

Abstract
Background
The complexity of delivering precision medicine to oncology patients has led to the creation of molecular tumourboards (MTBs) for patient selection and assessment of treatment options. New technologies like the liquid biopsy are augmenting available therapeutic opportunities. This report aims to analyse the experience of our MTB in the implementation of personalised medicine in a cancer network.
Materials and methods
Patients diagnosed with solid tumours progressing to standard treatments were referred to our Phase I unit. They underwent comprehensive next generation sequencing (NGS) of either tumour tissue or cell-free circulating tumour DNA (ctDNA) or both. The MTB expressed either a positive or negative opinion for the treatment of the patients with discovered druggable alterations inside a clinical trial, in an expanded access programme, with a compassionate use. Afterwards, discovered alterations were matched with OncoKB levels of evidence for the choice of alteration-specific treatments in order to compare MTB outcomes with a standardised set of recommendations.
Results
NGS was performed either on ctDNA or tumour tissue or in both of them in 204 patients. The MTB evaluated 173 of these cases. Overall, the MTB proposed alteration-specific targeted therapy to 72 patients (41.6%). 49 patients (28.3% of the total evaluated) were indicated to enter a clinical trial. In 29 patients with matched liquid biopsy NGS (lbNGS), tumour tissue NGS (ttNGS) and MTB evaluation, the MTB changed the treatment strategy coming from standardised recommendations based on lbNGS and ttNGS alone in 10 patients (34.5%), thanks to the evaluation of other clinical parameters. In our cohort, lbNGS was more likely, compared with ttNGS, to detect point mutations (OR 11, 95% CI 2.9 to 24.1, p<0.001) and all-type alterations (OR 13.6, 95% CI 5.5 to 43.2, p<0.001) from the same genes of matched patients.
Conclusions
Our MTB allows patients with refractory cancer to be included in clinical trials and improves the precision of clinical decisions compared with a standardised set of mutation-driven recommendations.
Keywords: molecular tumor board, liquid biopsy, Ngs, precision oncology, precision medicine
Key questions.
What is already known about this subject?
The use of both molecular tumour board and liquid biopsy has already been described in the clinical practice. It is acknowledged that molecular tumour boards can be used for the selection of treatment following the use of NGS panels from solid tumour tissue DNA. Also, it has already been shown that the next generation sequencing (NGS) performed in the circulating DNA can reach the same level of sensitivity for the detection of genetic alterations compared with the NGS performed on DNA coming from solid tumour biopsies or gross masses.
What does this study add?
We were the first to compare the impact of the molecular tumour board decision to a set of standardised list of evidence level for the choice of alteration-driven recommendations. Also, we were the first to describe the activity of a molecular tumour board which used the results of NGS performed on circulating DNA. Finally, this study shows a real-life comparison between NGS panels performed on circulating DNA and DNA from solid tumour biopsies or gross tumour masses.
How might this impact on clinical practice?
Physicians might want to implement molecular tumour boards in their clinical practice.
Physicians might choose to prefer the use of liquid biopsy DNA over the use of DNA from old formalin-fixed paraffin-embedded samples.
Introduction
With the advent in clinic of next generation sequencing (NGS) panels, an increasing amount of therapeutic choices have become available, particularly for patients with limited standard of care options.1 NGS panels can provide the treating physician with a variably extensive list of druggable alterations that can subsequently lead to the enrolment of that patient in a trial, treatment of that patient in an expanded access programme or, also, with an off-label indication. Treating physicians, however, are not always trained to interpret the reports of these analyses. Also, treatment options associated with newly discovered druggable alterations have often limited evidence of efficacy in the daily practice.2 As a result, the treating physician has to carefully evaluate the potential benefits of a clinical trial or of an off-label therapy taking into consideration other therapeutic choices, particularly best supportive care.
Targeted therapy in non-small cell lung cancer (NSCLC) based on identification of seven genomic targets has been shown to double overall survival in academic and community settings.3 4 However, the use of NGS panels in a broader array of cancer types has proven to improve the patients’ outcomes in some studies but not in others.5 The Molecular Screening for Cancer Treatment and Optimisation trial showed that patients treated with off-label alteration-driven treatments had improved progression free survival compared with their previous treatments.6 However, in the SHIVA trial, no difference were observed in overall survival for patients randomised to receive either alteration-driven targeted therapy or physician-choice chemotherapy.7 Taking into consideration, these contradictory clues we believe that, in order to fully exploit the potentials of NGS driven therapy, a formal entity bringing interdisciplinary expertise into evaluation of patients with advanced cancer should exist to indicate when alteration-driven treatment is advisable.
The complexity of delivering precision medicine to oncology patients in the context of a public healthcare system further suggested the need to create molecular tumour boards (MTBs) to assess when extensive molecular testing for tumour profiling was appropriate and, also, to discuss therapeutic chances of patients with newly discovered druggable alterations.8 It has already been shown in a diverse set of clinical scenarios that interdisciplinary tumour boards can result in significant changes in treatment decisions.9–11 However, aside from changes in treatment plans and increased enrolment of patients in clinical trials, the impact of MTBs on treatment outcomes has not yet been studied.12–14
We expected that the MTB could improve the precision of a single physician choice based on outcomes of high throughput molecular testing. The aim of this study was to retrospectively measure the impact of MTB discussion on physician decision-making regarding treatment in patients with NGS panels data available. To test it, we compared how evidence-based recommendations for druggable alterations match with the indications of our MTB. Further, we compared lbNGS and ttNGS ability to provide the physician with valid treatment options in patients with no further therapeutic options available.
Methods
Patient recruitment
All the patients referred to our Phase I unit of the oncology department of the Antwerp University Hospital from May 2013 to September 2017 were eligible for the participation in the study. During the visit, NGS sequencing was proposed to patients who could potentially benefit from finding of druggable alterations. The criterion for preferring lbNGS over ttNGS was the absence of sufficient tumour tissue and the clinical unfeasibility of a re-biopsy. In patients with both lbNGS and ttNGS, the former was proposed to the patient in case the latter did not provide convincing results. Patients with a life expectancy of less than 3 months or with other very valid therapeutic chances were excluded. No restrictions were applied related to age or tumour type. Notably, for some patients for which chemotherapeutic agents with an expected low clinical benefit were available, molecular analysis were considered anyway, and, eventually, these patients started a new alteration-driven treatment either immediately—thus stopping chemotherapy—or as soon as they progressed from chemotherapy; this decision was taken, of course, according to the opinion of the MTB. These molecular gene panels were not reimbursed by insurance companies in Belgium. On their first visit to our unit, patients were informed about the potentials and the limitations that these techniques could offer for the treatment selection. After ensuring that the patients had understood the information received, they signed the corresponding informed consent and then the tumour sample was sent for processing.
Circulating free tumour DNA sequencing
The commercial platform Guardant360 was used to detect alterations of 73 related cancer genes15 in cell-free circulating tumour DNA (ctDNA) in a Clinical Laboratory Improvements Act-licensed, College of American Pathologists-accredited, New York State Department of Health-approved laboratory (Guardant Health, Redwood City, California, USA). The methods for ctDNA isolation and sequencing are extensively described elsewhere.16 Briefly, two blood samples were collected for each patient in 10 mL Streck Cell-Free DNA Blood Collection (Streck) tubes; these tubes, kept in room temperature, ensure high-quality preservation of cell-free (cfDNA) longer (up to 7 days at room temperature) than EDTA tubes17 and they proved to maintain cfDNA quality in 3 days long shipping-like experimental conditions (an important consideration as our cancer centre is a continent away).18 CfDNA was isolated from plasma, concentrated with Agencourt Ampure XP beads and finally prepared for sequencing. The minimal amount required for library preparation was 5 ng.16 After molecular barcoding, massively parallel NGS was conducted on an Illumina HiSeq, followed by bioinformatic error suppression of false positives. After complete sequencing of critical exons in target genes, all four major types of genomic alterations were reported, single nucleotide variants (SNVs) in all genes, and insertions and deletions (indels), fusions and copy number amplifications in selected genes. Variant allele fraction was reported as the percentage of mutated DNA molecules divided by the total cfDNA molecules at a given genomic position.
Tumour tissue DNA sequencing
Tumorous DNA was extracted from patient resection specimens or biopsies by the following procedure. Based on H&E slides, the pathologist selected the paraffin block with the most dense tumour, designated a region of interest (ROI) and estimated the tumorous cell fraction from this ROI. Only samples with a tumorous cell fraction of >10% were included. Macrodissection of the designated area was performed from 10 unstained slides, and DNA was extracted by means of the QIAamp DNA Mini QIAcube Kit making use of the QIAcube device according to the manufacturer’s instructions (Qiagen). Subsequently, sample preparation and target capture were performed by means of the ClearSeq HS Target Enrichment System (Agilent Technologies), enabling detection of relevant regions from the following 47 genes: ABL1, AKT1, ALK, AR, ATM, BRAF, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FANCA, FANCC, FANCF, FANCG, FGFR1, FGFR2, FGFR3, FLT3, HRAS, IDH1, IDH2, JAK2, JAK3, KIT, KRAS, MAP2K1, MAP2K2, MAP2K4, MET, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PIK3R1, PTEN, RET, RUNX1, SMAD4, SMD, SRC, STK11, TP53, VHL and WT1. Pooled samples were sequenced on a MiSeq Personal Sequencer (Illumina) with a maximum of 24 samples (+external control) per run. Data analysis was performed with BaseSpace and SeqNext for calling and annotation of variants.
MTB meetings
The molecular tumour board (MTB) met every 2 weeks. Regular attendees consisted of medical oncologists, general and molecular pathologists, a bioinformatics expert, a molecular biologist, a geneticist and a nurse navigator. If needed, specialists in specific areas, such as gynaecologists, thoracic surgeons or similar were invited for further expertise in specific cases. Each case was discussed in multidisciplinary group sessions. Apart from clinical data, the MTB took into consideration the results from NGS panels and, when available, also data coming from other molecular analysis such as fluorescent in situ hybridisation, immunohistochemistry and PCR. For each case, the MTB discussed which was the best treatment and expressed a recommendation accordingly. For patients discovered with druggable alterations, the different indications comprised standard of care treatments, enrolment in a clinical trial, expanded access programmes or compassionate use. Patients without any evidence of druggable alterations were evaluated for alternative therapeutic chances and, eventually, referred to their reference centre for best supportive care.
Comparison of MTB and NGS outcomes
In order to evaluate the impact of MTB decisions, we decided to compare its outcomes with a set of standardised evidence levels for the treatment of patients with specific alterations. To this point, we downloaded the OncoKB19 actionable variants list that comprises (October 2017) a list of 390 recommendations for 77 actionable genes and six recommendations for resistance to therapy. Recommendations level goes from 1 to 4 and are reported in online supplementary table 1. We matched each alteration with its corresponding level of evidence for that particular cancer type. Afterwards, we determined which was the treatment with highest level evidence provided to each patient based on his/her actual alterations. In our work, this value represented a standardised score to assess the likelihood of the patient to undergo an alteration-driven treatment—logically, the higher is the level of evidence for an alteration-driven treatment associated to a patient’s mutation, the more appropriate would be for that patient to undergo that specific treatment. Nevertheless, this score was ‘blind’ far what concerns the evaluation of every single clinical scenario details: such an evaluation is supposed to be the adjunct contribution of our MTB.
esmoopen-2018-000398supp002.pdf (296.3KB, pdf)
Statistical analysis and data handling
Mean, median and/or 95% CI were reported whenever deemed appropriate. Absolute value and frequency were reported in descriptive tables. Mann-Whitney test was used to compare differences of distributions for independent non-parametric sets of data. Pearson χ2 was used to compare difference in proportions for independent cohorts; Fisher’s exact test was used in place of Pearson χ2 when dealing with small and/or very asymmetric subsamples. McNemar χ2 was used to compare difference in proportions for paired sample. Univariate linear regression was used to assess dependency of independent, continuous, normally distributed variables; log transformation was applied when deemed appropriate to better fit the linear model. All recorded data were imported and analysed in R V.3.4.1.20 Most of the graphics were elaborated with R package ggplot2.21
Results
Patients characteristics
We performed the liquid biopsy NGS (lbNGS) on 53 samples from 46 patients from November 2016 to September 2017 with Guardant 360 platform. lbNGS population was particularly enriched for NSCLC (n=15, 30.4%). Tumour tissue NGS (ttNGS) was performed on 195 samples from 186 patients. The median number of genes sequenced for each ttNGS sample was 47 (range: 1–96; in 75% of the patients the number of sequenced gene was between 44 and 49) (online supplementary figure 1). Most common cancer types in the ttNGS cohort were lung cancer (n=37, 19.9%), colorectal cancer (n=26, 14.0%) and pancreatic cancer (n=22, 11.9%). Comprehensive patients characteristics are listed in online supplementary table 2. Twenty-nine patients received both at least one ttNGS and one lbNGS determination.
esmoopen-2018-000398supp001.pdf (282.1KB, pdf)
lbNGS outcomes
We detected 321 different alterations from lbNGS samples. There were no lbNGS sequencing failures; however, in six samples (11.3%) no alterations could be detected (online supplementary figure 2). The median number of alterations observed in each sample was 3 (range: 0–41). The vast majority were point mutations (n=199, 62.0%) and amplifications (n=94, 29.3%) (online supplementary figure 3). Alterations of TP53 gene were the most frequent (n=39) followed by EGFR (n=25), APC (n=21), PIK3CA (n=20), KRAS (n=18) and MET (n=18) (online supplementary figure 4). The median amount of ctDNA (representing the variant allele fraction) detected in each sample was 0.054% of the total cfDNA (range: 0%–27.6%) (figure 1). Intriguingly, univariate analysis showed a significant positive correlation between the level of ctDNA and the number of alterations detected (1.97 increase in log of total alteration discovered for every unitary increase of log of % of ctDNA; adjusted R2=0.66; p=3.4e−12) (figure 1). Five patients with lung adenocarcinoma (LUAD) underwent two liquid biopsy and one patient with small cell lung cancer underwent three liquid biopsies, each at subsequent lines of progression. Number of alterations was bigger for lung squamous cancer than for LUAD (median difference of nine mutations; Mann-Whitney: p=0.026; two couples of paired samples values were each substituted with the respective average value of the two in order to obtain independent observations). Every alteration was matched with OncoKB evidence level for targeted therapy.19 Figure 2 shows the highest level recommendation that was found for each sample. An R1 alteration (alteration proven to induce resistance to an approved treatment and thus warranting discontinuation of that treatment) was found in 14 out of 53 (26.4%) liquid biopsy samples.
Figure 1.
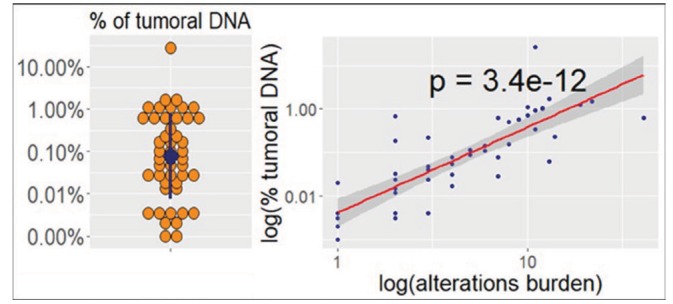
Distribution of cell free tumour DNA between liquid biopsy next generation sequencing samples and correlation with alterations burden. In the dot plot (left), the blue dot and the blue line represent, respectively, the median and the 25th to 75th percentiles range. In the scatterplot (right), X axis and Y axis are inverted.
Figure 2.
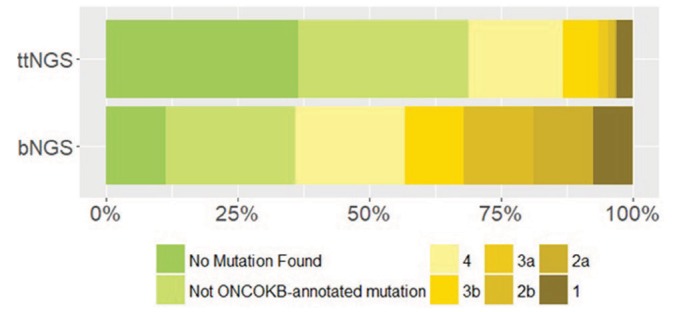
OncoKB evidence levels from liquid biopsy next generation sequencing (lbNGS) (n=53) and tumour tissue NGS (ttNGS) (n=195) in all available samples.
ttNGS outcomes
We detected 264 different alterations from ttNGS samples. The amount of samples with no alterations detected was 61 out of 195 (31.3%); probably, the higher frequency of negative results in ttNGS samples compared with LB was due the lower average number of genes sequenced. The median number of alterations detected with ttNGS was 1 (range: 0–21). TP53, KRAS and APC were the most frequently mutated genes—respectively, 39.8%, 18.9% and 16.4% of the samples in which each gene was sequenced (online supplementary figure 5). Figure 2 shows which was the highest level recommendation that could be given to each sample according to the OncoKB scale. Compared with ttNGS, lbNGS could provide more level ≥3b recommendations (43.4% vs 13.3%; P<0.0001) and level 1 recommendations trended to lower a lower value (7.5% vs 3.1%; Fisher’s exact test: p=0.22); the fact that less genes were sequenced in ttNGS samples compared with lbNGS samples probably plays a role in this difference.
Comparison of ttNGS and lbNGS in matched patients
In total, 29 patients received at least one LB and one NGS sequencing in their primary tumour tissue. In these 29 samples, the mean and the median of sequenced genes in the tumour tissue were, respectively, 34.6 and 47 and only four patients had less than 10 genes sequenced in their tumour tissue.
First, we measured the positive concordance for the detection of alterations in genes sequenced in the same patient with both lbNGS and ttNGS; due to the differences in the panels employed, a direct comparison of lbNGS and ttNGS concordance was possible only for a small subset of sequenced genes. Among these genes, a total of 53 point mutations were detected, respectively, 37 by lbNGS alone, five by ttNGS alone and 11 by both of them (OR 7.4 favouring the use of lbNGS, 95% CI 2.9 to 24.1; McNemar’s χ2: p=4.4×10−7). Thus, lbNGS and ttNGS could detect, respectively, 90.6% and 30.2% of the total point mutations (figure 3). This considerable difference was observed also when considering all kind of alterations (68, 5 and 18 alterations were detected, respectively, by lbNGS alone, ttNGS alone and both lbNGS and ttNGS; OR 13.6, 95% CI 5.5 to 43.2; McNemar’s χ2: p=3.4×10−15).
Figure 3.
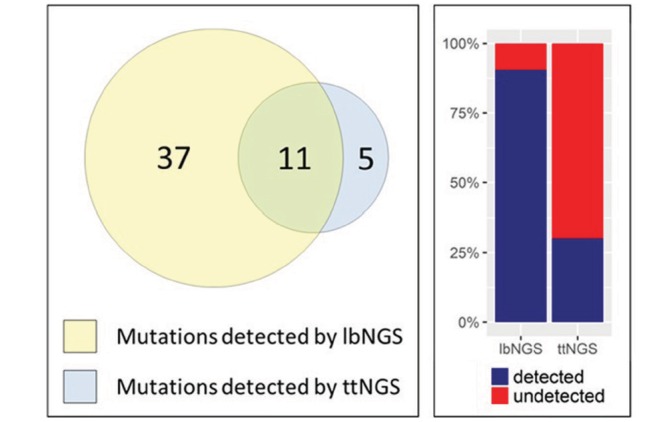
On the left, Euler-Venn diagram of point mutations detected by lbNGS and ttNGS on matched genes from the same patients; on the right, the percentage of point mutations detected by lbNGS and ttNGS on matched genes from the same patients. lbNGS, liquid biopsy NGS; NGS, next generation sequencing; ttNGS, tumour tissue NGS.
Second, we compared the ability of lbNGS and ttNGS of providing patients with an alteration-driven OncoKB-annotated recommendation. NGS on tumour tissue and LB were perfectly concordant for patients OncoKB level one recommendations (n=2); nevertheless, the chance of finding a ≥3b level recommendations was significantly higher in the lbNGS group (OR 11.0, 95% CI 1.6 to 473.4; McNemar’s χ2: p=0.006). Out of 29 patients with matched lbNGS and ttNGS, for 11 patients (37.9%) the LB provided an additional clinical benefit compared with tumour tissue NGS alone: these patients had no alterations detected, not OncoKB-annotated alterations detected or grade 4 alterations detected in the ttNGS but the LB could provide the patients with an OncoKB ≥3b recommendation for a targeted therapy (figure 4). Whereas for these 11 patients LB provided an additional clinical benefit compared with tumour tissue NGS alone, the opposite is true only for one patient (sample id A56133) (figure 4).
Figure 4.

Summary of indications of targeted therapy according to the MTB decision (second row), highest level OnoKB recommendations for LB-detected alterations (third row) and highest level OncoKB recommendations for tumour tissue-detected alterations (fourth row). lbNGS, liquid biopsy NGS; MTB, molecular tumour board; NGS, next generation sequencing; ttNGS, tumour tissue NGS.
Contributions of the molecular tumour board
In total, the MTB expressed an opinion for 173 unique patients. All the patients that underwent lbNGS and 150 out 195 (76.9%) of the ttNGS samples were evaluated by the MTB. For 72 (41.6%) of them, the MTB suggested initiation of a targeted therapy based on the molecular analysis of lbNGS, ttNGS or other available data. Forty-nine of these 72 patients (28.3% of the total) were suggested to enter a clinical trial; the remaining 23 patients were proposed either an expanded access programme or an off-label drug.
Of the 53 cases in the lbNGS cohort, the MTB could provide the patient with a treatment recommendation in 18 cases (34.0%) in which the liquid biopsy did not provide any ≥3b level recommendation (figure 5); moreover, among the 23 cases with a lbNGS ≥3b level recommendation, the MTB proposed an alternative treatment to the molecular-driven targeted therapy in almost one third of the patients (n=7; 30.4%). In the ttNGS cohort, the MTB could provide the patient with a treatment recommendation in 43 cases (28.6%). Even in this cohort, a remarkable proportion of patients with druggable molecular alterations were not considered for molecular driven target therapy—9 out of 24 (37.5%).
Figure 5.
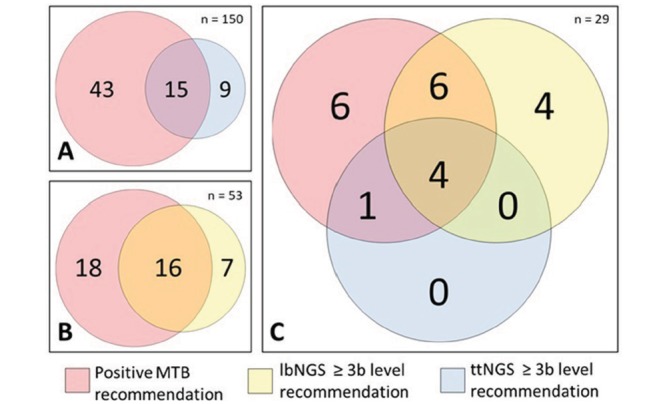
Euler-Venn diagram showing concordance of MTB decision with OncoKB recommendations for molecular driven targeted therapy; comparison of (A) MTB and ttNGS cohort, (B) MTB and lbNGS cohort and (C) matched MTB, lbNGS and ttNGS. lbNGS, liquid biopsy NGS; MTB, molecular tumour board; NGS, next generation sequencing; ttNGS, tumour tissue NGS.
In order to compare MTB, lbNGS and ttNGS recommendations in homogenous cohorts, we analysed the outcomes in 29 samples with matched MTB recommendations, lbNGS and ttNGS available (online supplementary table 3 lists NGS profiles and clinical features of the patients). In 17 cases (58.6%), the MTB, based on the molecular profile of each sample, expressed a positive opinion for the treatment of the patient with a targeted therapy—either with a standard of care treatment in a clinical trial or in an expanded access programme. MTB decisions had a measurable impact on the choice of the treatment: for 10 of the 29 patients (34.5%), the MTB expressed an opinion discordant with the result of the molecular analysis. In particular, for 4 of 16 patients (25.0%) with an OncoKB treatment evidence level ≥3b the MTB did not propose any targeted therapy; the MTB changed the treatment indications mostly because other therapeutic chances were actionable. At the same time, the MTB proposed to 6 of the 13 patients (46.2%) without any OncoKB treatment evidence level ≥3b to enter a clinicaltrial.
Discussion
In this paper, we report the impact of an interdisciplinary MTB on the treatment decision for patients with advanced solid cancers. Previous studies reported only MTB assessments without comparison to other standardised measures12 13; other papers compared the benefit of gene sequencing and subsequent target therapy.6 22 To our knowledge, we were the first to compare MTB decisions with a standardised set of evidence-based levels, thus making it possible to measure the impact of MTB decisions as a change of patient treatment.
In our experience, the majority of NGS-detected druggable alterations were not Food and Drug Administration/European Medicines Agency-approved for the treatment of the respective cancer type; this is consistent with previous reports.23 In this scenario, a balance has to be made between the expected benefit of a specific targeted therapy and other possible interventions—including palliative care. Our MTB could simultaneously evaluate all these aspects, melding the best evidence regarding each genetic alteration with a patient-specific approach. As a result, we could exploit one or more NGS panels to assign a remarkable number of patient to a clinical trial (28.3%), while, simultaneously, the use of molecular alterations for the patients’ clinical evaluation was implemented by the contribution of the MTB. Previous positive evidence for the use of NGS panels in improving patients outcomes in pan-cancer cohorts5 6 24 are somehow counteracted by negative results of the SHIVA trial.7 Intriguingly, in this last example, the function of the molecular board included only the choice of therapy for cases in which there were more than one actionable mutation. We believe that the MTB should, like in our case, include patients with a single alteration in order to indicate whether a druggable alteration is worth being targeted. It must be noted that our report represents a real world application of different technologies available for the discovery of druggable alterations in patients with cancer. It differs from previous trials as their aim was to assess an eventual superiority of alteration-driven treatment to standard of care. As these trials have opened a possibly revolutionising way of selecting treatments for patients with cancer, our aim was to describe the application of these methods in the real world and with the support of a multidisciplinary MTB. The relatively high number of patients considered for a clinical trial compared with previous studies is due to the facts that (1) some of the patients received a positive recommendation from the MTB but, subsequently, their clinical conditions deteriorated rapidly preventing them from participating and (2) for different patients a trial with immunotherapeutic agents was proposed.
In this heavily pretreated, advanced pan-cancer cohort, we found that lbNGS provided an additional clinical benefit compared with tumour tissue NGS alone in 37.9% of patients—mostly OncoKB ≥3 b. Despite a clear role of the differences between the panels used for lbNGS and ttNGS, this result was reinforced by the higher number of alterations detected by lbNGS in matched genes from matched patients. However, the sample size studied here was modest. Therefore, we feel that the difference in recommendations’ levels provided by the different NGS panels alone should not be regarded as a declaration of superiority of one of the two; it should, rather, be considered a further confirmation of feasibility and reliability of lbNGS panels in clinical practice. Also, we believe that the different timing of lbNGS and ttNGS in matched patients—ttNGS might have been performed on years old samples—and the selection bias for the use of lbNGS—patients with an inconclusive ttNGS were more likely to be chosen for the lbNGS—might have played a role in the sharp difference; nevertheless, we highlight once again that the value of this report resides in its adherence to the real-world practice, in which it is not important to evaluate two different platforms within comparable experimental conditions but rather it is crucial to provide the patient with the highest number possible of reliable alteration-driven therapeutic alternatives. Our report, along with other previous studies, poses the doubt as to whether the ttNGS has to be considered the gold standard and the lbNGS only a less sensitive method.
It has been reported that the lack of standard criteria to define actionability is a major challenge in the implementation of MTBs.8 Annotating evidence-based gene alteration panels could allow us to compare different NGS techniques outcomes both between them and with our MTB decisions. The choice of the OncoKB evidence level was made after an internal consultation; we do not assess that this list is superior to others that have been published.25 We encourage that other groups, in the future, rely on this approach to measure the effect of MTB implementation.
As an incidental finding, we report a positive and statistically significant correlation between the amount of ctDNA and the number alterations detected in each patient. We do not know whether this could depend on a real increase in ctDNA in highly mutated tumour or in a lack of sensitivity of the detection method in the presence of lesser amount of ctDNA. If the latter hypothesis was true, this result would suggest that patients with lower ctDNA levels would have less chance to have an alteration detected and therefore to enter a clinical trial. Nevertheless, we believe that this conclusion must be confirmed by an ad hoc study.
A major limitation of this study was the difference of the lbNGS and ttNGS cohorts; as such, in only 29 patients matched NGS panels could be compared. However, we focused on impact on treatment decisions and OncoKB levels of actionability as a superior standard for comparison between assays than gene-level concordance. Moreover, MTB evaluation for the treatment assessment lacked a control representing routine clinical and widespread practice for this kind of decisions—such as a blinded single clinical oncologist evaluation. The use of different commercial panels for ctDNA NGS and tumour tissue DNA NGS, while on one side represents a weak point as different number and type of genes were analysed for each patient, can be considered as a strength as these platforms have been extensively validated and it eases eventual future comparisons.
Conclusion
In conclusion, the use of MTB can allow a remarkable number of patient to undergo enrolment in a clinical trial or treatment with a targeted therapy while, simultaneously, delineating precise patient-specific therapeutic strategies.
Footnotes
Contributors: PM was responsible for the analysis and interpretation of data and writing of the paper. CR was responsible for the conception and design of the work. CR and RS contributed to the final approval of the version published. All authors contributed to the acquisition of data, drafting the work or revising it critically for important intellectual content.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Obtained.
Ethics approval: Committee for Medical Ethics UZA-UAntwerp (CME).
Provenance and peer review: Not commissioned; internally peer reviewed.
References
- 1. Van Allen EM, Wagle N, Stojanov P, et al. . Whole-exome sequencing and clinical interpretation of formalin-fixed. paraffin-embedded tumor samples to guide precision cancer medicine 2014;20:682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwaederle M, Chattopadhyay R, Kato S, et al. . Genomic alterations in circulating tumor DNA from Diverse Cancer Patients Identified by Next-Generation Sequencing. Cancer Res 2017;77:5419–27. 10.1158/0008-5472.CAN-17-0885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aisner D, Sholl LM, Berry LD, et al. . Effect of expanded genomic testing in lung adenocarcinoma (LUCA) on survival benefit: The Lung Cancer Mutation Consortium II (LCMC II) experience. J Clin Oncol 2016;34:11510. [Google Scholar]
- 4. Gutierrez ME, Choi K, Lanman RB, et al. . Genomic profiling of advanced non-small cell lung cancer in community settings: gaps and opportunities. Clin Lung Cancer 2017;18:651–9. 10.1016/j.cllc.2017.04.004 [DOI] [PubMed] [Google Scholar]
- 5. Belin L, Kamal M, Mauborgne C, et al. . Randomized phase II trial comparing molecularly targeted therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer: cross-over analysis from the SHIVA trial. Ann Oncol 2017;28:590–6. 10.1093/annonc/mdw666 [DOI] [PubMed] [Google Scholar]
- 6. Massard C, Michiels S, Ferté C, et al. . High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 Trial. Cancer Discov 2017;7:586–95. 10.1158/2159-8290.CD-16-1396 [DOI] [PubMed] [Google Scholar]
- 7. Le Tourneau C, Delord JP, Gonçalves A, et al. . Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–34. 10.1016/S1470-2045(15)00188-6 [DOI] [PubMed] [Google Scholar]
- 8. van der Velden DL, van Herpen CML, van Laarhoven HWM, et al. . Molecular tumor boards: current practice and future needs. Ann Oncol 2017;28:3070–5. 10.1093/annonc/mdx528 [DOI] [PubMed] [Google Scholar]
- 9. El Saghir NS, Charara RN, Kreidieh FY, et al. . Global practice and efficiency of multidisciplinary tumor boards: results of an american society of clinical oncology international survey. J Glob Oncol 2015;1:57–64. 10.1200/JGO.2015.000158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charara RN, Kreidieh FY, Farhat RA, et al. . Practice and impact of multidisciplinary tumor boards on patient management: a prospective study. J Glob Oncol 2017;3:242–9. 10.1200/JGO.2016.004960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newman EA, Guest AB, Helvie MA, et al. . Changes in surgical management resulting from case review at a breast cancer multidisciplinary tumor board. Cancer 2006;107:2346–51. 10.1002/cncr.22266 [DOI] [PubMed] [Google Scholar]
- 12. Lane BR, Bissonnette J, Waldherr T, et al. . Development of a center for personalized cancer care at a regional cancer center: feasibility trial of an institutional tumor sequencing advisory board. J Mol Diagn 2015;17:695–704. 10.1016/j.jmoldx.2015.07.003 [DOI] [PubMed] [Google Scholar]
- 13. Harada S, Arend R, Dai Q, et al. . Implementation and utilization of the molecular tumor board to guide precision medicine. Oncotarget 2017;8:57845–54. 10.18632/oncotarget.18471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rolfo CD, Machado Coelho A, Van Dam P, et al. . Multidisciplinary molecular tumour board: a tool to improve clinical practice and selection accrual for clinical trials in cancer patients. Ann Oncol 2016;27:54PD 10.1093/annonc/mdw363.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guardant health. Guardant 360 Gene Panel. http://guardanthealth.com/medical-professionals/#gene-panel (accessed 10th Oct 2017).
- 16. Lanman RB, Mortimer SA, Zill OA, et al. . Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One 2015;10:e0140712 10.1371/journal.pone.0140712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang Q, Henry NL, Paoletti C, et al. . Comparative analysis of circulating tumor DNA stability In K3EDTA, Streck, and CellSave blood collection tubes. Clin Biochem 2016;49:1354–60. 10.1016/j.clinbiochem.2016.03.012 [DOI] [PubMed] [Google Scholar]
- 18. Medina Diaz I, Nocon A, Mehnert DH, et al. . Performance of streck cfDNA blood collection tubes for liquid biopsy testing. PLoS One 2016;11:e0166354 10.1371/journal.pone.0166354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chakravarty D, Gao J, Phillips SM, et al. . OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017;2017:1–16. 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. R Core Team. R: A language and environment for statistical computing, 2018. [Google Scholar]
- 21. Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag, 2009. [Google Scholar]
- 22. Janne P, Garber J, MacConaill L, et al. . Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Nature medicine 2012;18:6373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwaederle M, Daniels GA, Piccioni DE, et al. . On the road to precision cancer medicine: analysis of genomic biomarker actionability in 439 patients. Mol Cancer Ther 2015;14:1488–94. 10.1158/1535-7163.MCT-14-1061 [DOI] [PubMed] [Google Scholar]
- 24. Von Hoff DD, Stephenson JJ, Rosen P, et al. . Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–83. 10.1200/JCO.2009.26.5983 [DOI] [PubMed] [Google Scholar]
- 25. Kurnit KC, Bailey AM, Zeng J, et al. . "Personalized Cancer Therapy": a publicly available precision oncology resource. Cancer Res 2017;77:e123–6. 10.1158/0008-5472.CAN-17-0341 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2018-000398supp002.pdf (296.3KB, pdf)
esmoopen-2018-000398supp001.pdf (282.1KB, pdf)