: theoretical prediction of steric pressure generated from lateral collisions versus fraction coverage for folded (blue trace) and unfolded (red trace) HSA on PS GUV. Symbols: qualitative measurement of percentage of GUVs forming tubules determined from confocal fluorescence images as a function of increasing percentage of Ni-NTA-DGS in the liquid-disordered phase in PS GUV. N = 3 independent experiments, >100 GUV per experiment. SD calculated from 3 independent trials. (D) Representative confocal fluorescence images of PS GUVs containing 10, 20, and 30 mol % Ni-NTA-DGS in the liquid-disordered domain after incubation with HSA (upper images) or HSA with DTT (lower images). Scale bar, 50 μm long. The green color represents Alexa 488-labeled His-tagged HSA, while the red color represents the Texas Red-doped liquid-disordered domain of GUV. The yellow color indicates overlap between the two. White arrows indicate tubules from GUV.
Upon incubation of the protein-bound GUV with DTT, highly flexible tubules with diffraction-limited diameters and dynamically fluctuating conformations were observed (Figure 4D, Movie S1). This observation indicates an increase in spontaneous curvature of the membrane caused by enhanced lateral pressure generated from collisions between unfolded membrane bound proteins.10,43,44 We varied the percentage of Ni-NTA-DGS lipid in the fluid domain of GUV to change the density of His-tagged protein bound to the membrane surface. The number of GUV that formed tubules was found to depend on the doping level of Ni-NTA-DGS in the liquid-disordered domain. The percentage of GUV forming tubules in the presence of folded and unfolded HSA was plotted relative to the percentage of Ni-NTA-DGS content in the liquid- disordered domain of lipid vesicles in Figure 4C. The resulting increase in tubule formation follows the trend predicted by the Carnahan—Starling equation,12 as shown in the overlaid curves in Figure 4C. These data demonstrate qualitatively that the unfolding transition of HSA together with high protein density on the liquid-disordered domain of lipid membrane affect the frequency of tubule formation. Observation of vesicles budding at PS GUV containing 30 mol % Ni-NTA-DGS (Figure 4D) further highlights the coupled impact on membrane geometry.
4. CONCLUSIONS
In summary, by reductively unfolding membrane bound HSA with DTT, we investigated the effect of an induced protein conformational change (unfolding) on membrane curvature. We found that the process of unfolding a model membrane bound protein can drive membrane bending. This study improves our understanding of the contribution of protein crowding and the role of protein conformational change on membrane curvature generation. Our results are important for developing peripheral membrane protein immobilization strategies, and they open new avenues for exploring the role of conformational changes in membrane bending driven by peripheral membrane—protein interaction.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (GM053640 to R.B.D.).
■ ABBREVIATIONS
- HSA
human serum albumin
- DTT
dithiothreitol
- DPhPC
1,2-diphytanoyl-sn-glycerol-3-phosphocholine
- DPPC
1,2-di-palmitoyl-sn-glycero-3-phosphocholine
- POPC
1-palmitoyl-2- oleoyl-sn-glycerol-3-phosphocholine
- Ni-NTA-DGS
1,2-dio- leoyl-sn-glycerol-3-[(N-(5-amino-1-carboxypentyl)imino-diacetic acid)succinyl] (nickel salt)
- Texas Red DHPE
Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanol-amine, triethylammonium salt
- PSL
phase separated liposome
- NPSL
nonphase-separated liposomes
- PS GUV
phase separated giant unilamellar vesicle
- NPS GUV
nonphase-separated giant unilamellar vesicles
- CD
circular dichroism
- DLS
dynamic light scattering
- TEM
transmission electron microscopy
- ACF
autocorrelation function
- ATR-FTIR
attenuated total reflection fourier transform infrared
Footnotes
■ ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.lang-muir.8b01136.
Supplementary movie (AVI)
Supplementary methods and supplementary data (PDF)
■ AUTHOR INFORMATION
Author Contributions
H.S. carried out the experiments, G.R. carried out the modeling, and H.S., G.R., and R.B.D. designed the research and wrote the article.
Notes
The authors declare no competing financial interest.
■ REFERENCES
- (1).Smith AW Lipid-protein interactions in biological membranes: a dynamic perspective. Biochim. Biophys. Acta, Biomembr 2012, 1818, 172–177. [DOI] [PubMed] [Google Scholar]
- (2).Kirchhausen T Bending membranes. Nat. Cell Biol 2012, 14, 906–908. [DOI] [PubMed] [Google Scholar]
- (3).Ferguson SM; De Camilli P Dynamin, a membrane remodelling GTPase. Nat. Rev. Mol. Cell Biol 2012, 13, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hofmann H; Soranno A; Borgia A; Gast K; Nettels D; Schuler B Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 16155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ferreon AC; Gambin Y; Lemke EA; Deniz AA Interplay of alpha-synuclein binding and conformational switching probed bysingle-molecule fluorescence. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 5645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shi Z; Sachs JN; Rhoades E; Baumgart T Biophysics of alpha-synuclein induced membrane remodelling. Phys. Chem. Chem. Phys 2015, 17, 15561–15568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jiang Z; de Messieres M; Lee JC Membrane remodeling by alpha-synuclein and effects on amyloid formation. J. Am. Chem. Soc 2013, 135, 15970–15973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McMahon HT; Boucrot E Membrane curvature at a glance. J. Cell Sci 2015, 128, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zimmerberg J; Kozlov MM How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol 2006, 7, 9–19. [DOI] [PubMed] [Google Scholar]
- (10).Stachowiak JC; Hayden CC; Sasaki DY Steric confinement of proteins on lipid membranes can drive curvature and tubulation. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 7781–7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen Z; Atefi E; Baumgart T Membrane Shape Instability Induced by Protein Crowding. Biophys. J 2016, 111, 1823–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stachowiak JC; Schmid EM; Ryan CJ; Ann HS; Sasaki DY; Sherman MB; Geissler PL; Fletcher DA; Hayden CC Membrane bending by protein-protein crowding. Nat. Cell Biol 2012, 14, 944–949. [DOI] [PubMed] [Google Scholar]
- (13).Busch DJ; Houser JR; Hayden CC; Sherman MB; Lafer EM; Stachowiak JC Intrinsically disordered proteins drive membrane curvature. Nat. Commun 2015, 6, 7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Snead WT; Hayden CC; Gadok AK; Zhao C; Lafer EM; Rangamani P; Stachowiak JC Membrane fission by protein crowding. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E3258–E3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Derganc J; Copic A Membrane bending by protein crowding is affected by protein lateral confinement. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1152–1159. [DOI] [PubMed] [Google Scholar]
- (16).Kozlov MM; Campelo F; Liska N; Chernomordik LV; Marrink SJ; McMahon HT Mechanisms shaping cell membranes. Curr. Opin. Cell Biol 2014, 29, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ahmad B; Muteeb G; Alam P; Varshney A; Zaidi N; Ishtikhar M; Badr G; Mahmoud MH; Khan RH Thermal induced unfolding of human serum albumin isomers: assigning residual alpha helices to domain II. Int. J. Biol. Macromol 2015, 75, 447–452. [DOI] [PubMed] [Google Scholar]
- (18).Santra MK; Banerjee A; Rahaman O; Panda D Unfolding pathways of human serum albumin: evidence for sequential unfolding and folding of its three domains. Int. J. Biol. Macromol 2005, 37, 200–204. [DOI] [PubMed] [Google Scholar]
- (19).Del Giudice A; Dicko C; Galantini L; Pavel NV Time- Dependent pH Scanning of the Acid-Induced Unfolding of Human Serum Albumin Reveals Stabilization of the Native Form by Palmitic Acid Binding. J. Phys. Chem. B 2017, 121, 4388–4399. [DOI] [PubMed] [Google Scholar]
- (20).Pande VS; Rokhsar DS Molecular dynamics simulations of unfolding and refolding of a beta-hairpin fragment of protein G. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Carter DC; He XM; Munson SH; Twigg PD; Gernert KM; Broom MB; Miller TY Three-dimensional structure of human serum albumin. Science 1989, 244, 1195–1198. [DOI] [PubMed] [Google Scholar]
- (22).Getz EB; Xiao M; Chakrabarty T; Cooke R; Selvin PR A comparison between the sulfhydryl reductants tris(2-carboxyethyl)- phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem 1999, 273, 73–80. [DOI] [PubMed] [Google Scholar]
- (23).Lee JY; Hirose M Partially folded state of the disulfide- reduced form of human serum albumin as an intermediate for reversible denaturation. J. Biol. Chem 1992, 267, 14753–14758. [PubMed] [Google Scholar]
- (24).Cleland WW Dithiothreitol, a New Protective Reagent for SH Groups*. Biochemistry 1964, 3, 480–482. [DOI] [PubMed] [Google Scholar]
- (25).Bordovsky SS; Wong CS; Bachand GD; Stachowiak JC; Sasaki DY Engineering Lipid Structure for Recognition of the Liquid Ordered Membrane Phase. Langmuir 2016, 32, 12527–12533. [DOI] [PubMed] [Google Scholar]
- (26).Angelova MI; Dimitrov DS Liposome electroformation. Faraday Discuss. Chem. Soc 1986, 81, 303. [Google Scholar]
- (27).Pattanayak R; Basak P; Sen S; Bhattacharyya M An insight to the binding of ellagic acid with human serum albumin using spectroscopic and isothermal calorimetry studies. Biochem Biophys Rep 2017, 10, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Arsov Z; Quaroni L Detection of lipid phase coexistence and lipid interactions in sphingomyelin/cholesterol membranes by ATR- FTIR spectroscopy. Biochim. Biophys. Acta, Biomembr 2008, 1778, 880–889. [DOI] [PubMed] [Google Scholar]
- (29).Hull MC; Cambrea LR; Hovis JS Infrared spectroscopy of fluid lipid bilayers. Anal. Chem 2005, 77, 6096–6099. [DOI] [PubMed] [Google Scholar]
- (30).Carnahan NF; Starling KE Equation of State for Nonattracting Rigid Spheres. J. Chem. Phys 1969, 51, 635–636. [Google Scholar]
- (31).Lauer SA; Nolan JP Development and characterization of Ni-NTA-bearing microspheres. Cytometry 2002, 48, 136–145. [DOI] [PubMed] [Google Scholar]
- (32).Dorn IT; Pawlitschko K; Pettinger SC; Tampe R Orientation and two-dimensional organization of proteins at chelator lipid interfaces. Biol. Chem 1998, 379, 1151–1159. [DOI] [PubMed] [Google Scholar]
- (33).Nagle JF Area Lipid of Bilayers from Nmr. Biophys. J 1993, 64, 1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Anand U; Ray S; Ghosh S; Banerjee R; Mukherjee S Structural Aspects of a Protein-Surfactant Assembly: Native and Reduced States of Human Serum Albumin. Protein J. 2015, 34, 147–157. [DOI] [PubMed] [Google Scholar]
- (35).Shivu B; Seshadri S; Li J; Oberg KA; Uversky VN; Fink AL Distinct beta-sheet structure in protein aggregates determined by ATR-FTIR spectroscopy. Biochemistry 2013, 52, 5176–5183. [DOI] [PubMed] [Google Scholar]
- (36).Gilmanshin R; Williams S; Callender RH; Woodruff WH; Dyer RB Fast events in protein folding: relaxation dynamics of secondary and tertiary structure in native apomyoglobin. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 3709–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Veatch SL; Gawrisch K; Keller SL Closed-loop miscibility gap and quantitative tie-lines in ternary membranes containing diphytanoyl PC. Biophys. J 2006, 90, 4428–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dimova R Recent developments in the field ofbending rigidity measurements on membranes. Adv. Colloid Interface Sci 2014, 208, 225–234. [DOI] [PubMed] [Google Scholar]
- (39).Vitkova V; Meleard P; Pott T; Bivas I Alamethicin influence on the membrane bending elasticity. Eur. Biophys. J 2006, 35, 281–286. [DOI] [PubMed] [Google Scholar]
- (40).Gizeli E; Glad J Single-step formation of a biorecognition layer for assaying histidine-tagged proteins. Anal. Chem 2004, 76, 3995–4001. [DOI] [PubMed] [Google Scholar]
- (41).Blanchette CD; Fischer NO; Corzett M; Bench G; Hoeprich PD Kinetic analysis of his-tagged protein binding to nickel-chelating nanolipoprotein particles. Bioconjugate Chem. 2010, 21, 1321–1330. [DOI] [PubMed] [Google Scholar]
- (42).Baumgart T; Hess ST; Webb WW Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 2003, 425, 821–824. [DOI] [PubMed] [Google Scholar]
- (43).Lipowsky R Spontaneous tubulation of membranes and vesicles reveals membrane tension generated by spontaneous curvature. Faraday Discuss 2013, 161, 305–331, discussion 419–359;. [DOI] [PubMed] [Google Scholar]
- (44).Stachowiak JC; Brodsky FM; Miller EA A cost-benefit analysis of the physical mechanisms of membrane curvature. Nat. Cell Biol 2013, 15, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.