

Abstract
Background
In order to start to understand the function of individual members of gut microbiota, we cultured, sequenced and analysed bacterial anaerobes from chicken caecum.
Results
Altogether 204 isolates from chicken caecum were obtained in pure cultures using Wilkins-Chalgren anaerobe agar and anaerobic growth conditions. Genomes of all the isolates were determined using the NextSeq platform and subjected to bioinformatic analysis. Among 204 sequenced isolates we identified 133 different strains belonging to seven different phyla - Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Verrucomicrobia, Elusimicrobia and Synergistetes. Genome sizes ranged from 1.51 Mb in Elusimicrobium minutum to 6.70 Mb in Bacteroides ovatus. Clustering based on the presence of protein coding genes showed that isolates from phyla Proteobacteria, Verrucomicrobia, Elusimicrobia and Synergistetes did not cluster with the remaining isolates. Firmicutes split into families Lactobacillaceae, Enterococcaceae, Veillonellaceae and order Clostridiales from which the Clostridium perfringens isolates formed a distinct sub-cluster. All Bacteroidetes isolates formed a separate cluster showing similar genetic composition in all isolates but distinct from the rest of the gut anaerobes. The majority of Actinobacteria clustered closely together except for the representatives of genus Gordonibacter showing that the genome of this genus differs from the rest of Actinobacteria sequenced in this study. Representatives of Bacteroidetes commonly encoded proteins (collagenase, hemagglutinin, hemolysin, hyaluronidase, heparinases, chondroitinase, mucin-desulfating sulfatase or glutamate decarboxylase) that may enable them to interact with their host. Aerotolerance was recorded in Akkermansia and Cloacibacillus and was also common among representatives of Bacteroidetes. On the other hand, Elusimicrobium and the majority of Clostridiales were highly sensitive to air exposure despite their potential for spore formation.
Conclusions
Major gut microbiota members utilise different strategies for gut colonisation. High oxygen sensitivity of Firmicutes may explain their commonly reported decrease after oxidative burst during gut inflammation.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4959-4) contains supplementary material, which is available to authorized users.
Keywords: Microbiota, Microbiome, Chicken, Whole genome sequencing, Metabolic pathway, Host-microbiota interactions, Anaerobe, Butyrate, Propionate
Background
Characterisation of gut microbiota is nowadays relatively simple due to recent advances in next generation sequencing. However, though DNA sequencing is useful for monitoring dynamic changes in microbiota composition, it does not enable the understanding of biological functions of individual microbiota members. Shotgun sequencing of total DNA or RNA/cDNA sequencing can partly indicate the metabolic potential of microbial communities but is limited in addressing biological functions of individual microbiota members [1–3]. Even in the cases of analysis of microbial communities with low complexity when long DNA contigs can be assembled and associated with a particular bacterium, such approaches do not allow for subsequent experimental verification of observed data due to the unavailability of pure bacterial cultures. Isolation of gut anaerobes in pure cultures is therefore the best way to examine the characteristics of individual microbiota members experimentally [4].
Isolation of bacterial gut microbiota members in pure cultures is usually an issue since the majority of bacterial isolates colonising the intestinal tract are strict anaerobes. Although it is commonly reported that between 10 and 50% of bacterial species colonising the intestinal tract can be grown in vitro, recent studies proposed that up to 90% of major gut colonisers may be grown in vitro if multiple culture conditions are tested [5, 6]. Despite this, the isolation of a particular gut anaerobe may remain an issue since even the most abundant microbiota members at species level only rarely form more than 1% of the total population [7, 8]. This means that the desired bacterial species may be represented by a single colony growing on an agar plate together with hundreds or thousands of others and the likelihood of picking up the particular species is indeed rather low [6].
Chickens represent a specific case for studies focused on host - microbiota interactions. Chickens in commercial production are hatched in a clean environment of hatcheries without any contact with adult hens and their colonisation is dependent on environmental sources. Moreover, newly hatched chickens can be colonised by microbiota of wide a range of compositions [7] and colonisation of the intestinal tract of commercially hatched chickens may therefore differ from the colonisation of intestinal tract of chickens which would hatch in a nest. Perhaps not surprisingly, commercially hatched chickens are highly sensitive to colonisation with different pathogens, e.g. Salmonella, nevertheless, their resistance can be increased by providing them with a complex microbiota of adult hens [9, 10].
In our recent studies, we characterised the development of gut microbiota in commercially hatched chickens throughout their whole life [8], identified proteins expressed by the main microbiota members [7] and verified that gut microbiota may protect chickens against Salmonella Enteritidis infection [9]. However, which bacterial species are the protective ones is not known and more detailed knowledge of individual microbiota members is needed. One way forward is to obtain well-characterised pure cultures of gut anaerobes [11]. In this study we therefore cultured hundreds of isolates of anaerobes from chicken caecum and sequenced more than 200 of them. Analysis of their genomic sequences showed that we collected isolates from 7 different phyla. The aims of the subsequent comparisons focused mainly on the representatives from phyla Bacteroidetes and Firmicutes was to reveal differences in (poly)saccharide utilisation, propionate and butyrate fermentation and interactions with the host including motility or the ability to degrade host derived proteins.
Results
Altogether 204 isolates were obtained in pure culture and sequenced. Since in several cases we sequenced isolates exhibiting higher than 99% sequence similarity between their genomes, the final number of different isolates decreased to 133 (Table 1, Fig. 1). The lowest sequencing coverage was 43 fold for Drancourtella massiliensis An12, the highest 1526 fold for Lactobacillus gasseri An197, and median coverage over all sequenced isolates was 312 fold (see Additional file 1 for all details). The isolates belonged to 7 different phyla – Firmicutes (84 isolates), Bacteroidetes (29 isolates), Actinobacteria (15 isolates), Proteobacteria (1 isolate each of Escherichia and Desulfovibrio), Verrucomicrobia (1 isolate of Akkermansia), Elusimicrobia (1 isolate of Elusimicrobium) and Synergistetes (1 isolate of Cloacibacillus). The similarity of whole sequences of 16S rRNA to the to the most homologous GenBank entries was lower than 94% for 15 isolates. Considering taxonomic recommendations [12], these isolates represent candidates for new genera and two Muribaculum-like isolates might belong to a novel bacterial family or even an order. Alternative analysis based on alignment of RpoB amino acid sequences [13] yielded similar clustering of individual isolates as that achieved by 16S rRNA comparison (Additional file 2).
Table 1.
List of strains isolated, sequenced and analysed in this study
| Phylum | Family | Species | ID | rRNA % sim.a | genome (bp) | GC (%) |
|---|---|---|---|---|---|---|
| Actinobacteria | Coriobacteriaceae | Collinsella intestinalis | An268 | 94 | 2,375,164 | 65.28 |
| Actinobacteria | Coriobacteriaceae | Collinsella intestinalis | An7 | 95 | 2,368,937 | 65.33 |
| Actinobacteria | Coriobacteriaceae | Collinsella intestinalis | An307 | 96 | 2,218,890 | 62.95 |
| Actinobacteria | Coriobacteriaceae | Collinsella tanakaei | An271 | 96 | 2,790,023 | 64.34 |
| Actinobacteria | Coriobacteriaceae | Collinsella tanakaei | An2 | 94 | 2,542,639 | 62.39 |
| Actinobacteria | Coriobacteriaceae | Enorma massiliensis | An70 | 99 | 2,374,054 | 62.09 |
| Actinobacteria | Coriobacteriaceae | Enorma timonensis | An5 | 96 | 2,299,978 | 65.99 |
| Actinobacteria | Coriobacteriaceae | Gordonibacter urolithinfaciens | An234A | 99 | 3,542,488 | 65.99 |
| Actinobacteria | Coriobacteriaceae | Gordonibacter urolithinfaciens | An232A | 94 | 3,457,314 | 65.16 |
| Actinobacteria | Coriobacteriaceae | Gordonibacter urolithinfaciens | An230 | 94 | 3,885,330 | 64.16 |
| Actinobacteria | Coriobacteriaceae | Olsenella uli | An188 | 96 | 2,138,506 | 68.68 |
| Actinobacteria | Coriobacteriaceae | Olsenella uli | An290 | 96 | 2,158,343 | 68.09 |
| Actinobacteria | Coriobacteriaceae | Olsenella uli | An285 | 96 | 2,413,556 | 66.98 |
| Actinobacteria | Coriobacteriaceae | Olsenella uli | An293 | 96 | 2,318,914 | 67.87 |
| Actinobacteria | Coriobacteriaceae | Olsenella uli | An270 | 96 | 2,014,154 | 67.55 |
| Bacteroidetes | Bacteroidaceae | Bacteroides clarus | An43 | 99 | 4,226,284 | 45.22 |
| Bacteroidetes | Bacteroidaceae | Bacteroides clarus | An189 | 99 | 4,166,078 | 45.36 |
| Bacteroidetes | Bacteroidaceae | Bacteroides dorei | An16 | 99 | 5,376,103 | 41.79 |
| Bacteroidetes | Bacteroidaceae | Bacteroides dorei | An41 | 99 | 5,463,463 | 42.00 |
| Bacteroidetes | Bacteroidaceae | Bacteroides ovatus | An161 | 99 | 6,700,861 | 42.08 |
| Bacteroidetes | Bacteroidaceae | Bacteroides salanitronis | An322 | 92 | 3,449,415 | 44.84 |
| Bacteroidetes | Bacteroidaceae | Bacteroides uniformis | An67 | 99 | 4,590,834 | 46.36 |
| Bacteroidetes | Bacteroidaceae | Bacteroides xylanisolvens | An109 | 99 | 5,713,108 | 41.57 |
| Bacteroidetes | Bacteroidaceae | Bacteroides xylanisolvens | An107 | 99 | 5,745,201 | 41.89 |
| Bacteroidetes | Bacteroidaceae | Mediterranea massiliensis | An20 | 91 | 3,968,548 | 49.41 |
| Bacteroidetes | Porphyromonadaceae | Barnesiella viscericola | An22 | 98 | 3,264,150 | 53.04 |
| Bacteroidetes | Porphyromonadaceae | Barnesiella viscericola | An55 | 98 | 3,040,531 | 51.37 |
| Bacteroidetes | Porphyromonadaceae | Butyricimonas paravirosa | An62 | 98 | 5,176,855 | 42.63 |
| Bacteroidetes | Porphyromonadaceae | Muribaculum intestinale | An289 | 83 | 2,336,263 | 49.09 |
| Bacteroidetes | Porphyromonadaceae | Muribaculum intestinale | An287 | 83 | 2,349,712 | 49.04 |
| Bacteroidetes | Porphyromonadaceae | Odoribacter splanchnicus | An45 | 99 | 4,610,398 | 43.75 |
| Bacteroidetes | Porphyromonadaceae | Odoribacter splanchnicus | An39 | 99 | 4,722,515 | 43.48 |
| Bacteroidetes | Porphyromonadaceae | Parabacteroides distasonis | An199 | 99 | 5,145,110 | 45.11 |
| Bacteroidetes | Porphyromonadaceae | Parabacteroides johnsonii | An42 | 99 | 4,430,164 | 45.03 |
| Bacteroidetes | Porphyromonadaceae | Parabacteroides merdae | An277 | 92 | 3,709,857 | 46.69 |
| Bacteroidetes | Rikenellaceae | Alistipes onderdonkii | An90 | 100 | 3,488,443 | 58.52 |
| Bacteroidetes | Rikenellaceae | Alistipes senegalensis | An31A | 97 | 2,626,508 | 61.85 |
| Bacteroidetes | Rikenellaceae | Alistipes senegalensis | An116 | 97 | 3,264,205 | 58.57 |
| Bacteroidetes | Rikenellaceae | Alistipes senegalensis | An66 | 97 | 3,064,564 | 59.37 |
| Bacteroidetes | Rikenellaceae | Alistipes shahii | An54 | 96 | 3,272,633 | 58.38 |
| Bacteroidetes | unclassified_"Bacteroidales” | Bacteroides salanitronis | An269 | 92 | 4,466,522 | 45.87 |
| Bacteroidetes | unclassified_"Bacteroidales” | Bacteroides salanitronis | An279 | 92 | 3,976,735 | 45.82 |
| Bacteroidetes | unclassified_"Bacteroidales” | Bacteroides salanitronis | An19 | 92 | 4,779,606 | 45.88 |
| Bacteroidetes | unclassified_"Bacteroidales” | Bacteroides salanitronis | An51A | 92 | 4,415,476 | 45.79 |
| Firmicutes | Enterococcaceae | Enterococcus cecorum | An69 | 100 | 2,010,800 | 36.66 |
| Firmicutes | Enterococcaceae | Enterococcus cecorum | An144 | 100 | 2,521,030 | 36.10 |
| Firmicutes | Lactobacillaceae | Lactobacillus gallinarum | An119 | 99 | 2,068,702 | 36.50 |
| Firmicutes | Lactobacillaceae | Lactobacillus gallinarum | An153 | 99 | 2,042,196 | 36.47 |
| Firmicutes | Lactobacillaceae | Lactobacillus gallinarum | An115 | 99 | 2,039,377 | 36.52 |
| Firmicutes | Lactobacillaceae | Lactobacillus gallinarum | An101 | 99 | 2,051,080 | 36.54 |
| Firmicutes | Lactobacillaceae | Lactobacillus gasseri | An197 | 99 | 1,786,561 | 34.60 |
| Firmicutes | Lactobacillaceae | Lactobacillus reuteri | An71 | 99 | 2,330,171 | 38.47 |
| Firmicutes | Lactobacillaceae | Lactobacillus reuteri | An166 | 99 | 2,293,009 | 38.50 |
| Firmicutes | Lactobacillaceae | Lactobacillus salivarius | An63 | 99 | 1,826,390 | 32.76 |
| Firmicutes | Lactobacillaceae | Lactobacillus salivarius | An84 | 99 | 2,115,320 | 32.74 |
| Firmicutes | Lactobacillaceae | Lactobacillus salivarius | An128 | 99 | 1,900,005 | 32.75 |
| Firmicutes | Erysipelotrichaceae | [Clostridium] spiroforme | An158 | 99 | 2,749,214 | 28.90 |
| Firmicutes | Erysipelotrichaceae | [Clostridium] spiroforme | An15 | 94 | 3,043,058 | 30.24 |
| Firmicutes | Erysipelotrichaceae | [Clostridium] spiroforme | An26 | 99 | 2,686,825 | 28.58 |
| Firmicutes | Erysipelotrichaceae | [Clostridium] spiroforme | An149 | 99 | 2,786,011 | 28.86 |
| Firmicutes | Erysipelotrichaceae | [Clostridium] spiroforme | An173 | 94 | 2,963,161 | 30.12 |
| Firmicutes | Erysipelotrichaceae | [Eubacterium] cylindroides | An64 | 99 | 1,822,768 | 34.80 |
| Firmicutes | Erysipelotrichaceae | [Eubacterium] cylindroides | An178 | 99 | 1,979,688 | 34.75 |
| Firmicutes | Erysipelotrichaceae | Massiliomicrobiota timonensis | An13 | 100 | 2,808,053 | 31.46 |
| Firmicutes | Erysipelotrichaceae | Massiliomicrobiota timonensis | An142 | 98 | 2,805,108 | 31.51 |
| Firmicutes | Erysipelotrichaceae | Massiliomicrobiota timonensis | An134 | 98 | 2,583,105 | 31.58 |
| Firmicutes | Erysipelotrichaceae | Massiliomicrobiota timonensis | An105 | 98 | 2,657,304 | 31.69 |
| Firmicutes | Erysipelotrichaceae | Massiliomicrobiota timonensis | An80 | 98 | 2,632,118 | 31.88 |
| Firmicutes | Clostridiaceae 1 | Clostridium perfringens | An68 | 99 | 3,279,733 | 28.07 |
| Firmicutes | Clostridiaceae 1 | Clostridium perfringens | An185 | 99 | 3,267,175 | 28.06 |
| Firmicutes | Lachnospiraceae | [Clostridium] glycyrrhizinilyticum | An169 | 96 | 4,874,615 | 49.44 |
| Firmicutes | Lachnospiraceae | [Clostridium] glycyrrhizinilyticum | An298 | 96 | 3,190,622 | 46.76 |
| Firmicutes | Lachnospiraceae | [Clostridium] glycyrrhizinilyticum | An76 | 96 | 3,392,322 | 50.34 |
| Firmicutes | Lachnospiraceae | [Clostridium] lactatifermentans | An114 | 95 | 2,765,027 | 31.21 |
| Firmicutes | Lachnospiraceae | [Clostridium] lactatifermentans | An75 | 99 | 3,241,294 | 44.78 |
| Firmicutes | Lachnospiraceae | [Clostridium] oroticum | An181 | 95 | 2,778,317 | 43.19 |
| Firmicutes | Lachnospiraceae | [Clostridium] saccharolyticum | An14 | 95 | 4,319,160 | 54.42 |
| Firmicutes | Lachnospiraceae | [Clostridium] saccharolyticum | An168 | 99 | 3,501,825 | 50.31 |
| Firmicutes | Lachnospiraceae | [Clostridium] saccharolyticum | An196 | 93 | 3,261,385 | 50.05 |
| Firmicutes | Lachnospiraceae | [Eubacterium] contortum | An118 | 95 | 3,480,620 | 52.25 |
| Firmicutes | Lachnospiraceae | [Eubacterium] fissicatena | An131 | 94 | 3,529,099 | 50.87 |
| Firmicutes | Lachnospiraceae | [Eubacterium] fissicatena | An138 | 94 | 3,702,317 | 50.44 |
| Firmicutes | Lachnospiraceae | [Eubacterium] hallii | An3 | 95 | 4,260,545 | 46.43 |
| Firmicutes | Lachnospiraceae | [Eubacterium] hallii | An11 | 95 | 3,929,296 | 46.78 |
| Firmicutes | Lachnospiraceae | Blautia coccoides | An46 | 94 | 3,844,348 | 44.62 |
| Firmicutes | Lachnospiraceae | Blautia producta | An81 | 95 | 4,340,741 | 44.56 |
| Firmicutes | Lachnospiraceae | Blautia schinkii | An249 | 93 | 3,969,115 | 45.08 |
| Firmicutes | Lachnospiraceae | Drancourtella massiliensis | An210 | 95 | 3,049,918 | 45.07 |
| Firmicutes | Lachnospiraceae | Drancourtella massiliensis | An177 | 95 | 2,986,142 | 44.76 |
| Firmicutes | Lachnospiraceae | Drancourtella massiliensis | An57 | 95 | 3,307,621 | 45.72 |
| Firmicutes | Lachnospiraceae | Drancourtella massiliensis | An12 | 96 | 3,637,323 | 46.18 |
| Firmicutes | Ruminococcaceae | Anaerofilum agile | An201 | 91 | 3,232,216 | 60.34 |
| Firmicutes | Ruminococcaceae | Anaeromassilibacillus senegalensis | An200 | 94 | 3,738,663 | 54.30 |
| Firmicutes | Ruminococcaceae | Anaeromassilibacillus senegalensis | An250 | 97 | 3,582,839 | 53.28 |
| Firmicutes | Ruminococcaceae | Anaeromassilibacillus senegalensis | An172 | 92 | 2,820,032 | 41.40 |
| Firmicutes | Ruminococcaceae | Anaerotruncus colihominis | An174 | 99 | 4,104,028 | 53.46 |
| Firmicutes | Ruminococcaceae | Anaerotruncus colihominis | An175 | 99 | 4,098,164 | 53.45 |
| Firmicutes | Ruminococcaceae | Anaerotruncus colihominis | An251 | 99 | 3,446,606 | 54.60 |
| Firmicutes | Ruminococcaceae | Butyricicoccus pullicaecorum | An179 | 99 | 3,474,625 | 53.61 |
| Firmicutes | Ruminococcaceae | Butyricicoccus pullicaecorum | An180 | 99 | 3,016,034 | 54.43 |
| Firmicutes | Ruminococcaceae | Faecalibacterium prausnitzii | An121 | 95 | 2,728,163 | 61.06 |
| Firmicutes | Ruminococcaceae | Faecalibacterium prausnitzii | An77 | 96 | 3,092,450 | 59.48 |
| Firmicutes | Ruminococcaceae | Faecalibacterium prausnitzii | An192 | 97 | 3,520,066 | 58.80 |
| Firmicutes | Ruminococcaceae | Faecalibacterium prausnitzii | An122 | 96 | 3,272,200 | 59.47 |
| Firmicutes | Ruminococcaceae | Faecalibacterium prausnitzii | An58 | 96 | 2,956,350 | 60.63 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An91 | 97 | 3,629,120 | 57.60 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An52 | 97 | 2,845,789 | 59.13 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An92 | 98 | 3,498,453 | 62.13 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An112 | 97 | 2,967,986 | 59.27 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An135 | 98 | 3,907,586 | 61.45 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An82 | 97 | 3,683,590 | 58.85 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An248 | 100 | 3,776,725 | 60.99 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An4 | 97 | 3,239,670 | 58.22 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An9 | 97 | 3,357,728 | 58.20 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An100 | 95 | 3,054,748 | 57.15 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An306 | 98 | 3,967,264 | 59.00 |
| Firmicutes | Ruminococcaceae | Flavonifractor plautii | An10 | 96 | 3,867,419 | 61.74 |
| Firmicutes | Ruminococcaceae | Gemmiger formicilis | An50 | 94 | 3,597,344 | 58.67 |
| Firmicutes | Ruminococcaceae | Gemmiger formicilis | An194 | 95 | 3,080,663 | 59.30 |
| Firmicutes | Ruminococcaceae | Gemmiger formicilis | An87 | 94 | 3,355,244 | 58.58 |
| Firmicutes | Ruminococcaceae | Gemmiger formicilis | An120 | 95 | 3,406,814 | 60.15 |
| Firmicutes | Ruminococcaceae | Pseudoflavonifractor capillosus | An176 | 96 | 2,574,287 | 58.19 |
| Firmicutes | Ruminococcaceae | Pseudoflavonifractor capillosus | An187 | 96 | 2,623,188 | 57.97 |
| Firmicutes | Ruminococcaceae | Pseudoflavonifractor capillosus | An44 | 96 | 2,764,660 | 57.41 |
| Firmicutes | Ruminococcaceae | Pseudoflavonifractor capillosus | An85 | 96 | 3,018,026 | 56.20 |
| Firmicutes | Ruminococcaceae | Pseudoflavonifractor capillosus | An184 | 96 | 3,617,258 | 59.87 |
| Firmicutes | Veillonellaceae | Megamonas hypermegale | An288 | 99 | 2,165,576 | 33.61 |
| Firmicutes | Veillonellaceae | Megasphaera elsdenii | An286 | 95 | 2,396,433 | 53.29 |
| Proteobacteria | Desulfovibrionaceae | Desulfovibrio desulfuricans | An276 | 92 | 3,230,576 | 55.37 |
| Proteobacteria | Enterobacteriaceae | Escherichia fergusonii | An190 | 99 | 5,352,565 | 50.48 |
| Synergistetes | Synergistaceae | Cloacibacillus porcorum | An23 | 93 | 2,902,045 | 57.89 |
| Verrucomicrobia | Verrucomicrobiaceae | Akkermansia muciniphila | An78 | 100 | 2,734,062 | 55.68 |
| Elusimicrobia | Elusimicrobiaceae | Elusimicrobium minutum | An273 | 94 | 1,505,722 | 51.73 |
aPercent similarity along the whole 16S rRNA sequence to the GenBank entry with the highest similarity
Fig. 1.

Phylogenetic tree with selected functional properties of 133 sequenced isolates obtained from chicken caecum based on the Bayesian analysis of the full-length sequences of 16S rRNA genes. Families within the phylum Firmicutes are shown in light blue, green and yellow. Families within the phylum Bacteroidetes are shown in shades of purple. Actinobacteria (family Coriobacteriaceae – Cor) are highlighted in red. In the cases when only one or two isolates were sequenced per phylum, these isolates are described by phylum name – Prot – Proteobacteria, Elu – Elusimicrobia, Ver – Verrucomicrobia, Syn – Synergistetes. In the remaining cases, branches with different families are highlighted with different colors. Rik – Rikenellaceae, Porp - Porphyromonadaceae, Bact – Bacteroidaceae, Lach – Lachnospiraceae, Clos - Clostridiaceae, Ery – Erysipelotrichaceae, Entcoc – Enterococcaceae, Lact – Lactobacillaceae, Rum – Ruminococcaceae, Veil – Veillonellaceae. BUK – butyrate kinase, BPT – phosphate butyryltransferase, BTR – butyryl-CoA transferase, AcCo – acetyl CoA pathway, LYS – lysine fermentation pathway, SUC – succinate fermentation pathway, M-MLN presence of methylmalonyl mutase, epimerase and decarboxylase required for conversion of succinate to methyl-malonyl CoA and propionate. AcGen – presence of genes required for reductive acetogenesis
When we compared the sequence of 16S rRNA genes of all 133 isolates with the operational taxonomic units (OTUs) combined from our previous studies [7, 8], rather unexpectedly, 7 isolates were excluded from the analysis by QIIME at the chimera removal step. Three isolates formed OTUs which we did not detect among the microbiota of the two studies. The rest of the isolates were assigned into particular OTUs. Nineteen isolates were assigned to 11 OTUs from the 100 most frequent OTUs, and out of the 1000 most common OTUs, we obtained 76 isolates belonging to 42 OTUs. Exact ranking of each individual isolate among the most frequent OTUs present in the chicken caecum can be found in Additional file 1.
Genome sizes ranged from 1.51 Mb in Elusimicrobium to 6.70 Mb in Bacteroides ovatus. Larger genomes were usually recorded in isolates from phylum Bacteroidetes (Additional file 1). Actinobacteria possessed small genomes ranging from 2.1 to 2.5 Mb. Genomes of Firmicutes ranged mostly from 3 to 4 Mb although genomes of Enterococcus, [Eubacterium] cylindroides and Lactobacillus were among the smallest ones with genome sizes around 2 Mb. Genomic GC content ranged from 28.0 to 62.1% in Firmicutes, from 41.6 to 61.9% in Bacteroidetes and from 62.1 to 68.7% in Actinobacteria. GC content of individual isolates belonging to the remaining 4 phyla ranged from 50.5 to 57.9% (Additional file 1).
Whole genome comparison
Network analysis based on the correlation of individual gene counts in individual isolates confirmed similarities observed by 16S rRNA gene alignment. Individual isolates from phyla Proteobacteria, Verrucomicrobia, Elusimicrobia and Synergistetes formed disconnected vertices of the network (Fig. 2). Firmicutes split into families Lactobacillaceae, Enterococcaceae, Veillonellaceae (genera Megasphaera and Megamonas) and order Clostridiales (families Erysipelotrichaceae, Ruminococcaceae and Lachnospiraceae) except for Clostridium perfringens. Members of the family Erysipelotrichaceae formed a slightly eccentric cluster at the periphery of other isolates from the order Clostridiales indicating their slightly different coding capacity when compared to isolates belonging to families Lachnospiraceae and Ruminococcaceae. All Bacteroidetes formed a disconnected network cluster showing similar genetic composition in all isolates. The majority of Actinobacteria formed a single network cluster except for the representatives of genus Gordonibacter showing that the genome of this genus differed from the rest of Actinobacteria sequenced in this study.
Fig. 2.

Network analysis of isolates from chicken caecum based on their gene composition. Firmicutes split into families Lactobacillaceae, Enterococcaceae, Veillonellaceae (genera Megasphaera and Megamonas) and the order Clostridiales except for Clostridium perfringens. Separation of Erysipelotrichaceae from Lachnospiraceae and Ruminococcaceae (all belonging to order Clostridiales) was observed. All Bacteroidetes formed a separated cluster showing similar genetic composition in all isolates. The majority of Actinobacteria formed a single cluster except for the representatives of the genus Gordonibacter indicating that genetic composition of these isolates differed from the rest of Actinobacteria
Basic biological processes
Since gut microbiota is formed mainly by representatives of phyla Bacteroidetes and Firmicutes, we specifically focused on the comparison of genomes of isolates belonging to these two phyla. Representatives belonging to phylum Bacteroidetes (Gram negative bacteria) coded for proteins required for the biosynthesis of a Gram negative cell wall while representatives of phylum Firmicutes (Gram positive bacteria) coded for proteins required for the biosynthesis of a Gram positive cell wall. However, Megamonas and Megasphaera (family Veilonellaceae, phylum Firmicutes, i.e. Gram positive bacteria) harboured genes required for the synthesis of Gram negative cell wall type (Fig. 3). Bacteroidetes and Firmicutes differed in their mode of transport across the cell wall and cytoplasmic membrane. In Bacteroidetes, genes belonging to Ton and Tol transport systems were the most frequent whilst Firmicutes encoded ABC transporters, ECF class transporters and TRAP transporters (Fig. 3). Genes enabling sporulation were specific to Gram positive Firmicutes except for Lactobacillaceae, Enterococcaceae, Veilonellaceae and two [Eubacterium] cylindroides isolates (Fig. 3). However, there were differences in the composition and distribution of genes necessary for spore formation among the isolates with sporulation potential. Members of the family Erysipelotrichaceae (Massiliomicrobiota sp. and [Clostridium] spiroforme) did not code for stage III sporulation proteins and Faecalibacterium, Anaerofilum and Gemmiger (family Ruminococcaceae) did not code for spore proteins though the rest of the genes required for sporulation were present in their genomes (Fig. 3). None of the Bacteroidetes isolates encoded proteins required for sporulation and their oxygen tolerance could be dependent on batABCDE operon (Bacteroides aerotolerance). batABE genes were present in all representatives of Bacteroidetes and batCD were present in all Bacteroidetes except for two Muribaculum isolates. When we examined the survival rate of the anaerobes after exposure to aerobic conditions experimentally, the most sensitive isolates were single isolates of Muribaculum intestinale (phylum Bacteroidetes), [Clostridium] glycyrrhizinilyticum, [Clostridium] saccharolyticum, Anaeromassilibacillus senegalensis and Flavonifractor plautii (all phylum Firmicutes) which did not survive an hour long exposure to the air. Bacteria which did not survive 8 or 24-h-long exposure to the air belonged mainly to the order Clostridiales. Within Clostridiales, 45% of the tested isolates did not survive 8-h long exposure to the air, and an additional 25% died between 8 to 24-h exposure. Only 25% of tested isolates from the order Clostridiales survived 24-h air exposure. On the other hand, representatives of Bacteroidetes and Actinobacteria were usually tolerant to sudden air exposure since 62 and 73% of tested isolates survived 24-h air exposure, respectively (Fig. 4 and Additional file 3). Bacteroides sp. encoded a high number of proteins involved in polysaccharide and monosacharide metabolism while the number of genes required for metabolism of di- and oligosaccharides was similar in both Bacteroidetes and Firmicutes (Fig. 3).
Fig. 3.

Distribution of genes in selected categories among representatives of major gut colonisers belonging to phyla Bacteroidetes and Firmicutes. X axes indicate the numbers of genes in a given category per genome. For a scalable figure see Additional file 5 online
Fig. 4.

Sensitivity of gut anaerobes to air exposure. Most of isolates from the order Clostridiales, phylum Firmicutes (n = 69) died during 1–8 long hour exposure to the air. On the other hand, majority of the tested representatives of Bacteroidetes (n = 29) and Actinobacteria (n = 15) survived 24-h-long exposure to air. For full information on survival of individual isolates see Additional file 3 online
Production of butyrate, propionate and acetogenesis
Short chain fatty acids and butyrate in particular, are acknowledged as important metabolites of bacterial origin [14, 15]. All genes required for butyrate production from pyruvate and acetyl-CoA were present in the genomes of Ruminococcaceae (genera Butyricicoccus, Pseudoflavonifractor, Flavonifractor, Anaeromassilibacillus, Anaerotruncus and Faecalibacterium) (Fig. 1). In addition, this pathway was also present in Megasphaera elsdenii, [Eubacterium] cylindroides, [Clostridium] lactatifermentans, [Clostridium] saccharolyticum and [Eubacterium] hallii, the latter three belonging to the family Lachnospiraceae (Fig. 1). Butyricimonas paravirosa was the only isolate from phylum Bacteroidetes coding for all genes required for butyrate production from pyruvate and acetyl-CoA. Genes coding for enzymes of complete lysine fermentation pathway leading to butyrate production were present in three genera of the phylum Bacteroidetes (Muribaculum, Butyricimonas and Odoribacter) and the majority of Flavonifractor isolates belonging to the phylum Firmicutes. Butyricimonas and Odoribacter also encoded the whole pathway required for the conversion of succinate and 4-hydroxybutyrate into butyrate (Fig. 1). Terminal steps in butyrate production were dependent on transferases transferring CoA moiety from butyryl-CoA to acetate, acetoacetate or 4-hydroxybutyrate, or phosphate butyryltransferase (PBT) and butyrate kinase (BUK). Surprisingly, we did not find butyrate kinase in all isolates using the PBT - BUK pathway for butyrate release from butyryl-CoA. In such a case, butyrate-phosphate may serve for substrate phosphorylation in enzymatic reactions similar to acetate-phosphate.
Propionate production via a succinate-methylmalonate pathway was characteristic of Bacteroidetes (Fig. 1) as genes for methylmalonyl-CoA mutase, epimerase and decarboxylase were detected in genomes of all isolates from this phylum. This pathway was quite rare in Firmicutes since methylmalonyl-CoA mutase, epimerase and decarboxylase were encoded only by Megamonas and two Flavonifractor isolates (Fig. 1).
Fermentation of carbohydrates results in the production of H2 which has to be removed from the community since its increased concentration suppresses glycolysis [16–18]. H2 can be removed by methanogens, acetogens or sulphate reducing bacteria. We did not isolate a single methanogen. Desulfovibrio (phylum Proteobacteria) encoded key genes for sulphate reduction to H2S (adenylylsulphate reductase, sulphate adenylyltransferase, dissimilatory sulphite reductase and sulphite reduction-associated complex DsrMKJOP). Potential for H2 removal by acetogenesis was recorded in [Clostridium] saccharolyticum, [Eubacterium] fissicatena and all Blautia isolates (B. coccoides, B. producta, B. schinkii) since all these bacteria encoded corrinoid iron-sulfur acetyl-CoA synthase and 5-methyltetrahydrofolate methyltransferase (Fig. 1).
Genes encoding proteins mediating interactions with the host
Since gut microbiota may interact with its host, finally we searched for the presence of genes which may facilitate such interactions (Additional file 4). Genes encoding collagenase precursor, hemagglutinin or hemolysin A were present in all 29 Bacteroidetes isolates but not in Firmicutes (Fig. 5). Hyaluronidase was present in 16 isolates from the phylum Bacteroidetes and five Firmicutes. Different heparinases were detected in 14 isolates from the phylum Bacteroidetes and seven representatives of Firmicutes – out of these all five Faecalibacterium isolates encoded heparinase II/III-like and outside this genus, heparinases were present only in two isolates from the phylum Firmicutes (i.e. Gemmiger formicilis and [Clostridium] glycyrrhizinilyticum). Chondroitinase was present in nine isolates from phylum Bacteroidetes (genera Alistipes and Bacteroides) but in no isolate from Firmicutes. Mucin-desulfating sulfatase was present in the genomes of 20 Bacteroidetes isolates (mainly in genera Alistipes, Parabacteroides and Bacteroides) but was absent from the genomes of Firmicutes. A gene for endothelin-converting enzyme 1 precursor was present in the genomes of all Bacteroidetes but not in Firmicutes. Except for two Odoribacter isolates and Parabacteroides distatonis, all the remaining representatives of the phylum Bacteroidetes encoded 3-oxo-5-α-steroid 4-dehydrogenase capable of modification of bile or steroid hormones. This gene was not detected in Firmicutes. Glutamate decarboxylase catalysing production of γ-aminobutyrate (GABA) and glutamate/GABA antiporter was present in 20 different Bacteroidetes isolates but only in two Firmicutes and these were both pathogenic Clostridium perfringens isolates. Histidine decarboxylase catalysing production of histamine was quite rare and was present only in two Bacteroides dorei isolates (Fig. 5). On the other hand, except for all Lactobacilli, the gene for fibronectin/fibrinogen-binding protein was present in all isolates from Firmicutes but in none representative from the phylum Bacteroidetes. Genes for flagellar motility were quite rare and were absent in all Bacteroidetes isolates. Complete flagellar operon was present only in two Flavonifractor isolates, one strain of Anaeromassilibacillus senegalensis and all three isolates of Anaerotruncus colihominis (Fig. 5).
Fig. 5.
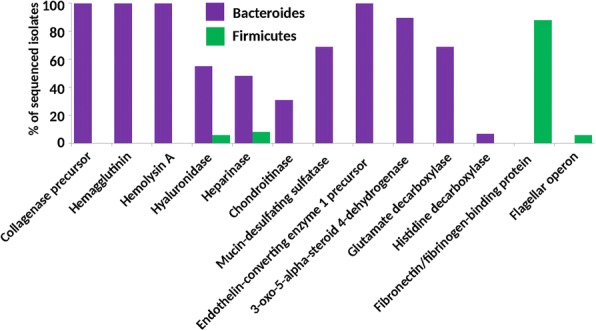
Distribution of genes encoding proteins mediating interactions of Bacteroidetes and Firmicutes with the host. The presence of genes which may allow for interactions with chicken host structures was determined in each isolate belonging to these two phyla and expressed as a percentage of positive isolates out of all the sequenced Bacteroidetes (29 isolates in a total) and Firmicutes (84 isolates in a total)
Discussion
In this study we sequenced, annotated and analysed genomes of 133 different anaerobes cultured from the chicken caecum. Several isolates represented species which are available in only a few pure cultures worldwide – a single manuscript reported a pure culture of Elusimicrobium [19] and only two papers reported culture of Cloacibacillus porcorum [20, 21]. Although such isolates are of clear potential for future experiments, in this report we mainly focused on the comparison of genetic potential of isolates belonging to two main phyla inhabiting the intestinal tract of chickens, Bacteroidetes and Firmicutes [8, 18].
Whole genome sequencing showed that representatives of Bacteroidetes and Firmicutes have a genetic potential to follow different strategies of gut colonisation which may explain their coexistence in the intestinal tract. Bacteroidetes encoded genes for the biosynthesis of a Gram negative cell wall, Gram positive anaerobes encoded genes for the biosynthesis of a Gram positive cell wall. Only Megamonas and Megasphaera, though belonging to Gram positive Firmicutes, encoded genes for the biosynthesis of Gram negative cell wall type, in agreement with previous report [22]. The Ton/ExbD transport system of Bacteroidetes has been identified earlier as highly expressed in vivo [7] and ABC, ECF and TRAP transporters were described as characteristic of Firmicutes [23–25]. Bacteroidetes were reported to increase in gut microbiota with high fiber content in their diet [26, 27] and to forage on host derived polysaccharides in the absence of fiber [28–30]. Consistent with this, Bacteroidetes and the genus Bacteroides in particular encoded a high number of genes required for polysaccharide metabolism [28, 30–33]. Since the polysaccharides of feed and host origin consist of various (amino)monosaccharides, Bacteroidetes encoded also a wider range of genes required for monosaccharide degradation than Firmicutes (Fig. 3).
Butyrate is produced mainly by Firmicutes. Carbohydrate fermentation to pyruvate and acetyl-CoA was the most frequent butyrate production pathway, as proposed earlier [34]. Butyrate production was mainly associated with Ruminococcaceae and less frequently with Lachnospiraceae or Erysipelotrichaceae [35]. Bacteroidetes are not the main butyrate producers as acetyl-CoA conversion to butyrate was only found in Butyricimonas. However, Bacteroidetes were capable of butyrate production by alternative pathways, e.g. from 4-hydroxybutyrate as recorded in Butyricimonas and Odoribacter or by lysine fermentation as recorded in Muribaculum, Butyricimonas and Odoribacter. This is in line with conclusions derived from human microbiota studies [35].
Gut microbiota interacts with the host. The potential of Firmicutes to interact with the chicken host seems to be less extensive in comparison to Bacteroidetes. Except for Lactobacilli, Firmicutes isolates encoded fibronectin/fibrinogen-binding protein [36, 37]. Interestingly, we detected chicken fibrinogen-domain containing proteins as tightly associated with gut microbiota [38]. Such proteins aggregate bacteria [39, 40] and enable association of different gut microbiota members, based on current results, preferentially those belonging to the phylum Firmicutes. Binding to chicken fibrinogen-domain containing proteins may result in the formation of random bacterial aggregates and those with the most optimal composition, e.g. butyrate producers releasing H2 with Blautia consuming H2 for acetate production thus allowing glycolysis in butyrate producers to proceed [16], will rapidly multiply and define final microbiota composition. Except for fibrinogen binding, representatives from the phylum Bacteroidetes had a greater potential to affect host behaviour than representatives from the phylum Firmicutes. Bacteroidetes encoded collagenase, hemagglutinin, hemolysin, hyaluronidase, heparinases, chondroitinase or mucin-desulfating sulfatase required for degradation of host structures. In addition, representatives from the phylum Bacteroidetes encoded endothelin-converting enzyme 1 precursor, which plays a significant role in cardiovascular diseases and Alzheimer’s disease in humans [41], 3-oxo-5-α-steroid 4-dehydrogenase capable of modification of steroid hormones or bile [42], or glutamate decarboxylase catalysing production of γ-aminobutyrate (GABA), a mediator within the enteric nervous system [43]. However, it will have to be elucidated whether these genes are expressed in vivo and whether their products may reach host structures.
Finally, the rather counterintuitive conclusion came from the prediction of survival following air exposure. Although Bacteroidetes should be more sensitive to air exposure than Firmicutes which are capable of spore formation, our data clearly showed that Clostridiales, including all important butyrate producers, were highly sensitive to air exposure. Due to the experimental design, we likely determined sensitivity of vegetative cells and not of spores. Despite this, the extreme sensitivity of vegetative cells of Clostridiales may explain their commonly reported disappearance in inflammatory bowel disease patients [44]. Inflammation leads to locally increased oxygen levels due to the oxidative burst of granulocytes and macrophages [45, 46]. Disappearance of Clostridiales including major butyrate producers can therefore be a consequence rather than a cause of inflammatory bowel diseases. Similarly, a reported increase in the abundance of Bacteroidetes or Megasphaera during inflammatory diseases [35, 47] may be a mere consequence of their higher resistance to oxygen and the disappearance of oxygen sensitive bacterial species from order Clostridiales. An overgrowth of facultative anaerobes like those from family Enterobacteriaceae in acute colitis also fits in the proposed scenario [47–49].
Conclusions
In this study we isolated and sequenced 133 different strains originating from chickens intestinal tracts belonging to seven different phyla. Analysis of their genomic sequences showed that butyrate production was mainly associated with Ruminococcaceae, and less frequently with Lachnospiraceae or Erysipelotrichaceae, all belonging to phylum Firmicutes. Representatives of phylum Bacteroidetes commonly encoded proteins (collagenase, hemagglutinin, hemolysin, hyaluronidase, heparinases, chondroitinase, mucin-desulfating sulfatase or glutamate decarboxylase) that may enable them to interact with their host. Even such a brief list of genes shows that representatives of Bacteroidetes and Firmicutes follow different strategies of gut colonisation which contributes to their coexistence in the intestinal tract.
Methods
Ethics statement
The handling of animals in this study was performed in accordance with current Czech legislation (Animal Protection and Welfare Act No. 246/1992 Coll. of the Government of the Czech Republic). Experiments performed in this study were approved by the Ethics Committee of the Veterinary Research Institute (permit number 4/2016) followed by the Committee for Animal Welfare of the Ministry of Agriculture of the Czech Republic.
Isolation and identification of caecal bacteria
The chickens were sacrificed under chloroform anesthesia by cervical dislocation. Whole caeca with their contents originating from 18 random healthy chickens or hens 4 to 40 weeks of age were removed during necropsy, chilled on ice and transported to an anaerobic chamber for further processing within one hour. The caeca were opened in an anaerobic chamber (10% CO2, 5% H2 and 85% N2 atmosphere; Concept 400, Baker Ruskinn, USA) and 0.5 g of content was squeezed into 4.5 ml pre-reduced PRAS dilution blank (0.1 g magnesium sulfate heptahydrate, 0.2 g monobasic potassium phosphate, 0.2 g potassium chloride, 1.15 g dibasic sodium phosphate, 3.0 g sodium chloride, 1.0 g sodium thioglycolate, 0.5 g L-cysteine, 1000 ml distilled water; final pH 7.5 +/− 0.2 at 25 °C) and mixed thoroughly. All samples were serially diluted in pre-reduced PRAS dilution blank and plated on Wilkins-Chalgren anaerobe agar (WCHA) (Oxoid) supplemented with 30% of rumen fluid. The rumen fluid was collected from cows by an oral probe, filtered through cheesecloth, centrifuged at 8000 g for 30 min and sterilised by filtration through a 0.22 μm filter. Aliquots of rumen fluid were stored at − 20 °C. WCHA was additionally supplemented with 5 mg/l hemin, 1 mg/l cellobiose, 0.5 g/l soluble starch, 1 mg/ml maltose, 0.2 ml vitamin K1 solution (0.1 ml of filter sterilized vitamin K1 in 20 ml 95% ethanol) and 0.5 mg/ml L-cysteine. Approx. 10 well-separated colonies of different morphology were selected from each agar plate after a five-day incubation at 37 °C and purified by subculture on WCHA. All isolates were stored at − 80 °C in pre-reduced PRAS dilution blank containing glycerol at 20% concentration and equal volume of sterile sheep blood. Sensitivity of pure anaerobe cultures to air oxygen exposure was tested exactly as described elsewhere [6]. Briefly, bacterial cultures were serially diluted in anaerobic chamber and plated on 4 copies of WCHA. One copy of WCHA was left in the anaerobic chamber to determine initial counts of each anaerobe. The remaining 3 copies of WCHA plates were placed into a standard aerobic 37 °C incubator and after 1, 8 and 24 h, a single copy of agar plate was returned back to the anaerobic chamber to check for growth restoration.
Whole genome sequencing
DNA was purified using DNeasy Blood & Tissue Kit (Qiagen). Sequencing library was prepared from 1 ng of RNA-free genomic DNA using the Nextera XT DNA Sample Preparation kit (Illumina) and whole genome sequencing was performed using the NextSeq 500/550 High Output Kit v2 and Illumina NextSeq 500 sequencing platform in the paired-end modus (2 × 150 bp). Raw sequencing reads were quality trimmed using Trimmomatic v0.32 [50] with the sliding window of 4 bp and average quality threshold value equal to 17. Minimal read length was set to 48 bp. Trimmed paired-end reads were assembled via de novo assembler IDBA-UD v1.1.1 [51] with k-mer sizes ranging from 20 to 110 with an increment of 15. Contigs with coverage lower than 10% of average coverage of L50 contigs were filtered out and the remaining contigs were scaffolded employing SSPACE scaffolder v3.0 [52, 53]. All scaffolds containing N’s in their sequences were split into N-free sequences. Finally, scaffolds with a length shorter than 500 bp were discarded.
Species definition and genome annotation
Species definitions used in this study are based on the BLAST comparison of whole 16S rRNA sequences against entries deposited in NCBI 16S rRNA sequence database performed on January 4, 2017. For clarity of the paper, we used the designation of the most similar bacterium based on the lowest E-value for description of our isolates, thus apparently ignoring the fact that in some cases there was 100% identity whilst in the opposite extreme, the sequence of 16S rRNA of one of our isolates was only 83% similar to the closest relative deposited in the NCBI 16S rRNA database (Additional file 1). All 16S rRNA sequences were compared also against RDP SeqMatch database (on January 10, 2017) which allowed for alternative taxonomy including classification of individual isolates into higher taxonomic units. In addition, ribosomal protein multilocus sequence typing (rMLST) [54] and GTDB organism identification (http://gtdb.ecogenomic.org/) databases were used to verify taxonomic classification. Taxonomic classification by these alternative protocols is provided in Additional file 1. Gene predictions and functional annotations were performed by RAST [55]. Assembled and annotated genomes as well as raw sequencing data were deposited in NCBI under accession number PRJNA377666 and genomes with comprehensive RAST annotation are available upon request.
Genome comparison
To exclude genomes of the same isolates picked up on independent occasions from the subsequent analyses, whole genome sequences of all isolates were mutually compared using the QUAST tool v3.1 [56]. Two genomes were considered as identical if they shared more than 99% of genome content and had less than 1 indel per 100 kb. A single isolate was selected as a representative of each group of identical isolates for all downstream analyses.
Whole gene content similarity clustering was compared using Gene Co-Expression Network analysis. This protocol detects genes with similar transcriptional regulatory program (potential members of the same pathway, protein complex, etc.). In our case, the gene expression vector for particular gene was replaced by gene copy-number vector for particular bacteria and the protocol therefore detected bacterial isolates with similar gene content. Interconnected bacteria shared more than 50% of genes based on gene name designation provided by RAST annotation. Whole gene content similarity of individual isolates was analysed in R. At first, matrix of Spearman’s correlation coefficients was calculated for all pairs of isolates using vectors of respective gene counts. The correlation matrix was then transformed to the adjacency matrix using threshold of + 0.5 as a cut-off for two vertices to be considered as connected. The undirected network was constructed from such a matrix using igraph package (http://igraph.org/r/), and edge betweenness based community structure detection algorithm was then employed to identify separate network modules. Communities with more than three members were considered nontrivial and were highlighted in a network plot.
Analysis of 16S rRNA genes
Trimmed reads were aligned against SILVA bacterial 16S rRNA database using SortMeRNA v2.1 [57] and extracted 16S rRNA reads were assembled via de Bruijn graph-based de novo assembler SPAdes v3.6.0 [58]. Finally, sequences coding for 16S rRNA genes were predicted employing barrnap tool v0.6 (https://github.com/tseemann/barrnap). For the purposes of phylogenetic analysis, 16S rRNA genes were aligned by ClustalW v2.1 [59] using default gap penalties, DNA weight matrix IUB and transition weight 0.2. The phylogenetic tree topologies were inferred employing Bayesian statistics via MrBayes v3.2.6 [60] using the parameters as follows: mixed model of nucleotide substitution, gamma-distributed rates among sites, four Monte Carlo Markov chains for 7,000,000 cycles which were sampled every 1000th generation and the first 25% of the samples were discarded as burn-in. The final tree topology was generated employing 50% majority-rule consensus. Average standard deviation of split frequencies was 0.0083, maximum standard deviation of split frequencies was 0.1095, average potential scale reduction factor was 1.000 and maximum potential scale reduction factor was 1.015. The final tree topology was visualized by iTOL v3.4.3 [61] and edited using Inkscape v0.91 (www.inkscape.org).
Additional files
List of 133 different isolates characterised in this study. The file contains taxonomical classification using NCBI and RDP databases based on whole sequence of 16S rRNA, similarity to the closest relative in the NCBI database, genome size, number of contigs into which the genome was assembled, genomic and 16S rRNA GC content, consensus 16S rRNA sequences with SNP positions indicated with lower case letters, 16S rRNA copy number estimated based on sequencing coverage and 16S rRNA copy number determined by RT PCR (only for the isolates in which sequencing coverage predicted more 10 copies of 16S rRNA genes). (XLSX 338 kb)
Phylogenetic tree of 133 sequenced isolates obtained from chicken caecum based on the Clustal alignment of the full-length sequence of RpoB proteins. Families within the phylum Firmicutes are shown in light blue, green and yellow. Families within the phylum Bacteroidetes are shown in shades of purple. The whole genome size and genomic GC content of each isolate is shown external to the dendrogram. (PDF 576 kb)
Aerobic survival of chicken gut anaerobes. (XLS 43 kb)
List of proteins encoded by individual anaerobes. (XLSX 7930 kb)
Distribution of genes in selected categories among representatives of major gut colonisers belonging to phyla Bacteroidetes and Firmicutes. X axes indicate the numbers of genes in a given category per genome. (PDF 23 kb)
Acknowledgements
Authors would like to thank Peter Eggenhuizen for English language corrections.
Funding
This work has been supported by project RO0517 from the Czech Ministry of Agriculture, AdmireVet project CZ.1.05/2.1.00/01.0006–ED0006/01/01 from the Czech Ministry of Education, Czech Science Foundation project 15-11688S and project QJ1310019 from the Czech Ministry of Agriculture. AC was supported by CEITEC 2020 (LQ1601) from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II. The funders had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Assembled and annotated genomes as well as raw sequencing data were deposited in NCBI under accession number PRJNA377666. Genomes with comprehensive RAST annotation are available upon request.
Abbreviations
- ABC transporter
ATP-binding cassette transporter
- batABCDE operon
Bacteroides aerotolerance operon
- BUK
Butyrate kinase
- ECF class transporter
Energy-coupling factor transporter
- GABA
γ-aminobutyrate
- GTDB
Genome taxonomy database
- NCBI
National Center for Biotechnology Information
- OTU
Operational taxonomic unit
- PBT
Phosphate butyryltransferase
- PRAS dilution blank
Pre-reduced anaerobically sterilized dilution blank
- RAST
Rapid annotations using subsystems technology
- RDP
Ribosomal database project
- rMLST
Ribosomal protein multilocus sequence typing
- TRAP transporter
Tripartite ATP-independent periplasmic transporter
- WCHA
Wilkins-Chalgren anaerobe agar
Authors’ contributions
MM participated in the sequence alignment, performed bioinformatic analysis and drafted the manuscript. DC performed the whole genome sequencing and sequence alignment. OP analysed sequencing data for protein coding genes and their functions. DK and TK subcultured bacterial isolates, and purified and checked the DNA for whole genome sequencing. AC was responsible for anaerobic culture, preliminary strain identification by mass spectrometry and maintenance of laboratory strain collection. IR participated in the design of the study, analysed data and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The handling of animals in this study was performed in accordance with current Czech legislation (Animal protection and welfare Act No. 246/1992 Coll. of the Government of the Czech Republic). Experiments performed in this study were approved by the Ethics Committee of the Veterinary Research Institute (permit number 4/2016) followed by the Committee for Animal Welfare of the Ministry of Agriculture of the Czech Republic. The animals used in this study originated from commercial sources. Their use for anaerobic gut microbiota culture was based on verbal consent of private owners due to continuous collaboration between authors’ laboratories and individual owners.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Matej Medvecky, Email: medvecky@vri.cz.
Darina Cejkova, Email: cejkova@vri.cz.
Ondrej Polansky, Email: polansky@vri.cz.
Daniela Karasova, Email: karasova@vri.cz.
Tereza Kubasova, Email: kubasova@vri.cz.
Alois Cizek, Email: cizeka@vfu.cz.
Ivan Rychlik, Phone: +420 533331201, Email: rychlik@vri.cz.
References
- 1.Gosalbes MJ, Durban A, Pignatelli M, Abellan JJ, Jimenez-Hernandez N, Perez-Cobas AE, Latorre A, Moya A. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One. 2011;6:e17447. doi: 10.1371/journal.pone.0017447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, Taylor TD, Noguchi H, Mori H, Ogura Y, Ehrlich DS, Itoh K, Takagi T, Sakaki Y, Hayashi T, Hattori M. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marx V. Microbiology: the return of culture. Nat Methods. 2016;14:37–40. doi: 10.1038/nmeth.4107. [DOI] [PubMed] [Google Scholar]
- 5.Lau JT, Whelan FJ, Herath I, Lee CH, Collins SM, Bercik P, Surette MG. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 2016;8:72. doi: 10.1186/s13073-016-0327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD. Culturing of 'unculturable' human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533:543–546. doi: 10.1038/nature17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polansky O, Sekelova Z, Faldynova M, Sebkova A, Sisak F, Rychlik I. Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Appl Environ Microbiol. 2016;82:1569–1576. doi: 10.1128/AEM.03473-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Videnska P, Sedlar K, Lukac M, Faldynova M, Gerzova L, Cejkova D, Sisak F, Rychlik I. Succession and replacement of bacterial populations in the caecum of egg laying hens over their whole life. PLoS One. 2014;9:e115142. doi: 10.1371/journal.pone.0115142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varmuzova K, Kubasova T, Davidova-Gerzova L, Sisak F, Havlickova H, Sebkova A, Faldynova M, Rychlik I. Composition of gut microbiota influences resistance of newly hatched chickens to Salmonella Enteritidis infection. Front Microbiol. 2016;7:957. doi: 10.3389/fmicb.2016.00957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rantala M, Nurmi E. Prevention of the growth of Salmonella infantis in chicks by the flora of the alimentary tract of chickens. Br Poult Sci. 1973;14:627–630. doi: 10.1080/00071667308416073. [DOI] [PubMed] [Google Scholar]
- 11.Duggett NA, Kay GL, Sergeant MJ, Bedford M, Constantinidou CI, Penn CW, Millard AD, Pallen MJ. Draft genome sequences of six novel bacterial isolates from chicken ceca. Genome Announc. 2016;4:e00448–e00416. doi: 10.1128/genomeA.00448-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, Whitman WB, Euzeby J, Amann R, Rossello-Mora R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–645. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 13.Adekambi T, Drancourt M, Raoult D. The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 2009;17:37–45. doi: 10.1016/j.tim.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Fleming SE, Fitch MD, DeVries S, Liu ML, Kight C. Nutrient utilization by cells isolated from rat jejunum, cecum and colon. J Nutr. 1991;121:869–878. doi: 10.1093/jn/121.6.869. [DOI] [PubMed] [Google Scholar]
- 15.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carbonero F, Benefiel AC, Gaskins HR. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat Rev Gastroenterol Hepatol. 2012;9:504–518. doi: 10.1038/nrgastro.2012.85. [DOI] [PubMed] [Google Scholar]
- 17.Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci U S A. 2013;110:13582–13587. doi: 10.1073/pnas.1312524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One. 2014;9:e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geissinger O, Herlemann DP, Morschel E, Maier UG, Brune A. The ultramicrobacterium "Elusimicrobium minutum" gen. Nov., sp. nov., the first cultivated representative of the termite group 1 phylum. Appl Environ Microbiol. 2009;75:2831–2840. doi: 10.1128/AEM.02697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Domingo MC, Yansouni C, Gaudreau C, Lamothe F, Levesque S, Tremblay C, Garceau R. Cloacibacillus sp., a potential human pathogen associated with bacteremia in Quebec and New Brunswick. J Clin Microbiol. 2015;53:3380–3383. doi: 10.1128/JCM.01137-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Looft T, Levine UY, Stanton TB. Cloacibacillus porcorum sp. nov., a mucin-degrading bacterium from the swine intestinal tract and emended description of the genus Cloacibacillus. Int J Syst Evol Microbiol. 2013;63:1960–1966. doi: 10.1099/ijs.0.044719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes LC, Poppleton D, Klingl A, Criscuolo A, Dupuy B, Brochier-Armanet C, Beloin C, Gribaldo S. Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes. Elife. 2016;5:e14589. doi: 10.7554/eLife.14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer M, Zhang QY, Hubbard RE, Thomas GH. Caught in a TRAP: substrate-binding proteins in secondary transport. Trends Microbiol. 2010;18:471–478. doi: 10.1016/j.tim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Rodionov DA, Hebbeln P, Eudes A, ter Beek J, Rodionova IA, Erkens GB, Slotboom DJ, Gelfand MS, Osterman AL, Hanson AD, Eitinger T. A novel class of modular transporters for vitamins in prokaryotes. J Bacteriol. 2009;191:42–51. doi: 10.1128/JB.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slotboom DJ. Structural and mechanistic insights into prokaryotic energy-coupling factor transporters. Nat Rev Microbiol. 2014;12:79–87. doi: 10.1038/nrmicro3175. [DOI] [PubMed] [Google Scholar]
- 26.Kubasova T, Davidova-Gerzova L, Merlot E, Medvecky M, Polansky O, Gardan-Salmon D, Quesnel H, Rychlik I. Housing systems influence gut microbiota composition of sows but not of their piglets. PLoS One. 2017;12:e0170051. doi: 10.1371/journal.pone.0170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivarsson E, Roos S, Liu HY, Lindberg JE. Fermentable non-starch polysaccharides increases the abundance of Bacteroides-Prevotella-Porphyromonas in ileal microbial community of growing pigs. Animal. 2014;8:1777–1787. doi: 10.1017/S1751731114001827. [DOI] [PubMed] [Google Scholar]
- 28.Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, Osterman AL, Gordon JI. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science. 2015;350:aac5992. doi: 10.1126/science.aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjdia A, Martens EC, Gordon JI, Berteau O. Sulfatases and a radical S-adenosyl-L-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J Biol Chem. 2011;286:25973–25982. doi: 10.1074/jbc.M111.228841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nihira T, Suzuki E, Kitaoka M, Nishimoto M, Ohtsubo K, Nakai H. Discovery of beta-1,4-D-mannosyl-N-acetyl-D-glucosamine phosphorylase involved in the metabolism of N-glycans. J Biol Chem. 2013;288:27366–27374. doi: 10.1074/jbc.M113.469080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang M, Chekan JR, Dodd D, Hong PY, Radlinski L, Revindran V, Nair SK, Mackie RI, Cann I. Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proc Natl Acad Sci U S A. 2014;111:E3708–E3717. doi: 10.1073/pnas.1406156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magnusdottir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, Greenhalgh K, Jager C, Baginska J, Wilmes P, Fleming RM, Thiele I. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017;35:81–89. doi: 10.1038/nbt.3703. [DOI] [PubMed] [Google Scholar]
- 34.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio. 2014;5:e00889. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anand S, Kaur H, Mande SS. Comparative in silico analysis of butyrate production pathways in gut commensals and pathogens. Front Microbiol. 2016;7:1945. doi: 10.3389/fmicb.2016.01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barketi-Klai A, Hoys S, Lambert-Bordes S, Collignon A, Kansau I. Role of fibronectin-binding protein a in Clostridium difficile intestinal colonization. J Med Microbiol. 2011;60:1155–1161. doi: 10.1099/jmm.0.029553-0. [DOI] [PubMed] [Google Scholar]
- 37.Hennequin C, Janoir C, Barc MC, Collignon A, Karjalainen T. Identification and characterization of a fibronectin-binding protein from Clostridium difficile. Microbiology. 2003;149:2779–2787. doi: 10.1099/mic.0.26145-0. [DOI] [PubMed] [Google Scholar]
- 38.Volf J, Polansky O, Varmuzova K, Gerzova L, Sekelova Z, Faldynova M, Babak V, Medvecky M, Smith AL, Kaspers B, Velge P, Rychlik I. Transient and prolonged response of chicken cecum mucosa to colonization with different gut microbiota. PLoS One. 2016;11:e0163932. doi: 10.1371/journal.pone.0163932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doolittle RF, McNamara K, Lin K. Correlating structure and function during the evolution of fibrinogen-related domains. Protein Sci. 2012;21:1808–1823. doi: 10.1002/pro.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu C, Soderhall K, Soderhall I. Two novel ficolin-like proteins act as pattern recognition receptors for invading pathogens in the freshwater crayfish Pacifastacus leniusculus. Proteomics. 2011;11:2249–2264. doi: 10.1002/pmic.201000728. [DOI] [PubMed] [Google Scholar]
- 41.Palmer JC, Kehoe PG, Love S. Endothelin-converting enzyme-1 in Alzheimer's disease and vascular dementia. Neuropathol Appl Neurobiol. 2010;36:487–497. doi: 10.1111/j.1365-2990.2010.01084.x. [DOI] [PubMed] [Google Scholar]
- 42.Langlois VS, Zhang D, Cooke GM, Trudeau VL. Evolution of steroid-5alpha-reductases and comparison of their function with 5beta-reductase. Gen Comp Endocrinol. 2010;166:489–497. doi: 10.1016/j.ygcen.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 43.Auteri M, Zizzo MG, Serio R. GABA and GABA receptors in the gastrointestinal tract: from motility to inflammation. Pharmacol Res. 2015;93:11–21. doi: 10.1016/j.phrs.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, Raes J, Verberkmoes NC, Fraser CM, Hettich RL, Jansson JK. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn's disease. PLoS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winter SE, Baumler AJ. A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes. 2011;2:58–60. doi: 10.4161/gmic.2.1.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berry D, Schwab C, Milinovich G, Reichert J, Ben Mahfoudh K, Decker T, Engel M, Hai B, Hainzl E, Heider S, Kenner L, Muller M, Rauch I, Strobl B, Wagner M, Schleper C, Urich T, Loy A. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012;6:2091–2106. doi: 10.1038/ismej.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One. 2011;6:e27949. doi: 10.1371/journal.pone.0027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 52.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 53.Hunt M, Newbold C, Berriman M, Otto TD. A comprehensive evaluation of assembly scaffolding tools. Genome Biol. 2014;15:R42. doi: 10.1186/gb-2014-15-3-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MC. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 58.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 60.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 61.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of 133 different isolates characterised in this study. The file contains taxonomical classification using NCBI and RDP databases based on whole sequence of 16S rRNA, similarity to the closest relative in the NCBI database, genome size, number of contigs into which the genome was assembled, genomic and 16S rRNA GC content, consensus 16S rRNA sequences with SNP positions indicated with lower case letters, 16S rRNA copy number estimated based on sequencing coverage and 16S rRNA copy number determined by RT PCR (only for the isolates in which sequencing coverage predicted more 10 copies of 16S rRNA genes). (XLSX 338 kb)
Phylogenetic tree of 133 sequenced isolates obtained from chicken caecum based on the Clustal alignment of the full-length sequence of RpoB proteins. Families within the phylum Firmicutes are shown in light blue, green and yellow. Families within the phylum Bacteroidetes are shown in shades of purple. The whole genome size and genomic GC content of each isolate is shown external to the dendrogram. (PDF 576 kb)
Aerobic survival of chicken gut anaerobes. (XLS 43 kb)
List of proteins encoded by individual anaerobes. (XLSX 7930 kb)
Distribution of genes in selected categories among representatives of major gut colonisers belonging to phyla Bacteroidetes and Firmicutes. X axes indicate the numbers of genes in a given category per genome. (PDF 23 kb)
Data Availability Statement
Assembled and annotated genomes as well as raw sequencing data were deposited in NCBI under accession number PRJNA377666. Genomes with comprehensive RAST annotation are available upon request.