

Abstract
Pulmonary alveolar proteinosis (PAP) is a rare syndrome of alveolar surfactant accumulation, resulting hypoxemic respiratory failure, and increased infection risk. Despite advances in our understanding of disease pathogenesis and the availability of improved diagnostics, the epidemiology and healthcare burden of PAP remain poorly defined. To determine the prevalence, and healthcare utilization and costs associated with PAP, we interrogated a large health insurance claims database containing comprehensive data for approximately 15 million patients in the United States. We also evaluated data from a referral-based diagnostic testing program collected over a 15-year period. The prevalence of PAP was determined to be 6.87 ± 0.33 per million in the general population, similar in males and females, and increased with age, however considering difficulties and delays in diagnosing this is likely a minimum estimate of true prevalence. PAP patients had significantly more comorbidities, health care utilization and associated costs compared to control patients precisely matched for age and gender. Between 2004 and 2018, 249 patients confirmed to have PAP were evaluated to identify the PAP-causing disease; 91.5% had autoimmune PAP, 3% had hereditary PAP caused by GM-CSF receptor mutations, 4% had secondary PAP, and 1.5% had congenital PAP. Considering the high diagnostic accuracy of serum GM-CSF autoantibody testing and predominance of autoimmune PAP, these results emphasize the importance of utilizing blood-based testing in PAP syndrome to identify the PAP-causing disease rather than invasive lung biopsies, resulting in earlier diagnosis, reduced morbidity and lower healthcare costs.
Keywords: Pulmonary alveolar proteinosis, Prevalence, Healthcare burden, GM-CSF autoantibody
Introduction
Pulmonary alveolar proteinosis is a rare syndrome characterized by progressive alveolar surfactant accumulation and hypoxemic respiratory failure that occurs in various diseases commonly categorized as primary, secondary or congenital PAP [1, 2]. Primary PAP accounts for the majority of cases and is caused by disruption of granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling, either by GM-CSF autoantibodies (i.e., autoimmune PAP) [3] or genetic mutations involving the GM-CSF receptor (i.e., hereditary PAP) [4]. Secondary PAP occurs in a heterogeneous group of conditions that reduce numbers or functions of alveolar macrophages and thereby surfactant clearance. Congenital PAP is caused by mutations in genes required for normal surfactant production [1, 2]. Despite pathogenic advances and improved diagnostics, the prevalence and healthcare burden of PAP remain poorly-defined: formal epidemiologic studies have not been previously reported. We evaluated the epidemiology and healthcare burden of PAP based on data from a large health insurance claims database and a referral-based PAP diagnostic testing program.
Methods
Study data were derived from the OptumInsight database [5], a repository of de-identified, Health Insurance Portability and Accountability Act-compliant data for 30 million unique healthcare-insured members (patients) at the time of the study from across the United States (US); tracked longitudinally to comprehensively capture data from inpatient, outpatient, emergency, and pharmacy-related health insurance claims (claims data). Patients were included in this study only if they had complete annual claims data in two or more consecutive years. Included patients were designated as having PAP in a study year if they had at least one claim containing an international classification of diseases (ICD)-9 code of 516.0 (the diagnostic code for PAP) in that year. Annual PAP prevalence was defined as the number of PAP patients identified divided by the total number of included patients in the same study year.
Relative healthcare utilization and costs were determined using a case-control approach. Cases comprised included patients with at least one ICD-9516.0-associated claim and controls were included patients without any ICD-9516.0 code-associated claims. Cases and controls were precisely matched one-to-one for age and gender. Only years with complete annual claims data were included in the analysis. Demographics, comorbidities, and annual per-patient healthcare utilization and costs were calculated for cases and controls and compared.
As part of a concurrent separate study, an independent cohort of patients were evaluated to determine the underlying cause of PAP syndrome. Patients were diagnosed with autoimmune PAP by the use of a serum GM-CSF autoantibody test with confirmation by the STAT5-phosphorylation index test to demonstrate inhibition of GM-CSF signaling [6, 7] while other PAP-causing diseases were diagnosed by DNA sequencing, receptor analysis, and other methods under an institutional review board-approved protocol at the Translational Pulmonary Science Center in Cincinnati. Numeric data are presented as mean ± SEM. Statistical analysis was performed using Prism 7.0 or SAS 12.3.
Results
The study population included 15,011,522 ± 175,519 patients (8,144,741 ± 84,094 females and 7,766,782 ± 91,560 males) annually between 1/1/2008 and 12/31/2012 comprising 5.16 ± 0.04% of the US population. PAP prevalence increased with age in bimodal distribution (Fig. 1a) but did not vary with gender (Fig. 1a, Table 1). The annual prevalence of PAP in the general population was 6.87 ± 0.33 per million (Table 1). Using this value and 308.7 million for the US population size [8], the number of PAP patients in the US was estimated to be 2120.
Fig. 1.
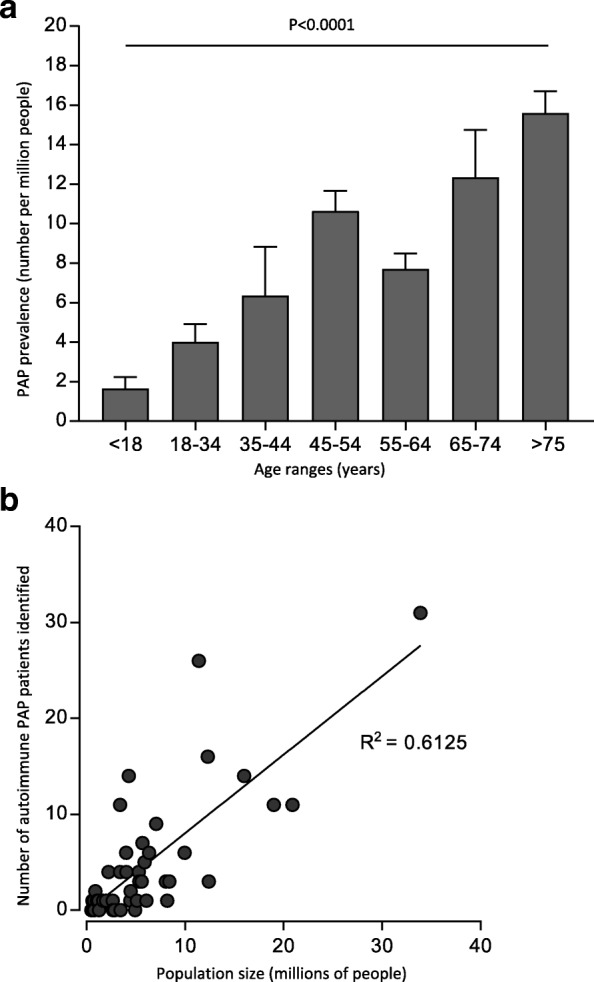
Prevalence of PAP syndrome and detection of autoimmune PAP in the United States (US). a Annual prevalence of PAP syndrome was determined retrospectively between January 1, 2008 and December 31, 2012 using data from the OptumInsight health insurance claims database. Bars represent the mean (±SD) prevalence stratified by age (left) or gender (right). Statistical comparisons were done with ANOVA or Student’s t-test, respectively. b Relationship between detection of autoimmune PAP and population size. Autoimmune PAP was identified among individuals with PAP syndrome across the US between 2004 and 2018 by serum GM-CSF autoantibody testing [6] with confirmation by blood-based STAT5 phosphorylation index testing to document impaired GM-CSF signaling [7] at the Translational Pulmonary Science Center in Cincinnati. Data are expressed as the number of individuals with autoimmune PAP by state plotted against the state population obtained from the 2010 US Census [8]. The correlation between GM-CSF autoantibody positive PAP patients and state population size was evaluated by Spearman correlation
Table 1.
Study Population and Annual Prevalence of PAP Syndrome in the USa
| Study year | Total patientsb | Total PAP patientsc | Total PAP prevalence per million patientsd |
| 2008 | 15,519,985 | 105 | 6.77 |
| 2009 | 15,686,604 | 127 | 8.10 |
| 2010 | 16,070,700 | 103 | 6.41 |
| 2011 | 15,764,422 | 109 | 6.91 |
| 2012 | 16,515,902 | 102 | 6.18 |
| Mean | 15,911,523 | 109 | 6.87 |
| Study year | Total female patientsb | Female PAP patientsc | PAP prevalence per million female patientse |
| 2008 | 7,953,672 | 51 | 6.41 |
| 2009 | 8,038,763 | 67 | 8.33 |
| 2010 | 8,233,635 | 57 | 6.92 |
| 2011 | 8,069,748 | 67 | 8.30 |
| 2012 | 8,427,886 | 59 | 7.00 |
| Mean | 8,144,741 | 60 | 7.39 |
| Study year | Total male patientsb | Male PAP patientsc | PAP prevalence per million male patientsf |
| 2008 | 7,566,313 | 54 | 7.14 |
| 2009 | 7,647,841 | 60 | 7.85 |
| 2010 | 7,837,065 | 46 | 5.87 |
| 2011 | 7,694,674 | 42 | 5.46 |
| 2012 | 8,088,016 | 43 | 5.32 |
| Mean | 7,766,782 | 49 | 6.31 |
Definition of abbreviations: PAP pulmonary alveolar proteinosis
aPatients are individuals represented by de-identified data in the OptumInsight database during the study period meeting the inclusion and exclusion criteria as defined in the methods
bIncludes all (or female/male patients as indicated) with complete annual claims data for at least two consecutive years
cIncludes all (or female/male patients as indicated) patients with at least one claim associated with an ICD-9516.0 code in the respective calendar year
dCalculated as the number of PAP patients in each year divided by the total number of patients included in that year
eCalculated as the number of female PAP patients in each year divided by the number of female patients included in that year
fCalculated as the number of male PAP patients in each year divided by the number of male patients included in that year
Comorbidities were higher in PAP than control patients as determined by the Charlson Comorbidity Index (CCI) [9] adapted for ICD-9-coded databases [10]. The CCI for PAP and control groups were 1.84 ± 2.48 and 0.55 ± 1.44, respectively (p < 0.0001) (Table 2). PAP patients had increased healthcare utilization and costs compared to controls attributable, respectively, to increased outpatient visits (17.30 ± 13.77 versus 10.40 ± 11.38; p < 0.01), emergency room visits (1.49 ± 1.17 versus 1.08 ± 0.27; p = 0.014), and longer hospital stays (15.96 ± 20.71 versus 5.40 ± 5.07 inpatient days; p = 0.027). The annual per-patient healthcare costs were 5-fold higher among PAP patients than controls ($54,865 ± 95,524 versus $10,214 ± 20,233, respectively; p < 0.001). This difference was due to increased costs of inpatient visits (2.7-fold, p = 0.04), outpatient visits (3.8-fold, p < 0.001), and prescriptions (4.75-fold, p < 0.001), but not differences in emergency room visit costs (p = 0.563).
Table 2.
Charlson Comorbidity Indexed Conditions in PAP Syndrome compared to control individuals
| Indexed Comorbidity | PAP | Control | p value |
|---|---|---|---|
| Myocardial Infarction | 9 (5.5%) | 4 (2.4%) | 0.157 |
| Congestive Heart Failure | 30 (18.3%) | 5 (3%) | < 0.001 |
| Peripheral Vascular Disease | 10 (6.1%) | 4 (2.4%) | 0.101 |
| Cerebrovascular Disease | 15 (9.1%) | 4 (2.4%) | 0.009 |
| Dementia | 0 (0%) | 1 (0.6%) | 0.317 |
| Chronic Pulmonary Disease | 71 (43.3%) | 16 (9.8%) | < 0.001 |
| Connective Tissue Disease | 14 (8.5%) | 1 (0.6%) | < 0.001 |
| Peptic Ulcer Disease | 3 (1.8%) | 2 (1.2%) | 0.652 |
| Mild Liver Disease | 15 (9.1%) | 2 (1.2%) | 0.001 |
| Diabetes without Complications | 36 (22%) | 20 (12.2%) | 0.019 |
| Diabetes with End-Organ Damage | 8 (4.9%) | 2 (1.2%) | 0.054 |
| Hemiplegia/Paraplegia | 1 (0.6%) | 0 (0%) | 0.317 |
| Renal Disease | 16 (9.8%) | 2 (1.2%) | < 0.001 |
| Non-metastatic Cancer Hematologic Malignancy | 15 (9.1%) | 6 (3.7%) | 0.042 |
| Moderate/Severe Liver Disease | 2 (1.2%) | 0 (0%) | 0.0156 |
| Metastatic Solid Tumour | 2 (1.2%) | 2 (1.2%) | > 0.9999 |
Between 2004 and 2018, 700 patients from 25 countries including 400 US patients participated in a laboratory-based research protocol to identify and study PAP-causing diseases. Among 249 US patients with confirmed PAP syndrome, 228 (91.5%) had a positive serum GM-CSF autoantibody test identifying autoimmune PAP, 7 (3%) had hereditary PAP caused by mutations in CSF2RA or CSF2RB (encoding GM-CSF receptor α or β chains), 10 (4%) had secondary PAP, and 4 (1.5%) had congenital PAP. The number of patients with a positive GM-CSF autoantibody test in the US varied by state (e.g., 1 in Vermont - pop. 608,827; 31 in California - pop. 33,871,648) and correlated with population density (Fig. 1b)(R2 = 0.6978, p < 0.0001).
Discussion
We determined the prevalence, healthcare utilization and costs of PAP in the US based on analysis of data from 5% of the US population, independent of bias related to age, gender, geographic location, environmental exposures, or ethnicity and PAP pathogenesis based on laboratory testing. Separately, we identied PAP-causing diseases in a cohort of patients referred to our site for tertiary evaluation and diagnosis. To our knowledge, these results comprise the first measurement of PAP prevelance in the US.
We believe our measurement of PAP prevalence in the US should be considered a minimum estimate and may underestimate actual prevalence for several reasons. First, since PAP typically presents as slowly progressive dyspnea of insidiouse onset, patients can remain asymptomatic for long periods before coming to medical attention. In fact, some patients are diagnosed incidentally, for example, when an abdominal CT scan identifies characteristic radiological findings in the lower lung fields. Further, in a report on the Japanese National PAP Registry, a large proportion (69/223, 31%) of PAP patients were asymptomatic and might have been missed if patient identification methods had not included mandatory health screening [11].
Second, symptomatic PAP patients are often misdiagnosed as asthma or pneumonia (before or after chest radiography, respectively) until failure to respond to ‘appropriate’ therapy prompts reconsideration and diagnostic testing. Notwithstanding, the PAP prevalence we determined among individuals of all ages in the US (6.87 per million) is similar to but slightly higher than the prevalence of PAP among adults in Japan (6.2 per million) [11]. Our observation that autoimmune PAP accounts for 91.5% of US PAP patients also agrees well with results from the Japanese National PAP Registry [11].
Our results indicated the prevalence of PAP syndrome increased with age across a broad range of ages including patients over 75 years old. It is important to note that our study did not address the incidence of PAP. Thus, it is possible and, perhaps likely, that the older individuals with PAP may have presented at a younger age.
Based on our minimum estimate of 2120 PAP patients in the US and experience of a relatively small number of referrals in an active diagnostic testing program (400 US patients over a 14-year period), we conclude that testing to diagnose specific diseases causing PAP syndrome is underutilized and that many patients with autoimmune PAP remain undiagnosed. Considering the high diagnostic accuracy of serum GM-CSF autoantibody testing [6] and prevalence of autoimmune PAP, the diagnostic insensitivity and morbidity associated with lung biopsy-based testing, and emerging treatment options for specific PAP-causing diseases, our results underscore the importance of increased use of blood-based testing in PAP patients, which can identify the PAP-causing disease, lead to earlier diagnosis, and reduce test-related morbidity and cost.
Acknowledgments
We thank Hongwei Wang, Wei Zhang, and Paul Kaplan for assistance with data acquisition and analysis. We thank our US PAP patients for their collaboration.
Funding
This work was supported by Sanofi, Inc., the Translational Pulmonary Science Center at Cincinnati Children’s Hospital Medical Center, the PAP Foundation, and grants from the NIH (R01 HL085453, U54 HL127672).
Availability of data and materials
The data that support the findings of this study are available from OptumInsight and Cincinnati Children’s Hospital and are available from the authors upon request and with permission of OptumInsight and Cincinnati Children’s Hospital.
Abbreviations
- CCI
Charlson Comorbidity Index
- DNA
Deoxyribonucleic acid
- GM-CSF
Granulocyte/macrophage colony-stimulating factor
- ICD
International Classification of Diseases
- PAP
Pulmonary alveolar proteinosis
- STAT5
Signal transducer and activator of transcription 5
- US
United States
Authors’ contributions
CM, RA and BCT designed the study, CM, RA, BCC, CC and BCT analysed the data. CM, RA and BCT wrote the manuscript and all authors approved it.
Ethics approval and consent to participate
The institutional review board of Cincinnati Children’s Hospital Medical Center a approved the study.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Cormac McCarthy, Email: Cormac.mccarthy@cchmc.org.
Ruzan Avetisyan, Email: Ruzan.Avetisyan@sanofi.com.
Brenna C. Carey, Email: Brenna.Carey@cchmc.org
Claudia Chalk, Email: Claudia.Chalk@cchmc.org.
Bruce C. Trapnell, Phone: 513.636.6361, Email: Bruce.Trapnell@cchmc.org
References
- 1.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 2.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 3.Sakagami T, Beck D, Uchida K, Suzuki T, Carey BC, Nakata K, Keller G, Wood RE, Wert SE, Ikegami M, Whitsett JA, Luisetti M, Davies S, Krischer JP, Brody A, Ryckman F, Trapnell BC. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med. 2010;182:49–61. doi: 10.1164/rccm.201001-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Sakagami T, Young LR, Carey BC, Wood RE, Luisetti M, Wert SE, Rubin BK, Kevill K, Chalk C, Whitsett JA, Stevens C, Nogee LM, Campo I, Trapnell BC. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med. 2010;182:1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Available from: https://www.optum.com.
- 6.Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, Koch DE, Stevens C, Inoue Y, Yamada Y, Trapnell BC. Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. J Immunol Methods. 2014;402:57–70. doi: 10.1016/j.jim.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, Smolarek T, Dishop MK, Wert SE, Whitsett JA, Grabowski G, Carey BC, Stevens C, van der Loo JC, Trapnell BC. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.US Census Bureau . 2010 Census demographic profile summary file. 2011. [Google Scholar]
- 9.Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40:373–383. doi: 10.1016/0021-9681(87)90171-8. [DOI] [PubMed] [Google Scholar]
- 10.Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45:613–619. doi: 10.1016/0895-4356(92)90133-8. [DOI] [PubMed] [Google Scholar]
- 11.Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, Kasahara Y, Tatsumi K, Hojo M, Ichiwata T, Tanaka N, Yamaguchi E, Eda R, Oishi K, Tsuchihashi Y, Kaneko C, Nukiwa T, Sakatani M, Krischer JP, Nakata K. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177:752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from OptumInsight and Cincinnati Children’s Hospital and are available from the authors upon request and with permission of OptumInsight and Cincinnati Children’s Hospital.