

Abstract
A 58-year-old man with limited cutaneous systemic sclerosis and myositis overlap complicated by interstitial lung disease presented with several months of progressive dyspnoea and weakness. CT chest revealed extensive pneumomediastinum that was not present on imaging 6 months before this study and appeared to be spontaneous, with no preceding trauma, infection or invasive procedures.
Keywords: interstitial lung disease, connective tissue disease
Background
Interstitial lung disease (ILD) is frequently described in a variety of rheumatological diseases, especially connective tissue diseases such as scleroderma and dermatomyositis/polymyositis. Spontaneous pneumomediastinum is an unusual and rare complication of ILD. Few case reports have described pneumomediastinum in the setting of systemic sclerosis. We believe this case is unusual in that it describes a limited cutaneous systemic sclerosis with myositis overlap and the occurrence of spontaneous pneumomediastinum associated with ILD. The case is also important because it highlights the need to consider pneumomediastinum as a cause of acute or subacute dyspnoea in patients with ILD or myositis.
Case presentation
A 58-year-old man had been diagnosed with limited cutaneous systemic sclerosis 13 years earlier based on the findings of sclerodactyly, telangiectasias, Raynaud’s phenomenon, gastro-oesophageal reflux and positive antinuclear antibody (1:640 with speckled pattern). He had suspicion for overlapping myositis given history of proximal weakness, though did not undergo a full workup as his symptoms improved with concurrent scleroderma treatment. Through 2016, he was maintained on mycophenolate mofetil 500 mg two times per day and low-dose prednisone with good control of his disease. Beginning in January 2017, he noted progressive dyspnoea on exertion and worsening lower extremity weakness. High resolution CT scan of the chest in April 2017 showed ground glass opacities without bronchiectasis in the lung bases, concerning for new ILD. There was no presence of subpleural cysts or bullae on the CT scan. Pulmonary function testing in June 2017 demonstrated forced expiratory volume in 1 s 47% predicted, forced vital capacity 40% predicted and DLCO 28% predicted, all of which were worsened from 1 year prior. At this time, his mycophenolate mofetil was increased in dose to 1500 mg two times per day from 500 mg two times per day and he was initiated on prednisone 40 mg daily, with plans for a slow taper. In September 2017, he was found to be hypoxaemic on ambulation and he was initiated on 2 L home oxygen therapy. Cardiac stress testing and echocardiography in 2016 had been unremarkable. Medical history was otherwise significant for coronary artery disease, hyperlipidaemia (on atorvastatin 40 mg) and hypertension. On presentation, he was short of breath with minimal activity of repositioning in bed and pulmonary examination was notable for mid-inspiratory crackles at lung bases. He had sclerodactyly of all digits of bilateral hands with erythematous cuticles without digital pits or ulceration. He had tightness of perioral region with decreased oral aperture and telangiectasias of the face. Musculoskeletal examination was notable for proximal weakness (4/5) of hip flexors and deltoids, with intact (5/5) distal muscle strength.
Investigations
CT (figure 1) showed significant pneumomediastinum tracking into the neck without clear aetiology. The lungs showed extensive diffuse ground-glass opacities most prominent in the bilateral lung bases and no honeycomb pattern. Patient was unable to withstand repeat pulmonary function testing while inpatient. Echocardiogram with bubble study showed preserved ejection fraction and evidence of intrapulmonary shunt; PET scan and ventilation/perfusion scan were unremarkable.
Figure 1.
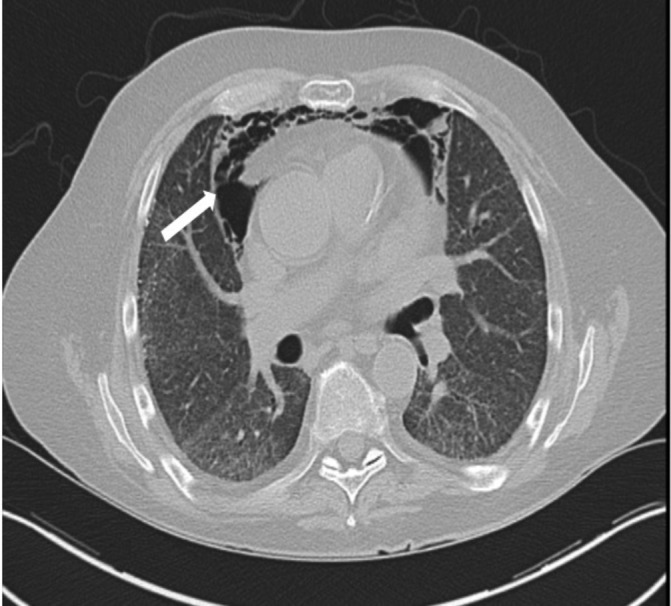
Arrow points to anterior pneumomediastinum. The lung fields show non-specific interstitial pneumonia pattern of pulmonary fibrosis without area of consolidation.
Laboratory tests were notable for mildly elevated erythrocyte sedimentation rate (23) and C reactive protein (3.3). Creatine kinase, liver enzymes and completes were within normal limits. His serology was notable for negative anti-double stranded DNA, Ro/La, antineutrophil cytoplasmic antibodies, rheumatoid factor and paraneoplastic panel. Myositis antibody panel revealed incidentally noted anti-RNA-polymerase 3 antibodies that was confirmed on ELISA. Prior panel also had presence of Mi-2 antibodies though not seen on repeat testing.
Thigh MRI revealed global pelvic and thigh muscle atrophy with multifocal sites of fatty replacement. Muscle biopsy showed mild acute and chronic neurogenic atrophy, striking perifascicular atrophy without myofiber degeneration, and lobulated myofibers on oxidative stains. There was no primary inflammation, but there was mild fascial oedema; taken together, these findings were consistent with neuromyopathy, and specifically, dermatomyositis without concomitant skin findings.
Differential diagnosis
The patient presented with subacute symptoms of dyspnoea, found to be caused by a pneumomediastinum on imaging, with likely contributions from progression of ILD as well. The cause of his atraumatic pneumomediastinum may have been a result of ruptured subpleural cyst or worsening of his ILD. Evaluation for infection, including bacterial and fungal (including pneumocystis pneumonia), is critical in the differential for all patients on immunosuppressing medications. Additionally, his ILD and myositis symptoms should prompt consideration of occult malignancy.
Outcome and follow-up
The patient was admitted for shortness of breath in the setting of pneumomediastinum. He had progressive hypoxia during the prolonged admission requiring high-flow nasal cannula and required intubation on hospital day 27 for hypoxic respiratory failure. He developed septic shock with Klebsiella pneumonia and rapidly progressive renal failure. After discussions with family, the patient was terminally extubated per the patient’s goals of care.
Discussion
Autoimmune and connective tissue diseases can present with multiple organ system involvement. Pulmonary involvement, particularly ILD, can present as a serious manifestation of diseases including polymyositis or dermatomyositis, rheumatoid arthritis, scleroderma, systemic lupus erythematous, Sjögren’s syndrome and mixed connective tissue disease.1 The aetiology for the development of ILD in scleroderma is not fully understood, but thought to be related to inflammation and vascular injury leading to fibrotic changes within the lung parenchyma.2 ILD is often an early manifestation of disease in scleroderma, usually presenting in the first 5 years of scleroderma symptom onset.3 In our case presentation, however, ILD was recognised 13 years after initial scleroderma diagnosis in this patient without antibodies to Scl-70. In addition to a diagnosis of systemic sclerosis, the patient we present also had overlap symptoms and imaging/histological findings of myositis. Like scleroderma, ILD is a common manifestation of myositis with prevalence between 20% and 78%.1
Though ILD is a relatively common manifestation of rheumatological diseases, spontaneous pneumomediastinum is a very rare complication. Few case reports describe the development of pneumomediastinum in systemic sclerosis.4–8 These case reports describe four spontaneous pneumomediastinum cases, one of which developed after pulmonary function testing.6 Four patients presented with acute onset of dyspnoea and one case report described an asymptomatic patient with pneuomediastinum noted on imaging.7 We report the first presentation of pneumomediastinum in scleroderma presenting as subacute dyspnoea.
Spontaneous pneumomediastinum is also a rare complication of myositis. Le Goff et al describe the only case series of patients with dermatomyositis and polymyositis presenting with pneumomediastinum, with 11 cases identified through a nationwide survey of French rheumatologists.9 Of note, one patient in this report had overlap symptoms of scleroderma and dermatomyositis. All patients in the study had underlying ILD. Five patients in this study had pneumomediastinum induced by a triggering factor including cough, pulmonary function testing and infectious pneumonitis. In combining their cases with prior published data, the study authors performed survival analysis of 31 patients. They reported a high mortality rate of 34% in patients with myositis and pneumomediastinum, with 25% of patients dying within the first month of presentation.9
Our case highlights the importance of considering pneumomediastinum as a cause of dyspnoea in patients with scleroderma and/or myositis who present with acute or subacute. This should be of particular concern in patients with known ILD. Published data in myositis patients reflect the importance of recognising pneumomediastinum given its high mortality rate.9
Learning points.
Interstitial lung disease is a risk factor for developing spontaneous pneumomediastinum in the context of connective tissue disease.
A common clinical feature of spontaneous pneumomediastinum is dyspnoea which may either be acute or subacute/chronic.
Patients with limited cutaneous systemic sclerosis with myositis overlap can develop spontaneous pneumomediastinum, and when this condition occurs, morbidity and mortality are high.
Footnotes
Contributors: EJD and KL conducted a literature review for this paper and EJD wrote the primary draft of the report. LH and HT revised and edited the case report to create the final draft.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ 2016;352:h6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Au K, Khanna D, Clements PJ, et al. Current concepts in disease-modifying therapy for systemic sclerosis-associated interstitial lung disease: Lessons from clinical trials. Curr Rheumatol Rep 2009;11:111–9. 10.1007/s11926-009-0016-2 [DOI] [PubMed] [Google Scholar]
- 3.Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390:1685–99. 10.1016/S0140-6736(17)30933-9 [DOI] [PubMed] [Google Scholar]
- 4.Haroon M, McLaughlin P, Henry M, et al. Spontaneous pneumomediastinum in a patient with anti-centromere antibody-positive limited scleroderma. J Clin Rheumatol 2011;17:42–3. 10.1097/RHU.0b013e3182055d5e [DOI] [PubMed] [Google Scholar]
- 5.Mohammad A, Boon Low T, O’Dwyer D, Low TB, O’Dwyer D, et al. Spontaneous Pneumo-mediastinum in systemic sclerosis a case report. Rheumatology 2007;46:1376–7. 10.1093/rheumatology/kem086 [DOI] [PubMed] [Google Scholar]
- 6.Jun JB, Song SY. The development of pneumomediastinum after pulmonary function testing in a patient with systemic sclerosis. Rheumatol Int 2007;27:1097–8. 10.1007/s00296-007-0369-7 [DOI] [PubMed] [Google Scholar]
- 7.Honne K, Maruyama A, Onishi S, et al. Simultaneous pneumatosis cystoides intestinalis and pneumomediastinum in a patient with systemic sclerosis. J Rheumatol 2010;37:2194–5. 10.3899/jrheum.100254 [DOI] [PubMed] [Google Scholar]
- 8.Almeida M, Dias LT, Fernandes SJS, et al. Spontaneous pneumomediastinum and subcutaneous emphysema in systemic sclerosis. Rheumatol Int 2007;27:675–7. 10.1007/s00296-006-0260-y [DOI] [PubMed] [Google Scholar]
- 9.Le Goff B, Chérin P, Cantagrel A, et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum 2009;61:108–18. 10.1002/art.24372 [DOI] [PubMed] [Google Scholar]