

Abstract
CD4+ T regulatory (Treg) cells are central to immune homeostasis, their phenotypic heterogeneity reflecting the diverse environments and target cells they regulate. To understand this heterogeneity, we combined single-cell RNAseq, activation reporter and TCR analysis to profile thousands of Tregs or Tconvs from mouse lymphoid organs or human blood. Treg and Tconv pools showed areas of overlap, as resting “furtive” Tregs with overall similarity to Tconv, or as a convergence of activated states. All Tregs express a small core of FoxP3-dependent transcripts, onto which additional programs are added less uniformly. Among suppressive functions, Il2ra and Ctla4 were quasi-constant, inhibitory cytokines being more sparsely distributed. TCR signal intensity didn’t affect resting/activated Treg proportions, but molded activated Treg programs. The main lines of Treg heterogeneity in mice were strikingly conserved in human blood. These results reveal unexpected TCR-shaped states of activation, providing a framework to synthesize previous observations about Treg heterogeneity.
Regulatory T cells (Tregs) are dominant negative regulators of many facets of the immune system, controlling immune responses and enforcing peripheral tolerance to self, symbiotic commensals and fetal antigens 1. In addition, some Tregs reside in non-lymphoid tissues, where they help control tissue homeostasis and sterile inflammation 2.
Tregs constitute a diverse constellation of cells 1,3,4. Their origins are diverse 5: many Tregs differentiate in the thymus, but others arise in the periphery from naive CD4+ T cells upon suboptimal exposure to antigen, in particular microbial. Their organismal locations vary: they reside in the T-cell zones of lymphoid organs, but also in B cell areas where they control antibody maturation and production (Tfr, T follicular regulators), in autoimmune or tumoral lesions, at body/microbiota interfaces. Their effector pathways are heterogeneous: Tregs utilize cell-surface inhibitors like CTLA4, inhibitory cytokines like IL-10, IL-35 or TGF-β, cytokine capture via the IL-2 receptor, purine-mediated suppression, or direct cytoxicity 6. These facets correspond to diverse Treg subphenotypes 1,3,4. Particular Treg subtypes have been recognized based on chemokine receptor expression like CXCR3 (CXCR3+ Tregs are particularly adept at suppressing Th1 responses 7–9) or CXCR5 (in T follicular regulatory cells (Tfr) 10,11), or activation markers (in “eTregs” or “aTregs”) 12–15. These more activated types of Tregs are particularly represented among extra-lymphoid Tregs in inflammatory sites 2.
Tregs and conventional CD4+FoxP3− T cells (Tconvs) have opposite immune functions but their molecular distinction can be complicated. Stable expression of FoxP3 is semantically eponymous for Tregs, and FoxP3 controls a substantial fraction of the characteristic transcriptional signature of Treg cells 16,17. However, it is not sufficient, and several other factors, not specific to Tregs but also present in Tconvs, are required by Tregs 5. Further blurring the Treg/Tconv distinction, FoxP3 itself can be expressed transiently upon activation in human 18 and mouse 19 Tconvs. Conversely, while the Treg phenotype is generally stable, Tregs can lose FoxP3 expression under stress, like IL-2 deprivation 20–22. Finally, Tregs can differentiate directly from Tconvs in tolerogenic contexts, in order to promote peaceful coexistence with commensal microbiota 23,24 or fetal antigens 25.
The T cell receptor (TCR) plays a central role in Treg life story 26. It is necessary for Treg differentiation, and the signals it delivers upon MHC-peptide recognition, conditioned by costimulatory and other modulators, rescues precursor cells from clonal deletion. Continued TCR presence and engagement by MHC molecules is required for suppressive activity and differentiation to an activated phenotype 27,28. The Treg TCR repertoire is skewed towards recognition of self-antigens, but is as broad as that of Tconvs 26,29.
Understanding Treg molecular diversity and definition, in relation to Tconv cells, is thus complex and confounded by the different states that both populations can adopt in response to various stimuli. Single-cell transcriptome analysis offers the potential to illuminate these questions, in an unbiased manner that does not rely on assumptions of cell-type identities 30–37. Although scRNAseq remains challenging due to the limiting sensitivity of detection, and the large dimensionality of the data, the approach has been transformative 38, e.g. in identifying novel cell-types 39, and in dissecting transcriptional differences that were previously masked by the “averaging” inherent to profiling RNA from pooled cells (e.g. 40,41).
Here, we apply scRNAseq to profile thousands of single Treg and Tconv cells, in mice and humans, to reveal the diversity of transcriptional phenotypes that can be adopted by Tregs. We concentrate on two driving questions: how Tregs and Tconvs are related; and how TCR-mediated signals affect Treg activation. For focus, we limit the present analysis to Treg and Tconv cells from lymphoid organs. The results reveal an unexpected degree of overlap between Tregs and Tconvs and provide a framework integrating many prior observations on effector Treg states.
RESULTS
CD4+ Treg and Tconv scRNAseq datasets
In order to maximize the power to find small (sub)populations of cells and significant gene correlations, we performed single-cell transcriptomics on thousands of single cells using InDrop, encapsulating single Treg and Tconv cells in microfluidic droplets 33,42. We sorted CD4+TCRβ+ GFP+ Treg and GFP− Tconv cells from Foxp3gfp mice (Fig. 1a) and encapsulated them separately at high efficiency (70–80%) prior to scRNAseq library construction 42. Over the course of this study, we analyzed 4,237 splenic Tregs (in 3 independent cell cohorts) and 1,093 splenic Tconvs (1 cohort matching Treg #1). In order to minimize batch effect and assess robustness of the results, each Treg cohort was analyzed independently, and the reproducibility of the cell or gene clusters confirmed in the replicates. We extended these data by scRNAseq of total splenic CD4+ T cells (n = 2508, containing Tregs and Tconvs) using a different microfluidic platform (10X Genomics) and library protocol, and by a plate-based approach where individually sorted Tregs were analyzed by CelSeq 32 (n = 200) (Supplementary Table 1).
Figure 1. Treg and Tconv cell scRNAseq datasets.
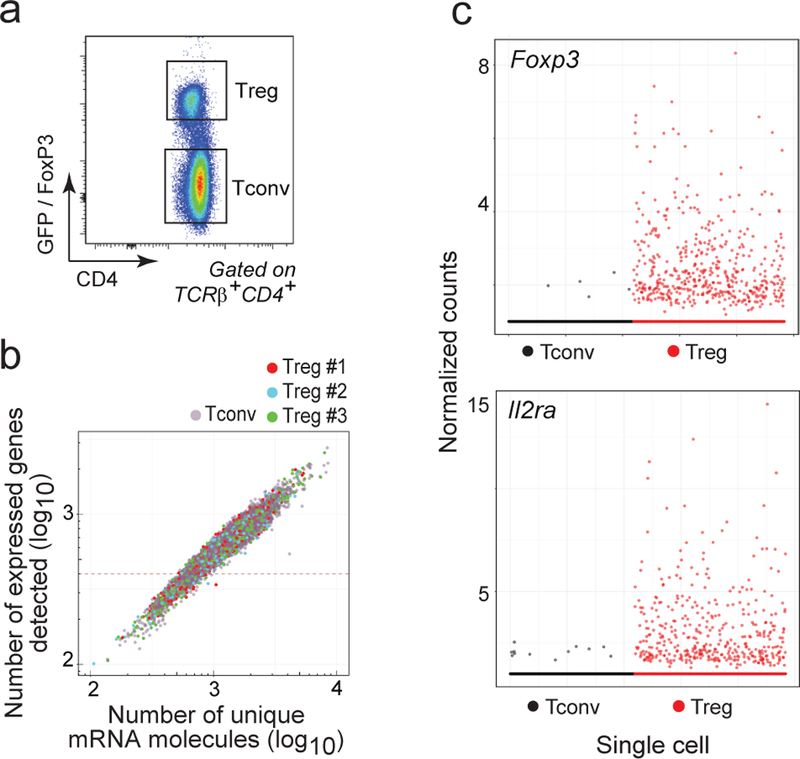
a. Flow cytometric plot of gated CD4+TCRβ+ Treg and Tconv cells from Foxp3.gfp mice sorted for scRNAseq library construction. Representative of 5 experiments.
b. Single cell transcriptome coverage, shown as the number of uniquely identified mRNA reads per cell (X-axis) vs the number of expressed genes per single cell, in 3 Treg biological replicate datasets and the Tconv dataset. Single cells with less than 500 transcripts sequenced were excluded from further analysis.
c. Scatterplot representing the expression (normalized counts) of Foxp3 and Il2ra in the single Treg (n = 708) and Tconv cells (n = 1098).
On average, 1,751 unique mRNA molecules representing 787 genes were detected per single cell in the primary InDrop datasets, altogether surveying 16,720 genes (Fig. 1b). All batches had similar coverage (Fig. 1b) and we observed good concordance between them for both the mean (Supplementary Fig. 1a) and the variation in gene expression (Supplementary Fig. 1b). One of the advantages of single-cell over population profiling is that rare contaminating cells from the sort can be identified, and we used for identification a naive Bayes algorithm that compares each single-cell transcriptome with each of the 249 immune-cell states profiled by the Immgen Consortium (here, 3.5% of the cells were identified as contaminants - mostly B and myeloid cells - and culled; Supplementary Fig. 1c). As a reality check, Foxp3 and Il2ra, two important Treg markers, were expressed throughout the different Tregs, and rarely in Tconv, as expected (Fig. 1c). More generally, the changes in gene expression between Treg and Tconv were conserved compared with a standard population RNAseq and substantially overlapped published Treg and Tconv specific signatures 17 (Supplementary Fig. 1d).
Transcriptional relationship between CD4+ Treg and Tconv cells
As discussed above, Treg and Tconv cells are ontogenetically related, and can derive from one another in some circumstances. Many profiling analyses at the population level have shown that a clear and reproducible gene expression signature distinguishes the two cell types, with Foxp3 the most extreme example of a Treg-specific gene. The averaging generated by population-level profiling can mask complexities in this relationship, so we explored our scRNAseq data specifically in this light. We focus here on splenic populations of unperturbed mice, where Treg and Tconv populations are generally agreed to be stable, as opposed to locations of cell stress, where instability has been observed 43.
We first used t-distributed stochastic neighborhood embedding (tSNE) 44 for dimensionality reduction to relate independently sorted splenic Treg and Tconv cells (sorted independently according to phenotype in the Foxp3-gfp mice; Fig. 2a,b). Although Treg and Tconv cells generally partitioned into two main areas (D and E in Fig. 2a), they were also somewhat imbricated: (i) some Treg cells were found in the main Tconv area (hereafter referred to as “furtive” Tregs) and vice versa (best seen in Fig. 2b, top row); and (ii) there were groups of cells in which Tregs and Tconvs comingled apart from the bulk Treg and Tconv pools (areas A, B and C, demarcated in Fig. 2a by bootstrap-optimized partition clustering). Differences in sequencing coverage 45 were unlikely to explain this clustering as the distribution of detected genes and number of reads were similar in the different areas (Supplementary Fig. 2a,b). In addition, similar patterns were observed on additional Treg and Tconv datasets generated with two other scRNAseq platforms: the 10x Genomics platform, or 96-well-sorted cells profiled by CEL-Seq (Supplementary Fig. 2 c-e).
Figure 2. Some Tregs and Tconv cells show closely related transcriptome.
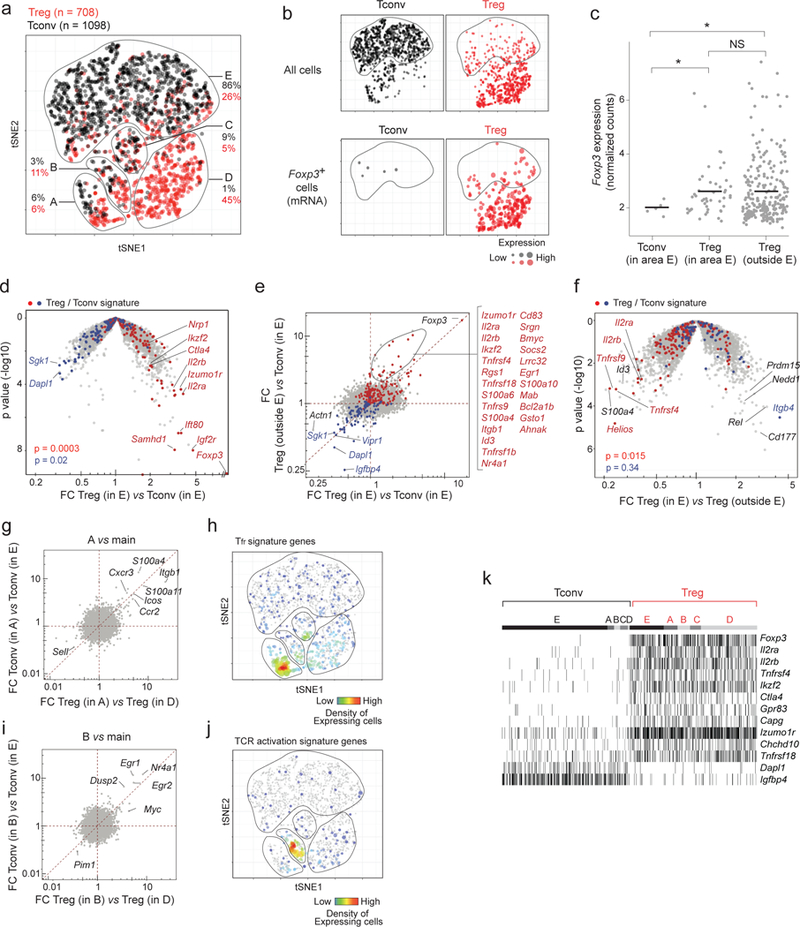
a. Two-dimensional tSNE plot of Treg (red dots) and Tconv (black) single cell transcriptomes (each dot represents one cell) with different areas (A-E) identified by unsupervised clustering. The proportions of Tregs and Tconvs in each area relative to the total number of Tregs and Tconvs is indicated.
b. Same tSNE plots as in a, but split to show Treg and Tconv cells separately (top row) and cells in which Foxp3 transcript was detected (bottom row; the size of each dot indicates the level of Foxp3 RNA expression.
c. Foxp3 expression (normalized counts) in “furtive” Tregs in area E in comparison to Tregs outside area E. Error bar: mean and 95% CI. * p < 0.05 (t-test)
d. Furtive Tregs show the usual biased transcriptome for Treg signature genes. Volcano plot (foldchange versus p value) comparing the gene expression profiles of furtive Treg versus main Tconv (both from area E of the plot). Up- and down-regulated Treg signature genes 17 are highlighted (red and blue respectively). χ2 test p.values
e. Treg/Tconv expression ratio for furtive Tregs in area E (x-axis) vs Treg/Tconv ratio outside area E (x-axis). Up- and down-regulated Treg signature genes are highlighted (red and blue respectively). Gene list are the genes gated on the plot.
f. Down-tuning of the Treg signature in furtive Tregs. Volcano plot comparing the gene expression profiles of furtive versus other Tregs (in and outside area E, respectively). Up- and down-regulated Treg signature genes are highlighted (red and blue respectively). χ2 test p values.
g. Tregs and Tconvs in area A differentially express the same set of genes compared to their respective main populations. Ratio of gene expression in Tregs in A vs D (x-axis) against the ratio of expression in Tconvs in A vs E (y-axis).
h. Same tSNE plot as in a, highlighting cells expressing Tfr signature transcripts 11.
i. Tregs and Tconvs in area B differentially express the same set of genes compared to their respective main populations. Ratio of gene expression in Tregs in B vs D (x-axis) against the ratio of expression in Tconvs in B vs E (y-axis).
j. Same tSNE plot as in a, highlighting cells expressing the most up regulated genes after TCR activation 49
k. Presence/absence (black/white) heatmap showing the core Treg transcripts expressed in all Tregs regardless of their area (each vertical line represents one cell).
Furtive Tregs represented 26% of splenic Tregs in these experiments. Foxp3 transcripts were detectable in many of them (Fig. 2b, lower row), at levels comparable with those of other Tregs (Fig. 2c). Cytometric sorting of Tregs into plates for scRNAseq allowed us to relate protein levels to the transcriptome in single-cells, which showed that furtive Tregs expressed FoxP3 protein at the same level as other Tregs (Supplementary Fig. 2e,f). Furtive Tregs were therefore unlikely to result from mis-expression of the reporter or from contaminating cells in the initial sort (plus, at 26% of the splenic Tregs profiled, furtive Tregs represented far more than the 3.5% contamination rate in the dataset). Canonical Treg signature genes were also over-expressed in these cells compared with surrounding Tconv cells (Fig. 2d). However, some of the Treg signature transcripts were shifted in those cells relative to bulk Tregs (Fig. 2e), including Il2ra and Ikzf2 (encodes Helios), an important TF in Treg cells 46,47. Furtive Tregs also over-expressed Rel, Cd177 and a few transcripts genes normally associated with Tconvs (Itgb4) (Fig. 2f).
In addition to these Tconv-like Tregs, we also observed some Tregs and Tconvs in discrete clusters at the interface (areas A-B-C in Fig. 2a), which were separated from the major Treg and Tconv populations and clustered together instead. In area A (Fig. 2g,h), both Tregs and Tconvs over-expressed a set of genes associated with residence in B cell follicles [i.e. T follicular helper (Tfh) and Tfr cells], including the characteristic Cxcr5 and Icos transcripts 48. In area B (Fig. 2i,j), Tconv and Treg cells both upregulated a set of genes that belong to the early response to TCR engagement in both cells (Nr4a1, Egr1, Egr2, Myc, Dusp2) 49, suggesting that TCR signals may drive similar programs in both Tregs and Tconvs.
While Treg and Tconv cells thus converge to similar subphenotypes defined by the integration of all transcripts through the tSNE algorithm, is there nevertheless an overarching set of transcripts that generally demarcates Treg and Tconv identities, independently of subphenotype variation? We systematically compared Tregs with their closest Tconvs (correlation distances) (Supplementary Fig. 3a) and identified a small geneset (Il2ra, Il2rb, Ikzf2, Ctla4, Capg, Tnfrsf4, Tnfrsf18, Izumo1r, Chchd10, Gpr83, and ex officio Foxp3) that is overexpressed by all Tregs irrespective of their location on the tSNE plot (Fig. 2k). The lack of expression of these genes in Tconv cells falling in areas A, B or C indicates that these are true Tconv cells, not activated cells transiently expressing FoxP3. These genes are all direct targets of FoxP3: they all contain enhancer elements that bind FoxP3 in chromatin immunoprecipitation experiments 50,51 (Supplementary Fig. 3b) and are up regulated upon ectopic FoxP3 expression in Tconvs.
Thus, these data show that while most Tregs are generally distinct from Tconvs, and a small core of Treg-identifying transcripts can be defined, specific subsets of the two cell types do have considerable overlap.
Treg signature: core transcripts and heterogeneity of expression
Switching the analysis to the gene axis, we revisited at the single-cell level the classic Treg signature identified by population transcriptomics 17,52. Collapsing the present single-cell data recapitulated the signature observed in previous microarray or population RNAseq datasets, confirming congruence between the techniques (Fig. 3a)
Figure 3. Treg signature expression in single Treg and Tconv cells.
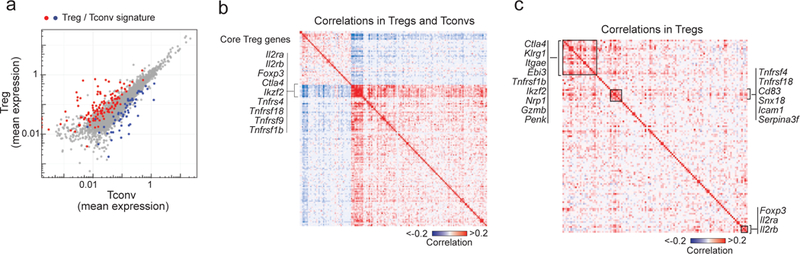
a. Mean expression in collapsed Treg and Tconv single-cell datasets (normalized counts). Canonical up- and down-regulated Treg signature genes 17 are highlighted (red and blue respectively).
b. Gene-gene correlation heatmap for the canonical Treg signature genes, calculated across all Treg and Tconv single-cell datasets.
c. Gene-gene as in b, but calculated in Treg single-cell datasets only.
The natural cell-cell variability in gene expression revealed in scRNAseq can be valuable to identify modules of coregulated genes 33,53,54. We thus searched for gene correlations in our data and asked whether the Treg signature behaves as a collection of discrete modules, as suggested by our earlier analyses 17. Within the entire dataset that includes both splenic Tregs and Tconvs, there was, as expected, a clear partitioning of Treg-up vs Treg-down signature transcripts (Fig. 3b), and a generally positive but weak correlation between Treg-up transcripts, except for a small cluster of highly correlated genes (Fig. 3b). This cluster encompassed the same core set of Treg-specific transcripts (Il2ra, Ctla4, Foxp3, etc) identified in Fig. 2 as shared by all Treg cells, independent of other variations. This result confirms that only this small core of over-expressed genes identifies Tregs, which variably express other genesets as a function of their location or functional subphenotypes, some of which can be shared with Tconv counterparts.
As expected, this tightly coregulated core set vanished when we tested for correlations within Tregs only, aiming to identify co-regulated components independently of their co-variation relative to Tconv (Fig. 3c). Little correlation was seen at all, and only three clusters were observed: a small but relatively tight one that included the TNFR family members Tnfrsf4 and Tnfrsf18 (encode OX40 and GITR, respectively), another that grouped Foxp3 and Il2ra, and a more loosely coordinated cluster that included several other molecules that mediate Treg function (Ebi3, Gzmb, Ctla4). Overall, this paucity of correlation of Treg signature transcripts within Tregs indicates that the different subphenotypes result from a diversity of regulatory influences, rather than a few dominant programs.
In conclusion, the Treg signature identified at the population level can be deconvoluted into a tight core of coregulated genes, likely tightly controlled by FoxP3, and otherwise heterogeneous genesets diversely expressed by individual Tregs.
Splenic Treg heterogeneity revolves around several different poles
In the analyses presented above, we parsed primarily Treg heterogeneity in relation to Tconv cells and to the classic Treg signature. Yet other transcriptional features are likely to be important in defining the functional identity of a given Treg cell, features that are not necessarily Treg-specific. We thus evaluated the distribution of all transcripts among splenic Treg cells. We centered further analyses on the 100 transcripts with the most variability among Tregs, above that expected from Poisson sampling statistics (Supplementary Fig. 4a), and thus the most informative 33 (these most variable genes were the same in the 3 batches of splenic Tregs) (Supplementary Table 2). Unsupervised partition clustering, using a bootstrap approach to optimize the number of clusters and assess their stability 55 (Supplementary Fig. 4b,c), grouped the splenic Tregs into 6 clusters (Fig. 4a on a subset of transcripts that most characterize the different groups) (Supplementary Table 3-4). These clusters should probably be thought of as discrete states in a continuum, rather than as strictly independent entities, as seen below. Importantly, the analysis was performed on one cohort of Tregs and these distinctions were reproduced in independent datasets of splenic Treg cells (Supplementary Fig. 4d), even though the relative proportions of cells in each cluster varied somewhat.
Figure 4. Resting and activated Treg states identified by scRNAseq.
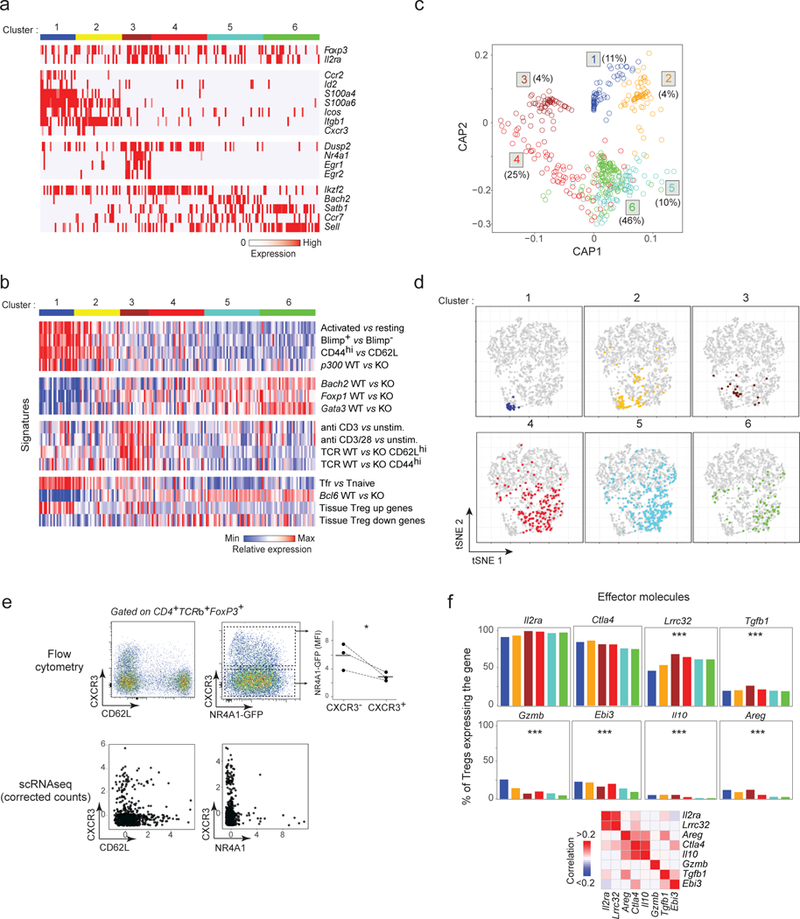
a. Biclustering heatmap of the expression of the most characteristic genes for each Treg cluster (down-sampled to equilibrate cluster size; each vertical line represents one cell)
b. Biclustering heatmap of expression signatures in Treg cells belonging to different clusters (cell and clusters aligned with a).
c. Circular a posteriori projection plot (CAP) 40. Each single cell is projected on a circular plan according to its likelihood to belong to any of the 6 clusters defined in a (n = 708). The proportion of cells in each cluster is indicated.
d. Same tSNE plots as in Fig. 2a, highlighting the location of individual Treg cells from each of the Treg clusters.
e. Cxcr3, Cd62l and Nr4a1 expression in single Treg cells determined by flow cytometry (top row, representative of 3 experiments) or by scRNAseq (corrected counts, bottom row). Top right: Nr4a1-GFP expression in CXCR3− and CXCR3+ Tregs (as gated),* p < 0.05 (t-test).
f. Proportion of cells in each Treg cluster (defined in a) that express different effector transcripts. Proportion calculated after correction for dropout events (see Methods). *** p < 10−4 (ANOVA).
Three main poles could be observed within this partitioning. First, there was a general gradient separating resting and activated Tregs, as judged by the frequency of transcripts typical of resting Tregs, Sell (encodes CD62L) and Ccr7. Tregs in clusters 4–6 seemed to be in a resting state, while those in cluster 1–3 cells were in an activated state (Fig. 4a, Supplementary Fig. 4d). Second, beyond this separation, cells in cluster 1–3 seemed to adopt two different poles of activated phenotypes. Cells in cluster 3 predominantly expressed a set of transcripts typical of early cell activation (Nr4a1, Egr1, Egr2, Myc, also Dusp2), and thus evoke TCR-mediated activation while cells in clusters 1–2 were characterized by a more diverse set of transcripts (Ccr2, Itgb1, S100a4, S100a6, Icos or Cxcr3).
To integrate these data with the body of existing observations on Treg heterogeneity 4,7,10–14, we probed a large set of signatures that distinguish Treg cells, retrieved from databases or curated in house (Figs. 4b, Supplementary Fig. 5). The first set distinguish activated and resting Tregs, and effectively confirmed that clusters 5 and 6 correspond to the most naïve Tregs, with signatures of a/eTregs defined by high Prdm1 (encodes BLIMP1) 56, or resulting from homeostatically-driven expansion 27,57. For the Ep300 (the gene encoding p300) knockout 58, the bias towards activated Tregs is consistent with the decreased proportion of activated CD62Llo Tregs in these mice. Interestingly, the transcriptional signatures of Tregs deficient in several transcriptional cofactors that support Treg differentiation (Foxo1, Bach2, Gata3) 59,60 were under-represented in cells from the activated clusters, congruent with the distribution of Bach2 expression predominant in resting clusters (Fig. 4a). The third set of signatures identified TCR-delivered signals, defined by direct engagement 49 or by acute gene ablation in vivo 27. These signatures clearly tagged cells of cluster 3 as those with the strongest response to TCR signals, consistent with their expression of early response genes. The Bcl6-dependent Tfr signature 11 was most clearly enriched in cluster 1, as was a generic signature that distinguishes Treg cells that localize to a variety of tissues and which we already knew to include a substantial component of cell activation. This juxtaposition would suggest that Tfr, which reside in the B cell areas of spleen and lymph nodes, are nonetheless very similar to cells that reside in non-lymphoid tissues and sites of sterile inflammation.
We projected the likelihood for each single cell to belong to any of these main states, based on the expression of the defining genes of Fig. 4a (circular a posteriori projections (CAP) plots) 40 (Fig. 4c). Most splenic Tregs projected cleanly in one of the main clusters, but with the best definition for activated clusters 1–3, as expected from the gene distinctions of Figs. 4a and S4d. Only cluster 4 remained less sharply defined, appearing almost like a transitional group between the resting clusters 5–6 and the TCR-responsive cluster 3. These assignments were again robust and applied effectively to independently derived Treg scRNAseq datasets (Supplementary Fig. 4e). We also mapped these clusters back onto the Treg/Tconv tSNE plot from Fig. 2 (Fig. 4d). The resting clusters mapped to the core Treg area, but also included many furtive Tregs. On the other hand, the activated clusters mapped to the more peripheral regions of overlap with Tconvs (areas A-C), indicating that Treg activation leads to some convergence with Tconvs.
Since this scRNAseq analysis identified a main resting->activated axis as well as clear distinctions between phenotypes of activated Tregs, it was important to validate independently these observations. We appliedstatistical methods to correct for the low sensitivity of the scRNAseq by calculating the technical dropout probability 53,61,62 (Fig. 4e). Using corrected counts, the scRNAseq data predicted CXCR3 to be predominantly displayed by CD62Llo activated T cells, and that CXCR3 and NR4A1 would be largely mutually exclusive. These predictions were confirmed, with expression of CXCR3 on CD62llo Tregs, and the highest levels of CXCR3 on NR4A1− Tregs (Fig. 4e). We do note a higher proportion of NR4A1+CXCR3lo cells than predicted from the scRNAseq data, perhaps owing to stability of the CXCR3 protein.
We then interrogated these data to determine the frequency and distribution of expression of Treg effector molecules 6 (Fig. 4f). We estimated their frequency of expression after dropout correction as in Fig. 4e, which were consistent with frequencies observed by intracellular flow cytometry analysis of splenic Tregs (not shown). Effector molecule expression was not mutually exclusive and we identified coexpressed effector molecules (Fig. 4f). As expected from the above, Il2ra and Ctla4 were by far the most frequent inhibitory molecules. In contrast, there was partial and reciprocally biased expression of Lrrc32 (encodes GARP, which presents processed TGF-β) and Tgfb1 on the one hand vs Gzmb and Ebi3. Finally, Il10 and Areg were preferentially represented in activated clusters but quite rare, consistent with their being deployed preferentially in tissue Tregs or at inflammatory sites. Thus, splenic Tregs adopt a continuum of states gravitating around one resting and two activated states with distinct phenotypic and effector functions.
Role of TCR signaling in influencing Treg states
The variable induction of the Nr4a1 gene module among activated Tregs suggested differences in how Tregs integrate TCR-derived signals. Signals from the TCR impact at multiple levels of Treg differentiation, activation and suppressive function, so we sought to further explore the role of TCR signaling intensity in shaping Treg states. NR4A1 is a transcription factor whose expression is induced rapidly upon T cell activation, proportionally to TCR signaling intensity 49,63. We took advantage of the Nr4a1-gfp reporter mouse, in which the level of GFP displayed by each cell reflects the intensity of TCR-delivered signal 64, and performed scRNAseq on Tregs sorted from 3 different zones of GFP expression (Fig. 5a). As expected, the frequency and levels of Nr4a1 conformed to the origin of the Tregs (Fig. 5a). Tregs from different Nr4a1 expression bins formed a trajectory from low to high expression (Fig. 5b), reproducing the main axis of Treg heterogeneity observed in Fig. 4.
Figure 5. Treg states associated with different TCR signaling strength.
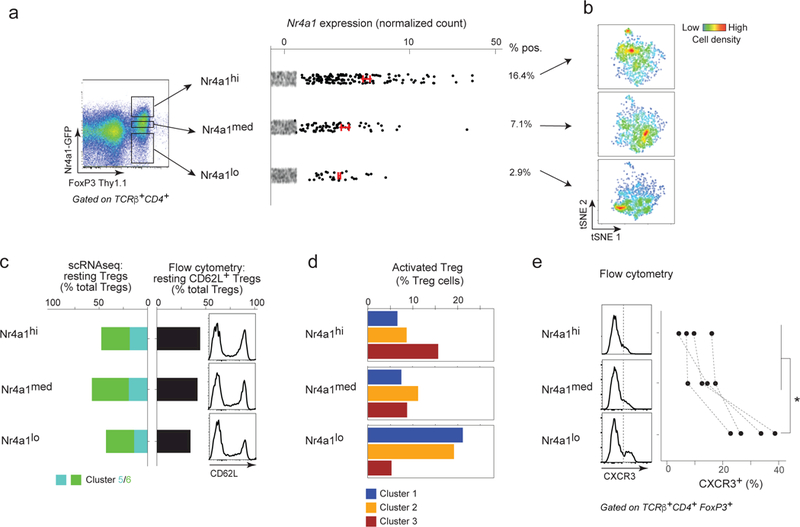
a. Foxp3-Thy1.1 versus Nr4a1-GFP expression in gated CD4+TCRβ+ T cells by flow cytometry showing the gating strategy used to sort Nr4a1-GFP-high, -medium and -low Treg cells before microfluidic encapsulation for scRNAseq library construction. Right: Nr4a1 expression in the corresponding Nr4a1-high, -medium and –low scRNAseq datasets (n = 1266, 1265 and 1567 respectively) (red bar: average and 95% CI in expressing cells).
b. Two-dimensional tSNE plot of Nr4a1-high, -medium and –low Tregs. The heat colors represent densities of cells.
c. Left: bar plot representing the proportion of Tregs belonging to the resting clusters 5 and 6 (defined in Fig. 4) in Nr4a1-high, -medium and –low Tregs. Left: in the scRNAseq datasets (cluster 5 and 6, in green and turquoise respectively). Right: expression of CD62L (flow cytometry, representative of n = 4 experiments) among the three Nr4a1 cell sets.
d. Bar plot representing the proportion of Treg cell belonging activated Treg clusters (defined in Fig. 4a)) in Nr4a1-high, -medium and –low Tregs.
e. CXCR3 expression in Nr4a1-high, -medium and –low Tregs by flow cytometry. Left: Representative CXCR3 histograms; Right: Proportion of CXCR3+ cells in Nr4a1-high, -medium and –low Tregs (as gated in the histograms, dashed line in the histograms). Gated on CD4+TCRβ+FoxP3+ cells (n = 4).
The data (1,265 to 1,567 cells per bin) were processed and analyzed in the probabilistic framework described for Fig. 4, assigning each cell according to its probability of belonging to clusters 1–6 (Supplementary Fig. 6). Two main observations stemmed from this analysis. First, and perhaps surprisingly since one might have expected that high TCR signals would promote an activated state, the proportion of cells falling in the resting pool (i.e. mapping to clusters 5 and 6) was essentially identical for all three bins of GFP expression (Fig. 5c). These results were confirmed by flow cytometry on other mice from this line, which showed an equivalent distribution of CD62L across windows of GFP expression (Fig. 5c). These data are consistent with prior observations that also found no relationship between Nr4a1-GFP activity and eTreg status 14.
On the other hand, and mirroring the biased representation of Nr4a1 transcripts among activated Treg clusters (Fig. 4), the distribution of activated Tregs varied markedly with Nr4a1-GFP intensity. The proportion of cells in cluster 3 increased with GFP, while the proportion of cells in clusters 1 and 2 cells decreased (Figs. 5d, Supplementary Fig. 6). These results were consistent with the distribution of Nr4a1 transcripts, highest in Tregs of cluster 3 in normal mice. In addition, and also consistent with the negatively correlated expression of Nr4a1 and Cxcr3 (Fig. 4), we found an under-representation of Cxcr3-containing Tregs in the GFP-medium and -high pools (Fig. 5e).
Thus, in physiological settings and in the absence of inflammatory challenge, the level of TCR signals does not influence the proportion of activated Tregs, but markedly skews their phenotypic choices.
Role of TCR specificity in influencing Treg phenotypes
We then used another approach to investigate the relationship between TCR signals and Treg subphenotypes. If they were connected, one would predict that Treg cells expressing an identical TCR clonotype (same V composition and rearranged CDR3 sequence) would share a transcriptional subphenotype. We adapted the InDrop protocol to obtain both the transcriptome and the TCRαβ variable region sequences from the same single cells (detailed in Methods). We sequenced the TCRs of Foxp3-gfp+ Treg cells from the spleen and colon, the latter because microbiota-driven differentiation and/or expansion of particular clones leads to a higher frequency of repeated clonotypes 23,24. Of the 32 and 138 Tregs from spleen and colon for which both TCRα and β.chains could be determined unambiguously, none was repeated in splenic Tregs, as expected since the repertoire of splenic Treg is very diverse. But 7 clonotypes (defined by identical TCRα and TCRβ nucleotide sequences) were found in two or more of the colonic Treg cells. With two exceptions, these clonotype pairs belonged to cells that mapped close to each other on the tSNE plot (Fig. 6a). In order to evaluate the significance of this apparent proximity, we calculated pairwise cell distances (as Pearson correlation) between the transcriptome of these shared-clonotype cells or between randomly sampled pairs of Tregs. Indeed, transcriptomes of Tregs that shared their TCRs were more closely correlated than by chance (p < 10−4), confirming the significance of the observation (Fig. 6b). Thus, Treg cells that display the same TCR clonotype tend to share transcriptional identity, in keeping with the conclusion that signals from the TCR can mold the subphenotype adopted by activated Tregs.
Figure 6. Tregs that express the same TCRαβ sequence have related transcriptional programs.
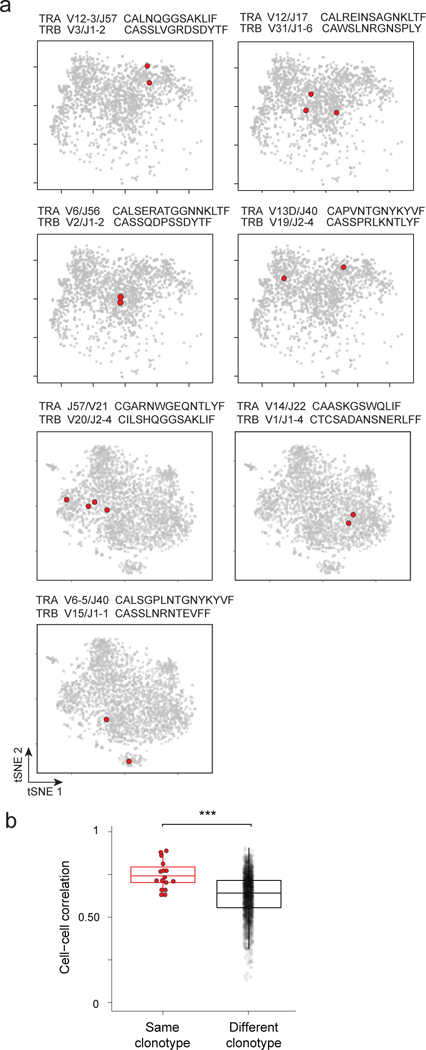
a. Two-dimensional tSNE plot of Tregs from the colon lamina propria, in 7 panels to highlight in red single Treg cells with the same rearranged TCRαβ sequences. V/J alleles and CDR3 sequences indicated above each panel. Data from 2 different experiments (top 4 and bottom 3 panels).
b. Cell-cell correlation between colonic Treg cells that express the same rearranged TCRαβ, or unrelated TCRs , *** p < 10−4 (t test p value).
Human and mice Tregs show similar patterns of heterogeneity
Lastly, we investigated whether the conclusions reached about mouse splenic Tregs would also apply in human cells. Blood Tregs and Tconvs were sorted (CD4+CD25highCD127lo and CD4+CD25−CD127hi respectively; Fig. 7a) from two healthy donors as previously described 65, and their transcriptomes were examined by scRNAseq (Supplementary Fig. 7a-d). Aspects of the heterogeneity and inter-relationships between Treg and Tconv cells, and of the genes underlying them, proved strikingly similar to the mouse in several important respects.
Figure 7. Similitudes in human and mouse Treg heterogeneity.
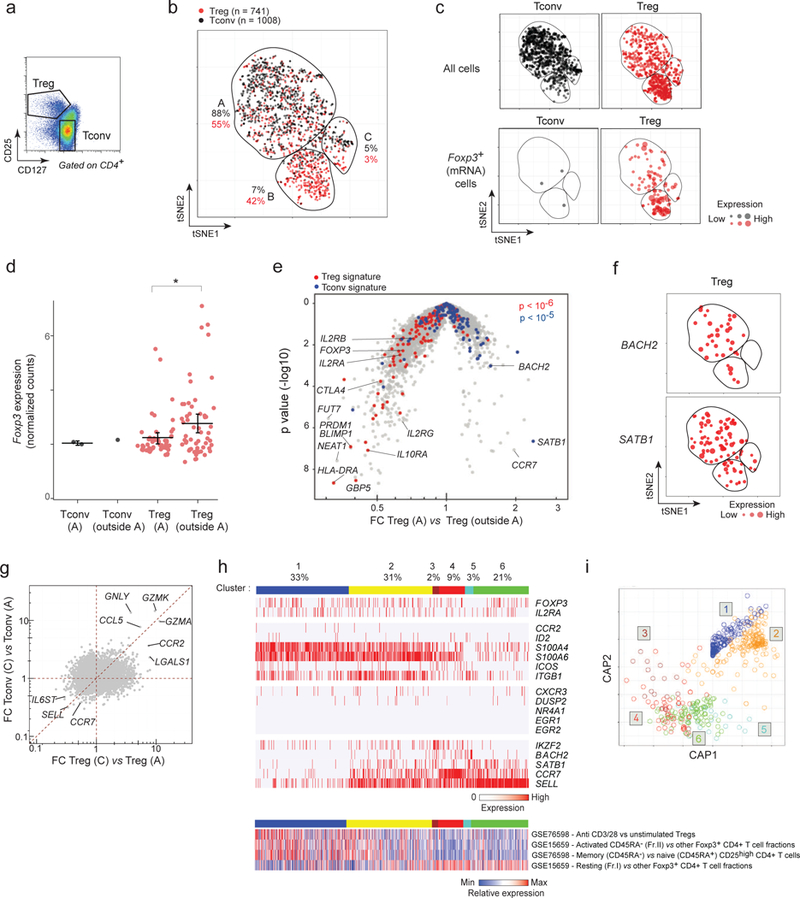
a. Flow cytometric plot of gated CD4+ Treg and Tconv cells in human blood defined by CD127 and CD25 expression, showing the sorting gates used before microfluidic encapsulation and scRNAseq library construction. Representative of cells isolated from 2 different donors.
b. Two-dimensional tSNE plot from transcriptomes of single human Treg (red dots, n = 741) and Tconv (black, n = 1008) single cells, with different areas (A-C) identified by unsupervised clustering. Each dot is an individual cell. The proportions of Tregs and Tconvs in each area relative to the total number of Tregs and Tconvs is indicated.
c. Same tSNE plots of human Treg and Tconv as in b, but split to show Treg and Tconv cells separately (top row) and cells in which FOXP3 transcript was detected (bottom row; the size of each dot indicates the level of FOXP3 RNA expression.
d. FOXP3 expression (normalized counts) in Tregs in area A (furtive Tregs) in comparison to Tregs outside area A (bar: mean and 95% CI. *: t.text p value < 0.05.
e. Volcano plot comparing the gene expression profiles of human furtive Tregs (in area A) vs all other Tregs (outside area A). Human up- and down-regulated Treg signature genes are highlighted (red and blue respectively). χ2 test p-values
f. tSNE plots as in b, highlighting BACH2- and SATB1-expressing Treg cells (red). Dot size: level of expression in each cell cell.
g. Fold change/fold change plot comparing the gene expression profiles of Tregs in C and A areas (x axis) with the gene expression profiles of Tconvs in C and A areas (y axis). Highlighted genes are genes specifically expressed by both Tregs and Tconvs in area C.
h. Biclustering expression heatmap of human blood Tregs, grouped into the equivalent of the 6 mouse clusters (defined in Fig. 4) using a likelihood model (see Methods), based on expression of human orthologs of the mouse genes (Top). Biclustering heatmap of published activated/resting signatures 12,66 (Bottom). Each vertical line represents one cell. The proportions of cells in each cluster is indicated.
i. Circular projection plot of the likelihood for each single human Treg cell to belong to any of the 6 clusters defined by homology to the mouse Treg clusters defined in Fig. 4.
First, the relative disposition of human CD4+ T cells from a tSNE representation (Fig. 7b,c) was similar to the mouse populations. Most Tregs and Tconvs clustered apart, but again furtive Tregs trespassed into the Tconv zone of the plot (Fig. 7b), actually in even higher proportions (55%) than for mouse splenocytes (26%). These furtive Tregs again expressed FOXP3 (Fig. 7c), albeit at lower levels than did other Tregs (Fig. 7d), and Treg signature transcripts were shifted in furtive Tregs compared with other Tregs (Fig. 7e). Particularly striking was the expression of the two Treg regulators, BACH2 and SATB1, mostly expressed in furtive Tregs (Fig. 7f). This over representation of BACH2 and SATB1 was also observed in mouse cells, albeit less strikingly. Another region of the tSNE spread exhibited tight co-mingling of Tregs and Tconvs (area C on Fig. 7b), reminiscent of area A-C in the mouse. Treg and Tconv cells in these areas still maintained their core transcriptional identity (FOXP3, IL2RA, HELIOS, CTLA4 for Treg, and THEMIS, ID2, IL7R for Tconv) but shared common transcriptional characteristics, uniquely expressing GZMA, GZMK, GNLY, CCR2 (Figs. 7g, Supplementary Fig. 7e).
Secondly, orthologs of the genes that clustered mouse Tregs (Fig. 4c) separated human Tregs, and we could project human Tregs to the mouse Treg clusters (Fig. 7h,i). The proportions of human blood Tregs in each cluster differed from the mouse splenic Tregs, with a higher presentation of the activated clusters. CCR7, SELL, SATB1 and BACH2 again characterized cells belonging to the resting Treg clusters (4, 5, and 6), while IKZF2 was high in cluster 3, and S100A4, S100A6 and ITGB1 were frequent in activated clusters 1 and 2. Integrating these data with the existing observations on human Treg heterogeneity, we analyzed the expression of published signature sets 12,66 in our single cells (similar to Fig. 4b). The sets distinguishing activated/memory and resting/naive Tregs were differently expressed in clusters 1–2 and 4–6, highlighting the conservation of activation signals between human and mouse Tregs. In a striking departure from mouse splenic Tregs, however, very few human blood Tregs expressed the set of transcripts dependent on TCR signaling (NR4A1, EGR1, or EGR2). This difference is not due to the tissue of origin (mouse blood Tregs have the same Nr4a1-GFP profile as splenic Tregs - Supplementary Fig. 7f), but could be due to different environmental influences on Treg activation in humans and mice. With this exception, however, Treg heterogeneity appears to follow very similar lines in mouse and human immune systems.
DISCUSSION
These results bring an unprecedented perspective on Treg diversity, and on what exactly defines a Treg cell, with two overarching considerations. First, Tregs are generally distinguishable from Tconvs within the CD4+ T cell space, but the two populations are multiply imbricated, with an important and previously unrecognized degree of overlap, whether among naïve cells or their more differentiated progeny. Second, signals from the TCR play unanticipated roles in shaping the different facets that activated Tregs can adopt. This study provides a unifying framework for diverse observations made on Tregs in the context of immune homeostasis.
Although scRNAseq lacks the sensitivity to measure the whole transcriptome of a single cell, we leveraged the power of performing scRNAseq on thousands of single cells using droplet microfluidics and applied statistical methods to take into account the technical dropout probability 53,61,62. Thus, conclusions are not drawn from a single cell but from several single cells with closely related programs, residing in the same area of the multidimensional space. With small aggregates of single cells, both technical noise and gene expression stochasticity are mitigated. This approach identifies different cell clusters and estimates the proportion of Tregs employing different suppression strategies.
Treg and Tconv cells have diametrically opposite functions, and this would suggest sharp boundaries to their transcriptional programs, a notion consistent with the hundreds of transcripts previously found to be differently expressed by population transcriptomics 17,52. However, revisiting this notion at the single-cell level showed that many of these transcripts were not ubiquitously expressed in Tregs. In fact, only a small core set of transcripts was uniformly expressed throughout all Tregs, and consistently missing or under-represented in Tconvs, and as such may truly define Treg identity. Some of these transcripts are involved in essential Treg pathways like Il2ra, which anchors the IL-2 paracrine loop (made by Tconvs and used in Tregs 67); others encode determining transcription factors (Foxp3 and Ikzf2), or several members of the TNF receptor family (Tnfrsf4 Tnfrsf18) 68. But these core Treg transcripts also include some whose function remains conjectural (Gpr83) 69, or have yet to be explored (Izumo1r, Capg or Wdr92). Conversely, Tregs consistently lacked the expression of Igfbp4 and Dapl1.
Beyond the core transcripts, Tregs and Tconvs appeared much more imbricated than the comfortable dichotomy of GFP profiles in Foxp3-gfp mice would suggest. The overall transcriptomes of furtive Tregs were very similar to those of the main Tconv population, while still maintaining their core identity (Foxp3 expression as RNA and protein). They formed a significant portion of Tregs (>20%) in both mouse spleen and human blood. They seemed to be “weaker” Tregs as they expressed less Foxp3 and Treg signature genes, and strongly expressed Satb1 and Bach2, two repressors of effector functions 59,70. They likely correspond to the poorly suppressive resting Tregs previously described 12. Because of their proportions and CD44loCD62Lhi naïve phenotype, it is unlikely that furtive Tregs represent activated Tconvs that transiently express FoxP3 upon activation 18,19, or, at the opposite end of the spectrum, Tregs in the process of losing FoxP3 and become activated effector T cells 71. One limitation to these speculations is that there is no rigorous way to infer precursor-product relationships from these data.
Also blurring the demarcation was the observation that some Treg and Tconv cells converged to similar transcriptome locations. These were activated cells (by CD44/CD62L criteria) and they expressed specific genesets overriding the core Treg/Tconv transcriptional difference. This confluence is reminiscent of the earlier reports that TFs characteristic of terminally differentiated Tconv states have also a critical role in Treg suppression (Tbx21, Rorc, Bcl6) 7,10,72–75. In one of those convergences, Tregs and Tconvs expressed transcripts known to characterize follicular T cells (in Tfh or Tfr) 11. It is possible that these Tregs and Tconvs converge transcriptionally as a consequence of residing in the same anatomical areas of the spleen, where they receive the same environmental cues 11,69.
These results are therefore consistent with a model defining Treg identity as modular 17. All Tregs express a very small core of Treg-specific transcripts, onto which additional programs are added, as a function of differentiation and/or location. More generally, our unbiased analysis of Treg heterogeneity revealed that Tregs form a continuum gravitating around three major poles. One axis of variation encompasses the previously known resting/activated difference, reflected by CD62L or CCR7 12–15. Two transcription factors, Bach2 and Satb1, which modulate Treg differentiation 47,51,70, were comparatively over-expressed in resting Tregs, and may help maintain this state; interestingly, they seemed to be reciprocally expressed in resting clusters 5 and 6, potentially suggesting redundant functions. However, most of the phenotypic divergence was revealed within the activated Treg compartment, with two orthogonal states of activation associated with differences in TCR signaling strength and suppressor molecule expression.
This single-cell data provided the opportunity to correlate TCR sequence and signaling intensity with the transcriptional output of each Treg. We expected that the activated Treg phenotype would be driven by the intensity of TCR signaling. Surprisingly, TCR signal intensity, as indicated by expression of Nr4a1, and confirmed across windows of activity of the Nr4a1-gfp reporter, did not correlate with the naïve/activated Treg ratio (reflected by the frequency of cluster 5 and 6 Tregs, or of Satb1 and Bach2 expression). This observation is consistent with the even distribution of CCR7+ Tregs across Nr4a1-gfp that was previously reported 14, but appears at odds with studies showing that activated Tregs electively disappear upon TCR ablation 27,28, a discrepancy which may merely reflect the difference between complete ablation and quantitative variation in transmitted signals. The puzzling disconnect between TCR-derived signals and activated status may also reflect different time-scales: Nr4a1 is an early-response gene, and the reporter likely responds to short-term stimuli, while the aTreg phenotype may integrate cues over a longer timeframe.
On the other hand, TCR signaling intensity seemed to have a strong influence shaping the different forms of activated Treg differentiation, and hence Treg effector functions. Tregs in cluster 3, with the signs of strongest TCR signals, preferentially expressed Cd24a, Tgfb1. Perhaps paradoxically, the cluster with the most strongly activated phenotype (cluster 1), which most actively expressed migratory molecules (Itgb1, Itae, Ccr2), had the lowest expression of the TCR-response (Nr4a1, Egr1, Egr2) cluster. It is possible that those Tregs are circulating to (or from) tissues where they encounter cognate antigen. Alternatively, it is known that TCR engagement triggers several signaling cascades 76 and that Nr4a1 induction is not equally dependent on all (e.g. independent of NF-AT) 77. Thus, low Nr4a1, Egr1, Egr2 induction may reflect a particular balance of TCR signaling paths, in cluster 1 and 2 Tregs. In addition, we cannot rule out that the different Treg clusters embody different time periods of activated Treg differentiation, and that TCR signals are dampened by negative feedback in Tregs of clusters 1 and 2.
In keeping with the notion that the TCR is a critical component shaping Treg fate, Tregs that share the same TCR were more transcriptionally similar than the norm. These similarities did not apply to all clonotypes, in keeping with the observation that Tregs in TCR transgenic mice can occupy different organismal and phenotypic niches (24,78 and our unpublished data). The similar quality and intensity of TCR engagement by MHC-peptide ligands may induce similar fine programs in Tregs that share the same clonotype. Secondly, the shared antigenic specificity may drive these Tregs to the same anatomical location, where they integrate the same environmental cues (MHC-peptide, cytokines, metabolic constraints, etc). Third, this phenomenon could reflect influences received early during Treg differentiation or peripheral activation, which imprint specific programs that persist after emigration to peripheral organs.
In comparing human and mouse Tregs at the single-cell level, we observed many similarities. In both species, Treg and Tconv cells were distinguishable by the ubiquitous expression of a small core set of transcripts (FOXP3, IKZF2, TNFRSF1B, IL2RA, IL2RB). Furtive Tregs were found in both species, more frequently in humans, likely explaining why CD45RA− FOXP3lo Tregs in humans have very low suppressive activity 12. As in mice, some activated human Tregs and Tconvs co-expressed similar transcriptional programs that overrode the canonical Treg:Tconv difference. Activated Tregs adopted diverse phenotypes in both species, albeit with the striking difference that the population with evidence of high TCR signals was essentially missing in human blood. This might not be a difference between species but between locations, as Tregs circulating in the blood may be devoid of MHC/TCR contacts, long enough that the early-response program tunes down.
In conclusion, this study provides a general model for lymphoid Treg identity and heterogeneity that is largely conserved between humans and mice. It also sheds light on a new and unexpected mechanism for the TCR in setting Treg fates. ScRNAseq analysis of complex T cell population provides unique opportunities to identify TCR sequences and other mechanisms modulating T cell fate in health and disease, and manipulating these states ultimately opens avenues to restore homeostasis.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Online Methods
Mice
C57BL/6J mice were obtained from The Jackson Laboratory. Foxp3-IRES-GFP/B6 1 mice were maintained in our colony. All experimentation was performed following animal protocol guidelines of Harvard Medical School (reviewed and approved HMS IACUC protocol 02954). Foxp3-IRES-Thy1.1 2 mice were crossed with the Nur77-GFP reporter mice 3.
Human blood
Blood was from 2 healthy 32-year-old donors, one male and one female (protocol IRB15–0504).
Flow cytometry
Spleen and blood. Blood was collected by cardiac puncture from euthanized mice and coagulation was prevented by adding heparin (20–50 U/mL blood) (Heparin Sodium Salt, Sigma-Aldrich H3393). A single cell suspension was obtained from murine splenocytes after physical dissociation with a 40 μm mesh (Facon). Red blood cell lysis was performed using 500 uL of ACK (Lonza) for 2 min on ice. Antibody staining was done in ice-cold buffer (DMEM without phenol red-2% FBS) for 5 min at a dilution of 1/100 using with antibodies against CD4 (GK1.5; BioLegend), CD25 (PC61; BioLegend), TCRβ (H57–597; BioLegend), CXCR3 (clone SA011F11, Biolegend), CD62L (MEL-14; BioLegend). Tregs were isolated as CD4+ TCRβ+ Foxp3-GFP+, CD4+ TCRβ+ Foxp3-Thy1.1+ (for the Nur77 experiments). Tconvs were isolated as CD4+ TCRβ+ Foxp3-GFP-.
The protocol of Sefik et al. 4 was used to isolate CD4+TCRβ+FoxP3GFP+ Tregs from the colonic lamina propria. Colonic tissue was dissociated using the following steps. Epithelial cells were removed by treating the tissue with RPMI containing 1 mM DTT, 20 mM EDTA and 2% FBS at 37°C for 15 min, then mincing and dissociating in a collagenase solution (1.5mg/ml collagenase II (Gibco), 0.5mg/ml dispase and 1%FBS in RPMI) with constantly stirring at 37°C for 45 min. Single cell suspensions were then filtered and washed with 4% RPMI solution. CD45+ cells were enriched before sorting using microbeads (Miltenyi Biotec).
Human Tregs and Tconvs were isolated from peripheral blood mononuclear cells (PBMC) as described by Ferraro et al. 5. An equal volume of room-temperature PBS/2mM EDTA was mixed to 15 mL of blood and carefully layered over 14 mL Ficoll-hypaque solution (GE Healthcare). After centrifugation for 30min at 900g (with no break) and room temperature, the mononuclear cell layer was washed 3 times with excess HBSS (Gibco) (10min at 400g) and resuspended in 2mL of HBSS. An FBS gradient was then performed to remove platelets and debris by layering the 2 mL cell suspension over 8mL FBS and centrifuging for 10min at 300g at room temperature. The pellet was resuspended in FACS buffer (phenol red-free DMEM, 2% FBS, 0.1% azide, 10mM Hepes PH 7.9) for antibody-staining in ice for 10min with: anti-CD4 (clone RPA-T4; Biolegend), anti-CD25 (clone BC96; Biolegend), anti-CD127 (clone A019D5; Biolegend), FcBlock (home-made). Treg and Tconvs were sorted as DAPI- CD4+ CD25+ CD127low and DAPI-CD4+ CD25- CD127high, respectively.
Single-cell RNAseq (InDrop)
Library construction
scRNAseq was done following the InDrop protocol described in length by Zilionis et al. 6. This technique sequences polyA mRNAs from thousands of single cells in a timely (< 30 min) and highly efficient manner (>70–80% of the input cells can be sequenced). Unique Molecular Identifiers are used in order to minimize the noise introduced during amplification. Only 3’ ends are sequenced, keeping strand information.
40–80,000 Tregs or Tconvs were first sorted by flow cytometry in complete medium (RPMI 10% FBS). Just before encapsulation, cells were spun for 5 min at 4°C 500g and resuspended in PBS-15% OptiPrep Density Gradient Medium (OptiPrep; Sigma-Aldrich) at a concentration of 80,000 cells/mL. This density gradient prevents sedimentation during encapsulation and allows a uniform flow of cell to enter the microfluidic device. Encapsulation is done by flowing 4 different inputs in the device: cells, reverse transcriptase (RT) buffer, primer hydrogels, and oil (water in oil droplets). Primer hydrogels contain reverse transcriptase (RT) primers with a unique single cell barcode. From 5’ to 3’, the primer contains a T7 promoter for amplification (in vitro transcription), the PE1 sequencing primer, a unique single cell barcode (16–19bp) (147,456 possible barcodes), a UMI (6bp) and a polyT sequence.
Around 5,000 single cells were then encapsulated in droplets of 3–4 nL containing a primer hydrogel bead and the reverse transcriptase (RT) buffer (SuperScriptIII, Invitrogen). Encapsulation was performed under 30 min in order to maintain cell viability and the emulsion was collected in a 500uL tube.
RT is performed immediately after encapsulation. Firstly, the primers were released from the gels by exposing the emulsion to UV for 7 min. RT was done at 50°C for 2h followed by enzyme inactivation for 15 min at 70°C. Emulsions were then split in small aliquots to have no more than 3’000 cells per tube, broken down by adding adding 20% 1H,1H,2H,2H-perfluorooctanol (PFO) in HFE-7500 oil (3M Novec 7500 Engineered Fluid, Novec) and kept at −80°C.
Samples were thawed on ice and spun at 4°C for 5 minutes at 19,000g to pellet cell debris. Excess primers and hydrogels beads were removed by filtration through a nucleic acid purification column and enzyme digestion (ExoI, HinfI). After purification of the DNA/RNA duplex with 1.2X AMPure beads (Beckman), second-strand cDNA synthesis was performed (NEB) for 2.5 h at 16°C. The library was then amplified using T7 in vitro transcription for 15h at 37°C (HiScribe T7 High Yield RNA Synthesis Kit, NEB). After purification, half of the amplified RNA was used for further processing. First, the aRNA was fragmented for 3 min at 70°C (magnesium RNA fragmentation kit, Ambion) and purified using AMPure Beads (1.2X). Then reverse transcription with random hexamers was done for 1h at 42°C (PrimeScript Reverse Transcriptase, Takara Clontec). A final PCR was done to amplify the library and add the P5-P7 and Illumina index primers (Kapa 2× HiFi HotStart PCR mix, Kapa Biosystems). The number of cycles was chosen by first running a qPCR on 1/20th of the RT reaction. The optimum was between 11 and 14 cycles.
Library size was analyzed with a Tapestation (High Sensitivity D1000 ScreenTape, Agilent technologies), quantified by qPCR and sequenced using NextSeq 500 and custom primers (read 1: 40bp, index: 7bp, read 2: 51 bp).
Data processing and normalization (InDrop).
Indrop sequencing data were processed as described by Zilionis et al. 6. Read 1 contains transcript information. Read 2 contains the unique molecular identifier (UMI) (6bp), single-cell barcode (16–19bp) and a conserved sequenced named W1. Fastq reads were first filtered for quality (>80% with Sanger Q>20) and on the expected structure of the reads: paired reads were kept only if read 2 contained the W1 and polyT sequence and if read 1 did not contain them. Single cell demultiplexing was performed against the possible barcode space and only reads mapping unambiguously and with less than 2 mismatches were kept. For each single cell library, the reads were then mapped against the mouse mm10 transcriptome (complemented with the sequence of the Foxp3-cd90.1- or Foxp3-gfp transgenes) using tophat2 v2.0.10 (--library-type fr-firststrand) 7. Reads mapping to multiple regions or having a low alignment score (MAPQ <10) were filtered out (flag 256). Duplicated reads were filtered as follows using UMIs and custom scripts (see Zemmour_Code/CorrectAndCountUMIsEditDist): for each set of reads mapping to one gene, we kept all reads having UMIs with a pairwise string distance >=2 (Levenshtein distance for taking into account indels and substitution). A final gene-table with genes in rows and cells in columns was then saved. A CellDataSet object from the Monocle package was used to store the data (see Zemmour_Code/Zemmour_Code.Rmd: **Make a CellDataSet object (from Monocle) **).
A first quality control was performed after demultiplexing by calculating the proportion of total reads that mapped to the most abundant single cell barcodes (see Zemmour_Code/Zemmour_Code.Rmd: **QC Plots**). Good libraries had >70% of reads mapping to a small set of single cell barcodes (500–3000) as expected from successful encapsulation. A second quality control involved the distribution of mapping rates and sequencing coverage. Good libraries were normally distributed with an average 80% mapping rate (5% SD) and more than 90% UMI duplicated reads (indicating sequencing saturation).
Single cells with more than 500 genes were kept for the analysis. Total UMI counts normalization was performed and scaled to the median UMI count (see Zemmour_Code/Zemmour_Code.Rmd: **Normalization and filtering of genes and cells**).
Single-cell RNAseq (CEL-Seq)
scRNAseq using the CEL-Seq protocol was done following the protocol described in length in 8. Briefly, Splenic Treg (CD4+ TCRβ+ FoxP3-GFP+) and Tconv (CD4+ TCRβ+ FoxP3-GFP-) cells were index-sorted with a BD FACSAria II in 96-well hard-shell PCR plates filled with 4.4uL of lysis buffer (HSP963, BioRad) containing 0.125 µl of RNAseOut (40 U per 1 µl stock; 10777–019, Invitrogen), 0.25 µl of reverse-transcription primer (25 ng per 1 µl stock) and 4 µl of RNAse-free water (AM9932; Ambion). Index-sorting allowed to record for each sorted single cell the fluorescence intensity of each flow cytometry marker used. After cell sorting, the plates were quickly covered with an aluminum seal (AlumaSeal96; F96100; Excel Scientific), then were vortexed for 10 s, centrifuged for 1 min at maximum speed (>2250g at 4 °C), frozen on dry ice and kept at −80 °C for up to 3 weeks. RNA was then denatured for 3 min at 70°C and reverse transcribed using ArrayScript Reverse Transcriptase (AM2048 Ambion) and mRNA Second strand synthesis module (E6111L; NEBNext). Single-cell cDNA libraries containing different barcodes were pooled into one tube and in vitro transcription was done using MEGAshortscript T7 transcription kit (AM1354; Ambion) for 14h. RNA was then fragmented, ligated to the 3’ Illumina adapter and reverse transcribed. Libraries were amplified by PCR and sequenced on a HiSeq 2500.
Data processing is described in length in 8. It followed similar steps as InDrop, filtering reads for quality, mapping to the mm10 transcriptome and filtering duplicated reads out using Unique Molecular Identifiers.
Single-cell RNAseq (10X Genomics)
7000 splenic CD4+ cells from C57/Bl6 mice were sorted by flow cytometry into a well of a chilled 96-well plate filled with 14 ul PBS-0.06% BSA. Cells were then encapsulated in one lane of the 10X Chromium Instrument and libraries were constructed with the Single Cell 3’ Reagent Kit (V2 chemistry) (https://support.10xgenomics.com/single-cell-gene-expression/library-prep). Libraries were sequenced on NextSeq 500 platform (26/8/0/98, Read1/i7/i5/Read2). Data was analyzed using the Cell Ranger Pipeline (https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome).
Algorithm to find contaminant cells
Contamination cells/doublets were identified in the gene table using the following criteria. Firstly, we compared each single cell transcriptome to the Immgen dataset (www.immgen.org) and calculated the likelihood for each single cell to be any of the Immgen cell types (see Zemmour_Code/Zemmour_Code.Rmd: **Immgen cell type prediction **). The Immgen matrix of gene expression was used to provide prior probabilities (probability to express gene i in cell type j = pij ) and we calculated for each single cell C the likelihood to be of cell type j (Lcj) (multinomial model). Log posterior probabilities were derived by normalizing so that they sum to 1 for each single cell.
Contaminant cells were flagged when a T cell type was not amid the top 5 most likely cell types. Secondly, we performed a PCA on the dataset using the most variable genes. Contaminant cells were flagged as they clustered in the first PCs. Cells usually matching a combination of these two criteria were removed from the analysis. Thirdly, we analyzed the distribution of UMIs and genes in each single cell. Single cells with more than 6,000 UMIs were also filtered out (possible doublets) (see Zemmour_Code/Zemmour_Code.Rmd: **Removing Contaminant cells**).
Gene expression comparison between groups of single cells
Differential gene expression was performed using the DESeq statistical model9 (see Zemmour_Code/Zemmour_Code.Rmd: **Differential gene expression function**). The distribution of counts Cij for each gene i in group j is modeled by a negative binomial of parameter (mean) and dispersion θi
First, the dispersion was estimated using the background model according to 10 in which a linear model is fitted between the and for each gene i. For each gene i, the dispersion , was estimated, being the fitted variance of gene i. A generalized linear model was then fit for each gene i in each group j [glm(formula = gene ~ groups, data = tmp, family = negative.binomial())] and we used an analysis of variance (anova) to compare the coefficients in each model and get a p-value. FDR was calculated to correct for multiple testing.
To identify the core Treg transcripts independently of other variables like activation, we compared matched Tregs and Tconvs (see Zemmour_Code/Zemmour_Code.Rmd: **Core Treg genes**). For each single Treg cell (n = 708), we compared its 50 closest Tregs and 50 closest Tconvs (correlation distance) and deemed significant genes with a p value < 0.05. Core Treg genes scored as significantly different in > 80% of the comparisons.
tSNE representation and clustering
Data was visualized using the t-Distributed Stochastic Neighbor Embedding (t-SNE) dimensionality reduction algorithm 11 (see Zemmour_Code/Zemmour_Code.Rmd: **t-SNE**). A PCA was first performed on the top 100 most variable genes that were expressed in more than 1% of the cells. The Fano factor (variance/mean) was used as a metric for variability because of its independence to the mean in Poisson distributed data. The number of significant PCs were determined by comparison to PCA over a randomized matrix as described by Klein et al. 12. Two-dimensional tSNE was run using the Rtsne function 10, on the significant PCs, setting the seed for reproducibility and using the following parameters (perplexity = 50, max_iter = 1000).
Clustering was done independently of the tSNE projections based on k-means and the method described by Krijthe et al. 13, using the clusterboot function in R and kmeansCBI 14 on the significant PCs (see Zemmour_Code/Zemmour_Code.Rmd: **Clustering**). Cluster stability was assessed by bootstrapping and analyzed with the Jaccard index (average similarity between the closest clusters during bootstrapping) and silhouette plots (difference between the average distance for each single cell to cells in the same cluster versus cells in the closest cluster). The number of clusters (k) was optimized by running the algorithm with different k and choosing the largest k with the most stable clusters (all clusters with a Jaccard index > 0.6). Cluster coherence was also assessed by visualizing them on the tSNE plots.
Circular A Posteriori Projection plots
In order to compare the different single cell dataset replicates, we calculated the likelihood for each single cell to belong to the spleen Treg clusters defined in Fig. 4 (see Zemmour_Code/Zemmour_Code.Rmd: **Circular A Posteriori Projection plots**). The average expression within each cluster of Fig. 4 was used as prior probabilities and we used a multinomial mixture model as described by Jaitin et al. 15 (and above for the contamination algorithm) to calculate the log posterior probabilities for each single cell to belong to any of the spleen clusters. For the human data, we used the ortholog genes (www.informatics.jax.org/downloads/reports/index.html#homology)
These probabilities were visualized as described by Jaitin et al. 15 using Circular A Posteriori (CAP) plots. The probabilities were projected on a 2-dimensional sphere using radial positions reflecting the relationship between clusters (a = (pi/2, pi/4, 3*pi/4, −3*pi/4, -pi/4, -pi/2) for clusters 1 to 6). For each single cell c and cluster k, the x and y coordinates were then calculated as
Treg signatures and single cell score
Genes signatures were curated from published datasets (references in Supplementary Fig. 4) and computed by comparison between two conditions (e.g. WT vs KO). Data were downloaded from GEO. Only datasets containing replicates were used. To reduce noise, genes with a coefficient of variation between biological replicates > 0.6 in either comparison groups were selected. Up- and down-regulated transcripts were defined as having a fold change in gene expression > 1.5 or < 2/3 and a t.test p-value < 0.05 . Others signatures were obtained from Godec et al. 16.
A signature score for each single cell was computed by summing the counts for the up-regulated genes and subtracting the counts for the down-regulated genes. Z-score were plotted in the heatmap (Fig. 4b) (see Zemmour_Code/Zemmour_Code.Rmd: **Treg signatures and single cell score**).
Correcting technical dropouts in scRNAseq to estimate frequency of expression
Frequency of expression is underestimated in single cell RNAseq studies because of the low sensitivity of the technique. However, several statistical methods have been developed to estimate technical dropouts 8,17,18 (see Zemmour_Code/Zemmour_Code.Rmd: **Correcting technical dropouts in scRNAseq to estimate frequency of expression**). Genes were first distributed in 25 bins of expression according to the population average. Then, for each single cell c, we calculated the fraction of genes that were expressed in each bin (count >=1) and fit a logistic regression to predict the dropout probability for each gene in the single cell. Goodness of fit was assessed by calculated the area under the ROC curve (AUC). 99% of the cells had an AUC between 0.88 and 0.9. Dropout-corrected gene expression for each single cell c and gene i (CorrectedCounti,c) was calculated using these dropout probabilities (dropouti,c) as weight and the average expression of the 10 closest genes (by correlation distance) () as expected counts.
Frequency of expression was calculated using the dropout-corrected gene expression (CorrectedCount > 0).
Paired single cell TCRαβ sequencing
Single-cell TCRαβ sequencing was done from 1 batch of splenic Tregs and 2 batches of colonic Tregs isolated from SPF mice. Single-cell TCRαβ sequencing was done on the same material as for the whole-transcriptome library construction, taking a small amount of material after the RNA amplification and before the fragmentation step. The protocol starts by taking an aliquot of the aRNA library before fragmentation as it retained full transcript sequence and performing a reverse transcription using TRAV and TRBV external primers (Supplementary Table 5). The sequences of the external and internal primers were obtained from ref 19 for TRBV and ref 20 for TRAV. The Illumina PE1 sequencing primer was added to the 5’ end of the internal primers (Supplementary Table 5). TCRα and TCRβ RT reactions were performed separately following the protocol for SuperScriptIII (Invitrogen): 1uL of the aRNA was mixed with 1uL of RT primers (external TRAV or TRBV primers) (10uM), 1uL of dNTP and 10uL of RNAse-free water and incubated at 65°C for 5 min before adding 4uL of SuperScriptIII (Invitrogen) buffer 5X, 1uL of DTT, 1 uL of RNAseOut (Invitrogen) and incubating for 50 min at 50°C, then for 70°C for 15 min. cDNA library was purified and size selected using 0.5X AMPure SPRI bead. The first PCR uses internal primers and adds PE1 sequencing primers. A qPCR was performed before to optimize the number of cycles needed for PCR1 using 1/10 of the previous purified cDNA (1uL), 5uL water, 10uL KAPA ReadyMix 2X (Kapa Biosystems), 2uL SYBRgreen (Invitrogen), 1uL of primers (RTP_TRAV or RTP_TRBV) (10µM), 1uL of P7_Rd2_idxN (10uM). Cycle parameters were: 95°C 3min; 40 cycles of: 98 °C 20s -> 60°C 30s -> 72°C 1min --> fluorescence read. The number of optimized cycles corresponded to the middle of the exponential phase (to which log2(10) = 3.3 cycles are subtracted, since the PCR uses 9 times more cDNA than the qPCR). PCR1 was then performed as follows: cDNA (10uL), 11uL water, 25uL KAPA ReadyMix 2X (Kapa Biosystems), 2uL of primers (RTP_TRAV or RTP_TRBV) (10µM), 2uL of P7_Rd2_primer_idxN_R (10uM). Cycle parameters were: 95°C 3min; X cycles of: 98 °C 20s -> 60°C 30s -> 72°C 1min. The PCR product was then purified and size selected using 0.5X AMPure SPRI bead. A second PCR incorporates the P5 Illumina sequence and done as follows: purified PCR1 product (5uL), 16uL water, 25uL KAPA ReadyMix 2X (Kapa Biosystems), 2uL of primers P5_Rd1 (10uM), 2uL of P7_Rd2_idxN (10uM). Cycle parameters were: 95°C 3min; X cycles of: 98 °C 20s -> 60°C 30s -> 72°C 1min. The number of cycles for PCR2 were optimized using a qPCR as described above (<10 cycles). PCR3 product was then purified and size selected twice using 0.5X AMPure SPRI beads. Library size was assessed using a High sensitivity D5000 ScreenTape (Agilent). TCRα and TCRβ libraries have an expected size of 1470 and 1230bp, respectively. Library concentration was determined by qPCR. Sequencing was done using paired-end NanoMiseq Read1: 265 bp, index:7 bp, Read2: 51 bp.
Read 1 contains the TCR transcript sequence. Read 2 contains the unique molecular identifier (UMI) (6bp), single-cell barcode (16–19bp) and a conserved sequenced named W1(similarly to the whole transcriptome analysis). As for the transcriptome analysis, Fastq reads were first filtered for quality (>80% with Sanger Q>20) and on the expected structure of the reads: paired reads were kept only if read 2 contained the W1 and polyT sequence and if read 1 did not contain them. Single cell demultiplexing was performed against the possible barcode space and only reads mapping unambiguously and with less than 2 mismatches were kept. TCRα and TCRβ alignment (V,D,J alignment and CDR3 identification) was done individually for each single cell against the mouse IMGT database (www.imgt.org) using the Mixcr 1.8.1 program 21: mixcr.jar align -f --library local -nw -OminSumScore=0 --loci $loci -OjParameters.parameters.floatingRightBound=False -OdParameters.absoluteMinScore=0 --species mmu --report $line.$loci.aln.log $line.fq $line.$loci.aln.vdjca; mixcr.jar exportAlignmentsPretty --skip 0 --limit 10 $line.$loci.aln.vdjca $line.$loci.aln.pretty.txt; mixcr.jar exportAlignments -f -p full --no-spaces $line.$loci.aln.vdjca $line.$loci.aln.txt; mixcr.jar assemble -f $line.$loci.aln.vdjca --report $line.$loci.asmbl.log $line.$loci.asmbl.clns; mixcr.jar exportClones -f -p full --no-spaces $line.$loci.asmbl.clns $line.$loci.asmbl.txt. Only sequences with a score > 100 were kept for further analysis. TCRα, TCRβ sequences and whole transcriptome data were matched to the same single cell barcode unambiguously with less than 2 mismatches. Only single cell barcodes with both TCRα and TCRβ sequences and for which the transcriptome was available were kept for further analysis. Single cells were grouped in clonotypes when sharing the same TCRα and TCRβ sequences (V,D,J and CDR3 sequence).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr E. Sefik for help with colon profiling, and K. Hattori, C. Araneo K. Waraska, M. Thorsen, and R. Steen for help with mice, profiling, sorting, and sequencing. This work was supported by NIH grants AI051530, AI116834 and AI125603 to CB/DM. DZ and EK were supported by a PhD fellowship from the Boehringer Ingelheim Fonds. All authors declare no competing interest.
Footnotes
AUTHOR CONTRIBUTIONS
DZ, EK and RZ performed the experiments; DZ, AMK, CB and DM designed the study, analyzed and interpreted the data; DZ, DM and CB wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Data availability
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo (accession nos. XXXXX).
REFERENCES
- 1.Josefowicz SZ, Lu LF, & Rudensky AY Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panduro M, Benoist C, & Mathis D Tissue Tregs. Annu. Rev Immunol 34, 609–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feuerer M, Hill JA, Mathis D, & Benoist C Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat. Immunol. 10, 689–695 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Campbell DJ & Koch MA Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 11, 119–130 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo CT & Li MO Transcriptional control of regulatory T cell development and function. Trends Immunol. 34, 531–539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vignali DA, Collison LW, & Workman CJ How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch MA et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall AO et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37, 511–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan TG, Mathis D, & Benoist C Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc Natl Acad Sci U S A( 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung Y et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17, 983–988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linterman MA et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17, 975–982 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyara M et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Kleinewietfeld M et al. CCR6 expression defines regulatory effector/memory-like cells within the CD25(+)CD4+ T-cell subset. Blood 105, 2877–2886 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Smigiel KS et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp. Med 211, 121–136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cretney E, Kallies A, & Nutt SL Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 34, 74–80 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Fontenot JD et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Hill JA et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Gavin MA et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A 103, 6659–6664 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyao T et al. Plasticity of foxp3(+) T cells reflects promiscuous foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36, 262–275 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Setoguchi R, Hori S, Takahashi T, & Sakaguchi S Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 201, 723–735 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubtsov YP et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zemmour D et al. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci U S A 114, E3472-E3480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cebula A et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497, 258–262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lathrop SK et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samstein RM et al. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell 150, 29–38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li MO & Rudensky AY T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16, 220–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine AG, Arvey A, Jin W, & Rudensky AY Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15, 1070–1078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vahl JC et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 41, 722–736 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Hsieh CS, Lee HM, & Lio CW Selection of regulatory T cells in the thymus. Nat Rev Immunol 12, 157–167 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Tang F et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Ramskold D et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimshony T, Wagner F, Sher N, & Yanai I CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666–673 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Islam S et al. Highly multiplexed and strand-specific single-cell RNA 5’ end sequencing. Nat. Protoc. 7, 813–828 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Prakadan SM, Shalek AK, & Weitz DA Scaling by shrinking: empowering single-cell ‘omics’ with microfluidic devices. Nat Rev Genet. 18, 345–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanay A & Regev A Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner A, Regev A, & Yosef N Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol. 34, 1145–1160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villani AC et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baron M et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 3, 346–360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zilionis R et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc. 12, 44–73 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Sawant DV & Vignali DA Once a Treg, always a Treg? Immunol Rev 259, 173–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Maaten L & Hinton G Visualizing high-dimensional data using t-SNE. Journal of Machine Learning Research 9, 2579–2605 (2008). [Google Scholar]
- 45.Heimberg G, Bhatnagar R, El-Samad H, & Thomson M Low Dimensionality in Gene Expression Data Enables the Accurate Extraction of Transcriptional Programs from Shallow Sequencing. Cell Syst. 2, 239–250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim HJ et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 350, 334–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu W et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol. 13, 972–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sage PT & Sharpe AH T follicular regulatory cells. Immunol Rev 271, 246–259 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Wakamatsu E, Mathis D, & Benoist C Convergent and divergent effects of costimulatory molecules in conventional and regulatory CD4+ T cells. Proc Natl Acad Sci U S A 110, 1023–1028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samstein RM et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitagawa Y et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol 18, 173–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gavin MA et al. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol 3, 33–41 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Meredith M, Zemmour D, Mathis D, & Benoist C Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol 16, 942–949 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martins AJ et al. Environment tunes propagation of cell-to-cell variation in the human macrophage gene network. Cell Syst. 4, 379–392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hennig C Cluster-wise assessment of cluster stability. Comput Stat Data Anal. 52, 258–271 (2007). [Google Scholar]
- 56.Cretney E et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 12, 304–311 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Feng Y et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature 528, 132–136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med 19, 1173–1177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roychoudhuri R et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature 498, 506–510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ouyang W et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491, 554–559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shalek AK et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kharchenko PV, Silberstein L, & Scadden DT Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu ZG et al. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature 367, 281–284 (1994). [DOI] [PubMed] [Google Scholar]
- 64.Moran AE et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp. Med. 208, 1279–1289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferraro A et al. Interindividual variation in human T regulatory cells. Proc Natl Acad Sci U S A 111, E1111-E1120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pesenacker AM et al. A Regulatory T-Cell Gene Signature Is a Specific and Sensitive Biomarker to Identify Children With New-Onset Type 1 Diabetes. Diabetes 65, 1031–1039 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Fontenot JD, Rasmussen JP, Gavin MA, & Rudensky AY A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol 6, 1142–1151 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Mahmud SA et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol 15, 473–481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Z et al. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beyer M et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol. 12, 898–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou X et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 10, 1000–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chaudhry A et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng Y et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sefik E et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohnmacht C et al. The microbiota regulates type 2 immunity through RORγ+ T cells. Science 349, 989–993 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Malissen B & Bongrand P Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu. Rev Immunol 33, 539–561 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Ashouri JF & Weiss A Endogenous Nur77 Is a Specific Indicator of Antigen Receptor Signaling in Human T and B Cells. J Immunol 198, 657–668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malchow S et al. Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 44, 1102–1113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES
- 1.Bettelli E et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature . 441, 235–238 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Liston A et al. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl. Acad Sci U S. A . 105, 11903–11908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moran AE et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp. Med . 208, 1279–1289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sefik E et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferraro A et al. Interindividual variation in human T regulatory cells. Proc Natl Acad Sci U S A 111, E1111-E1120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zilionis R et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc . 12, 44–73 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol . 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meredith M, Zemmour D, Mathis D, & Benoist C Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol 16, 942–949 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Love MI, Huber W, & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol . 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grun D et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015). [DOI] [PubMed] [Google Scholar]
- 11.van der Maaten L & Hinton G Visualizing high-dimensional data using t-SNE. Journal of Machine Learning Research 9, 2579–2605 (2008). [Google Scholar]
- 12.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krijthe,J.H. Rtsne: T-Distributed Stochastic Neighbor Embedding using a Barnes-Hut Implementation (https://github.com/jkrijthe/Rtsne, 2015).
- 14.Hennig,C. fpc: Flexible Procedures for Clustering. R package version 2.1–10 (https://CRAN.R-project.org/package=fpc, 2015).
- 15.Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godec J et al. Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity 44, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shalek AK et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kharchenko PV, Silberstein L, & Scadden DT Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker FJ, Lee M, Chien YH, & Davis MM Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci U S A 99, 9374–9379 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burzyn D et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolotin DA et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12, 380–381 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Heng TS, Painter MW, & Immunological Genome Project Consortium The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Hill JA et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity . 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Samstein RM et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.